

 , Ludmila Nedosugova 2, Tatiana Kirichenko 1, Vasily Sukhorukov 1, Alexandra Melnichenko 1, Alexander Orekhov 1,*
, Ludmila Nedosugova 2, Tatiana Kirichenko 1, Vasily Sukhorukov 1, Alexandra Melnichenko 1, Alexander Orekhov 1,*1 Laboratory of Angiopathology, Institute of General Pathology and Pathophysiology, 125315 Moscow, Russia
2 Sechenov First Moscow State Medical University (Sechenov University), 119991 Moscow, Russia
Abstract
The pathogenesis of type 2 diabetes mellitus (T2DM) is based on the development of insulin resistance, which is a disruption to the ability of the tissues to bind to insulin, leading to a general metabolic disorder. Mitochondria are the main participants in cellular energy metabolism, meaning their dysfunction is associated with the development of insulin resistance in T2DM. Mitochondrial function is affected by insulin resistance in various tissues, including skeletal muscle and the liver, which greatly influence glucose homeostasis throughout the body. This review studies mitochondrial dysfunction in T2DM and its impact on disease progression. In addition, it considers the causes underlying the development of mitochondrial dysfunction in T2DM, including mutations in the mitochondrial genome, mitochondrial DNA methylation, and other epigenetic influences, as well as the impact of impaired mitochondrial membrane potential. New therapeutic strategies for diabetes that have been developed to target the mitochondria will also be presented.
Keywords
- type 2 diabetes
- insulin resistance
- mitochondrial dysfunction
- glucose
Diabetes mellitus (DM) is a chronic metabolic disease caused by insufficient production and secretion of insulin, and in the case of type 2 diabetes mellitus (T2DM), an inability of the tissues to respond adequately to insulin. The result of these changes is a high glucose concentration in the blood, eventually leading to various complications [1]. The World Health Organization (WHO) defines diabetes as “a metabolic disorder of multiple etiologies characterized by chronic hyperglycemia with impaired carbohydrate, fat, and protein metabolism as a result of defects in insulin secretion” [2]. The pathogenesis/etiology of this form of diabetes is complex and includes many known and unknown factors, which can finally be described as a combination of genetic (polygenic) predisposition and strong environmental influences [3].
Type 2 diabetes is the most common form of diabetes and occurs in 90% of diabetes cases. The main pathological sign associated with the development of T2DM is the occurrence of insulin resistance, which initially leads to an increase in insulin production that binds to excess free glucose; however, due to insulin resistance, this is ineffective and results in an increase in blood glucose concentration and cell starvation. The risk group for type 2 diabetes is people over 40 years of age. Recently, there has been an increase in the prevalence of T2DM in young people due to a general decrease in physical activity among the population and an increase in adolescent obesity.
Subsequently, T2DM is one of the most common non-communicable diseases today. Thus, according to estimates, in 2019, the number of adult patients diagnosed with type 2 diabetes was about 463 million people, which is more than 9% of the total adult population worldwide. By 2030, this number is projected to increase to 578 million, which is 10.2% of the total global adult population, and by 2045 it is projected to increase to 700 million, which represents 10.9% of the total adult population of the world [4]. The incidence and prevalence of T2DM varies by geographic region: More than 80% of T2DM patients are residents of developing countries. This leads to additional barriers to providing these patients with therapies. T2DM is associated with an increased risk of mortality, which is 15% higher than in healthy people [5].
Hyperglycemia has also arisen in TDM2 patients and is the cause of several severe complications. Moreover, T2DM is associated with cardiovascular disease, blindness, kidney failure, and even lower limb amputation. Acute complications in T2DM are manifested in the form of hypoglycemia, diabetic ketoacidosis, and diabetic coma. Annually, up to 5% of middle-aged patients with T2DM receive a diagnosis associated with the occurrence of cardiovascular diseases [5].
Mitochondria are indispensable cellular organelles for cellular energy metabolism since they possess a primary role in the oxidation of glucose and lipids and the synthesis of adenosine triphosphate (ATP). Recently, mitochondrial dysfunction has been considered in the context of secondary T2DM complications [6]. Since mitochondrial dysfunction is present in various tissues and contributes to the pathogenesis and complications of diabetes in multiple ways, mitochondrial-targeted therapeutic intervention may represent a promising approach to treating various causative and secondary defects of T2DM simultaneously.
Initially, the role of mitochondria in cellular metabolism should be considered since changes in metabolism are key steps in determining the role mitochondria play in the pathogenesis of T2DM. The importance of mitochondria in processes such as oxidative phosphorylation, glutaminolysis, fatty acid oxidation, and pyruvate oxidation is discussed in detail in this section.
The primary function of mitochondria is the production of ATP through oxidative phosphorylation. Oxidative phosphorylation is a process during which successive redox reactions are performed alongside enzymes that form an electron transport chain (ETC) in the inner mitochondrial membrane, a result of which is the formation of a proton gradient and the generation of energy during the reverse proton transfer, which is stored in the form of ATP. Electron carriers (nicotinamide adenine dinucleotide hydrogene (NADH) and flavin adenine dinucleotide dihydrogene (FADH2)) are formed in the mitochondrial matrix during the tricarboxylic acid (TCA) cycle and precede oxidative phosphorylation [7]. These carriers transfer electrons to the ETC, consisting of large protein complexes—enzymes (I–IV)—and two additional carriers: Coenzyme Q and cytochrome C. Complex I is the first and largest of the complexes, consisting of 45 main subunits. Complexes I and II mediate the transfer of two electrons from electron carriers NADH/FADH2, respectively, to coenzyme Q, which carries mobile electrons. The latter can also receive electrons from the breakdown of many nutrients, for example, during the oxidation of fatty acids. Complex III is an adapter that receives two electrons from the reduced coenzyme Q and transfers one electron to cytochrome C. Complex IV completes the respiratory chain by accepting electrons from cytochrome C to reduce oxygen to water completely [8]. Redox reactions in the ETC lead to conformational changes in protein complexes, which facilitate the transfer of protons from the matrix into the intermembrane space to produce a transmembrane potential [9]. As a result of the proton gradient, the energy in the transmembrane potential is used by ATP synthase to form ATP from adenosine diphosphate (ADP). In addition to the formation of ATP, additional processes depend on the transmembrane potential, including the importation of mitochondrial proteins through the inner mitochondrial membrane [7].
Glutamine is considered the most abundant amino acid in nature. The process of glutamine uptake by cells occurs with the help of special transmembrane transporter proteins [10]. Once in cells, glutamine is used in translational processes to synthesize proteins and nucleotides, creating a chemical potential for the uptake of other metabolites. [11]. Thus, the essential amino acid leucine is absorbed by the cell through the LAT1 protein carrier, while the secretion of glutamine into the intercellular space is also required for the transfer to occur [12].
The greatest amount of glutamine inside the cell is transformed into glutamate
and ammonium ions during a reaction involving the enzyme glutaminase. Part of the
mitochondrial glutamate is exported to the cytosol, where it attaches to the
glutamate pool, and the other part of the glutamate will be further converted to
Fatty acid (FA) oxidation is a mitochondrial aerobic process that breaks down
fatty acids into acetyl-CoA. Fatty acids are involved in this pathway as CoA
derivatives through nicotinamide adenine dinucleotide (NAD) and flavin adenine
dinucleotide (FAD) [13]. The importation of mitochondrial FAs is a defining step
in fatty acid oxidation (FAO) and demonstrates how metabolic compartmentalization
adapts to the cellular state. Since long-chain FAs cannot cross mitochondrial
membranes, the mitochondria have developed a complex set of reactions and
transport activities that allow FAs to gain access to the mitochondrial
FFAs are extremely important in cellular homeostasis since FAs form the main
energy substrate of cytotoxic T cells, necessary for their survival and the
functioning of CD8
Pyruvate is formed from a number of sources, depending on nutrient and tissue availability, including the main source of glucose and lactate. In healthy tissue, the fate of pyruvate depends on the availability of oxygen and the respiratory capacity of mitochondria [17]. In a state of sufficient oxygen levels, pyruvate is synthesized during glycolysis and passes through the IMM via the mitochondrial pyruvate transporter (MPC) [18]. Pyruvate is further catabolized within the mitochondria in the TCA cycle. During hypoxia, mitochondrial respiration is suppressed, causing cells to adaptively transfer electrons to pyruvate through lactate dehydrogenase (LDH), forming lactate in the cytosol [10]. This pathway is involved in the muscles, the intestines, and the kidneys during exercise. Within mitochondria, pyruvate is introduced into the TCA cycle through reactions involving two enzymes: the pyruvate dehydrogenase complex (PDC), which is responsible for the synthesis of acetyl-CoA, and pyruvate carboxylase (PC), which catalyzes oxaloacetate [19].
Having understood the role of mitochondria in normal cellular metabolism, it is important to note the metabolic disorders that occur in T2DM and their consequences. Subsequently, this will make it possible to determine where mitochondrial dysfunction occurs in the pathogenesis of T2DM.
Increased glucose production in the liver is one of the reasons that patients with T2DM develop hyperglycemia. Insulin controls glucose production in the liver and promotes glucose utilization in the skeletal muscles [20]. In patients with T2DM, there is an increase in gluconeogenesis and, to a lesser extent, glycogenolysis in the liver. Factors contributing to the acceleration of glucogenesis are the increased production of lactate, alanine, and glycerol, which are gluconeogenic precursors, as well as hyperglucagonemia, accompanied by an increase in fatty acid oxidation. In addition, disruption of postprandial glucose homeostasis is associated with reduced suppression of hepatic glucose production following carbohydrate intake [21].
An increase in glycogenolysis and a decrease in hepatic glucose uptake by
glucagon result in a hyperglycemic phenotype, which is determined by insulin
deficiency and insulin resistance (IR). In healthy individuals, fasting plasma
glucose levels remain unchanged throughout the day. This constant glucose level
is highly dependent on the balance between glucose produced in the liver and
glucose absorbed by peripheral tissues [21]. In T2DM, fasting hyperglycemia
correlates to a lesser extent with increased hepatic glucose production due to
decreased hepatic sensitivity to insulin. However, this is largely due to a
reduction in insulin secretion and an increase in glucagon secretion. Although
basal levels of immunoreactive insulin and glucagon may be normal in T2DM
patients, testing the islet function at appropriate glucose levels reveals
various abnormalities in insulin and glucagon secretion due to decreased
The dysfunction of
In the later stages of the disease, there is a disturbance in the second phase
of insulin secretion, although this disturbance can be reduced by introducing
strict glycemic control. This phenomenon is called desensitization or glucose
toxicity of
Other defects in the
Metabolic changes in patients with T2DM are characterized by a high level of
glucogenesis, yet a decrease in glycogenolysis. In patients with IR, there is a
disruption in glucose transport, as well as the transmission of insulin signals
in tissues, which is accompanied by the production of inflammatory markers in the
adipose tissue [27]. The mediators that transmit signals to the
In addition to dysglycemia, the concomitant conditions of IR are dyslipidemia, hyperinsulinemia, obesity, and arterial hypertension, which means IR is a key sign of the development of metabolic syndrome and CVD. Moreover, it is believed that genetic predisposition worsens the disease in patients with T2DM. One of the most important etiological causes of T2DM is central obesity, which is thought to be exacerbated by genetic predisposition; however, it is worth noting that diet and exercise can reduce the effects of IR [29].
The main regulators of glucose metabolism in skeletal muscles are hexokinase, glycogen synthase, and the insulin-dependent glucose transporter GLUT4. At the same time, the emerging disorders of glycogen production in skeletal muscles play an important role in the development of IR in T2DM patients [20]. Impairment of the ability of skeletal muscles to bind circulating insulin is one of the factors in the development of IR, which can develop 10–20 years before the immediate diagnosis of T2DM [21].
Having identified general issues regarding the functions of mitochondria in cellular metabolism and metabolic disorders in T2DM, it is advisable to consider the role that the development of mitochondrial dysfunction in T2DM directly plays on energy metabolism in different tissues.
Insulin-resistant patients may have overt type 2 diabetes when pancreatic
Diabetes is characterized by persistently elevated blood sugar levels, which promote increased production of reactive oxygen species (ROS) through various processes, including glucose auto-oxidation, polyol pathway activation, and the formation of advanced glycation end products (AGEs) [31]. As soon as glucose arrives in the cells, it is oxidized by either the glycolytic pathway or the pentose phosphate pathway, which produces biosynthetic molecules and nicotinamide adenine dinucleotide phosphate (NADPH). ROS are produced during glycolysis, although the antioxidant defense mechanism in the cell normally effectively neutralizes them. Conversely, overly high blood glucose levels cause an increase in radical generation, which inhibits antioxidant mechanisms and damages DNA. Glyceraldehyde-3-phosphate dehydrogenase (GAPD) is inactivated by an increase in the concentration of DNA repair enzymes such as poly-ADP-ribose polymerase-1 (PARP-1). This causes an accumulation of metabolites, including glyceraldehyde-3-phosphate (GAP), glucose 6-phosphate (G-6-P), and fructose 6-phosphate (F-6-P). Increased concentrations of these metabolites have an impact on how oxidative stress progresses: GAP activates protein kinase C (PKC), whereby G-6-P and F-6-P can follow the polyol route, and auto-oxidation of GAP and G-6-P can result in the generation of AGE precursors [32]. Notably, as the quantity of glucose increases, the hexokinase enzyme gets saturated and cannot catalyze the synthesis of G-6-P. As a result, aldose reductase converts glucose to sorbitol, which sorbitol dehydrogenase (SDH) then further converts to fructose. Excess NADPH is used in this process, which provides GPx with a substrate to make glutathione (GSH) [33]. Thus, oxidative stress is also facilitated by the suppression of antioxidant enzymes in this pathway. Furthermore, increased fructose synthesis resulting from the activation of SDH levels in hyperglycemic circumstances leads to PKC activation and oxidative stress.
Elevated levels of ROS activate the mitochondrial protein uncoupling protein 2 (UCP2), which leads to
the leakage of protons through the inner mitochondrial membrane. This, in turn,
leads to a decrease in ATP production, which is necessary for insulin secretion
[30]. On the other hand, ROS causes damage to the phospholipids in the
mitochondrial membrane, which increases its permeability and promotes the leakage
of cytochrome C into the cytosol, ultimately activating the apoptosis of
pancreatic
Impaired mitochondrial function has been demonstrated in the muscles with
impaired insulin resistance in type 2 diabetic patients [35]. It is believed that
defective mitochondrial fatty acid metabolism in skeletal muscle affects insulin
signaling pathways, leading to insulin resistance [36]. Disruption of
mitochondrial fatty acid
The liver plays a critical role in the development of insulin resistance in T2DM
[38]. Some studies indicate that the resulting mitochondrial dysfunction in liver
cells may directly cause hepatic insulin resistance [39]. For example, a decrease
in the level of mitochondrial
Adipose tissue is an endocrine organ that is important in energy metabolism
[40]. The main regulators of adipose tissue metabolism are compounds called
adipocytokines, the best known of which are adiponectin, TNF-
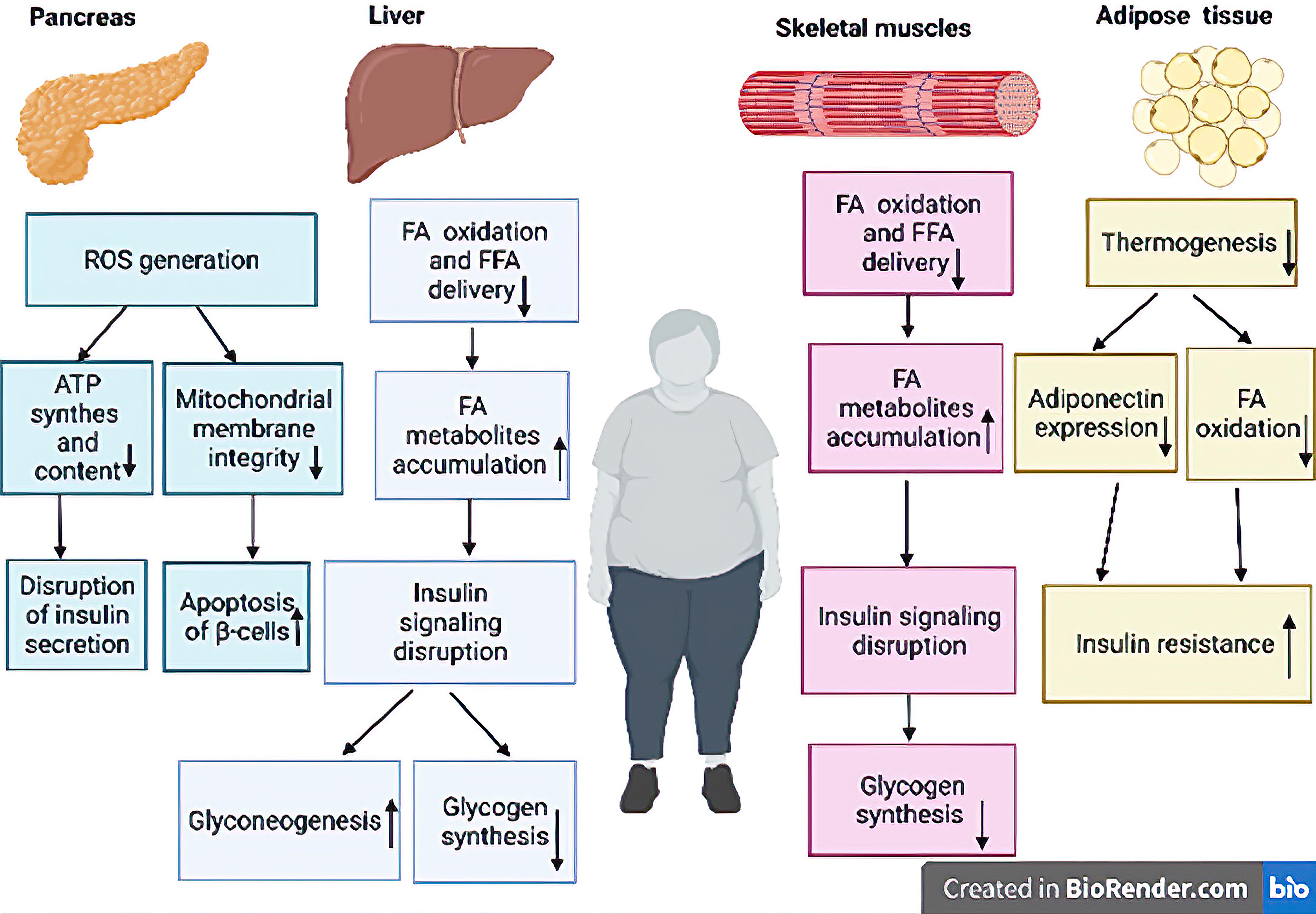 Fig. 1.
Fig. 1.Pathogenesis of T2DM based on mitochondrial dysfunction in different tissues. T2DM, type 2 diabetes mellitus; ROS, reactive oxygen species; ATP, adenosine triphosphate; FA, fatty acid; FFA, free fatty acid.
The occurrence of mitochondrial dysfunction can affect the pathogenesis of
diabetes mellitus in different tissues differently, which leads to the onset of
certain adverse events. Thus, in the pancreas, mitochondrial dysfunction is one
of the factors leading to impaired insulin secretion and increased apoptosis of
Having considered the significance of mitochondrial dysfunction in the pathogenesis of T2DM, the question arises regarding the factors that cause mitochondrial dysfunction to develop. These factors are carefully analyzed in the following section.
Mitochondrial dysfunction can be directly related to a change in the state of some factors immediately associated with mitochondrial function. Hence, a previous study [44] analyzed a culture of monocyte cells taken from patients with T2DM and found alterations in both the morphology of the mitochondria—a decrease in their volume resulting from an increase in mitochondrial division; their functional state associated with the hyperpolarization of the mitochondrial membrane. Chronic hyperglycemia and insulin resistance are thought to alter the mitochondrial structure and function, leading to an increase in small, hyperpolarized mitochondria that produce elevated levels of ROS, resulting in reduced ATP synthesis productivity and increased activation of inflammation. In another study [45], it was shown that even the treatment of myocardial mitochondria in patients with T2DM with diazoxide (which is an activator of potassium channels) did not lead to the depolarization of the mitochondrial membrane, in contrast to the negative control, which could possibly be associated with mitochondrial potassium channel dysfunction.
Based on the fact that ATP production is a necessary source of energy, including
for the secretion of insulin, and is provided by the work of five large protein
complexes consisting of 90 subunits, 13 of which are encoded by the mitochondrial
gene, the depletion of mtDNA in
A number of studies have shown that the degree of DNA methylation in the nuclear-encoded ETC and OXPHOS genes, cytochrome c oxidase polypeptide 7A1, NADH dehydrogenase 1 beta-subcomplex subunit 6, and PPARGC1A negatively correlates with changes in gene expression in insulin-resistant skeletal muscles [51]. Another study showed that mitochondrial DNA can also be methylated in a manner similar to prokaryotic DNA methylation [52]. In a rat model with type 2 diabetes, it was found that severe retinopathy was associated with an increase in the proportion of methylated mitochondrial DNA [53]. Additional epigenetic changes may be associated with non-coding RNAs. Thus, the study [54] showed that some LncRNAs can cause changes in mitochondrial homeostasis in diabetic retinopathy.
Based on the above analysis, it is clear that mitochondrial dysfunction is a significant factor in the development of T2DM. In this regard, several therapeutic strategies have been proposed to restore mitochondrial function in T2DM.
One of the broadest classes of mitochondria-targeted therapeutic agents includes compounds whose activity is associated with modulating the activity of ETC components. Among them, imeglimin is the most promising. Imeglimin is a newly developed antidiabetic compound that restores mitochondrial function in various organs of patients, including skeletal muscle, liver, and pancreas [55]. However, the exact mechanism of action of imeglimin on mitochondria remains unknown. In a number of studies on hepatocytes, it was found that imeglimin acts as a competitive inhibitor of ETC complex I and helps restore complex III activity, thus, improving the efficiency of oxidative phosphorylation [56]. Improvement in mitochondrial function under the action of imeglimin has also been confirmed in studies demonstrating the prevention of mitochondrial permeability-induced cell death in endothelial and islet cell models [57].
Another potential mitochondrial target for treating metabolic disorders is the mitochondrial pyruvate transporter (MPC), which mediates the importation of pyruvate into the mitochondrial matrix [58]. Impaired pyruvate uptake in the mitochondria is associated with mitochondrial dysfunction and is noted in the pathogenesis of T2DM.
Thiazolidinediones (TZDs), also called glitazones, are a class of peroxisomal
proliferation-activated receptor (PPAR)-
Recently, significant research has focused on developing molecules capable of
enhancing the transcription of nuclear and mitochondrial genes encoding subunits
in the ETC chain to improve the efficiency of oxidative phosphorylation. A
promising strategy is targeting peroxisomal proliferator-activated receptors
(PPARs), which are a group of transcription factors associated with regulating
various cellular functions, including mitochondrial metabolism and energy
homeostasis. Among the downstream PPAR targets is the PPAR-
Sirtuin 1 (Sirt1) is another potential target for modulating PGC-1
Since oxidative stress has been shown to play a role in the pathogenesis of T2DM, as already described in the previous paragraphs, the administration of antioxidants can potentially be used to treat T2DM. To date, several clinical studies have explored the use of antioxidants in treating T2DM, such as quercetin, resveratrol, and vitamin C. However, the results of these studies were found to be contradictory, which may be due to the difficulty in assessing the antioxidant capacity in insulin-sensitive tissues, such as skeletal muscle and adipose tissue, meaning it requires more research [63]. At the same time, pharmaceuticals aimed at increasing antioxidant protection in cells, such as NADPH oxidase inhibitors, xanthine oxidase inhibitors, and superoxide dismutase (SOD) mimetics, may also be promising for the treatment of T2DM [63]. Additionally, mitochondrial-targeting antioxidants, such as MitoQ and thyron, are worth investigating since they can directly penetrate the mitochondria and bind ROS at their site of origin, thus, preventing the development of oxidative stress at its inception [64].
A generalized scheme of therapeutic strategies aimed at restoring mitochondrial function in T2DM is presented in Table 1.
| Therapeutic strategy | Targeting |
| OxPhos modulation | ETC complex III functional recovery |
| Inhibition of complex I ETC | |
| Decline of ROS production | |
| Metabolites transport control | Inhibition of MPC |
| Mitochondrial biogenesis | Activation of PPAR |
| Activation of PGC-1 | |
| Activation of Sirt1 | |
| Oxidative stress inhibition | Direct binding of ROS in the cell |
| Direct binding of ROS in the mitochondria | |
| SOD mimicking | |
| Inhibition of NADPH oxidase | |
| ETC, electron transport chain; MPC, mitochondrial pyruvate transporter; PPAR,
peroxisome proliferator-activated receptor; PGC-1 | |
Recently, many studies have focused on studying therapeutic strategies that target mitochondria. The most promising strategies are modulators of oxidative phosphorylation, compounds that control the transport of metabolites, and agents aimed at mitochondrial biogenesis. Among the modulators of oxidative phosphorylation, the most actively studied drug is imeglimin, which is currently undergoing clinical trials [63]. It has been shown that treatment with imeglimin led to an increase in the redox potential of mitochondria in primary hepatocytes without adverse effects on mitochondrial respiration [63]. In this regard, imegimine is a promising antidiabetic drug with potential properties to improve the efficiency of oxidative phosphorylation, possibly through inhibiting the mitochondrial permeability transition pore (mPTP), and antioxidant activity. However, the exact mechanism of action remains to be elucidated.
Another described a potential strategy for treating T2DM is PPAR agonists. The most studied drug is GW501516, which mimics the effects of exercise by activating the same signaling pathways. However, during clinical trials, a high carcinogenicity of this drug was revealed. Despite the negative effect, this may be a promising approach in the development of new drugs for treating diabetes, with several PPAR agonists currently being tested as potential agents, including thiazolidinediones [37].
In addition to the strategies already described, one can also add potential agents that target mitochondria, namely, modulators of mitochondrial membrane properties, coenzyme Q10 (CoQ10) analogs, mitochondria-associated endoplasmic reticulum (ER) membrane modulators (MAM), and AMP kinase protein (AMPK) activators. Despite the effectiveness of these drugs in vitro, in vivo studies remain ineffective, which casts doubt on the viability of these strategies. In any case, further studies on the mechanisms of action and cell fate are needed in general [65].
Further, many issues on this topic that are not discussed in this article
require careful study. For example, investigating how nutrition and exercise
affect mitochondrial function in type 2 diabetes is very important for
understanding the pathogenesis of T2DM. The literature has examined how adopting
a low-calorie diet and boosting physical activity in T2DM patients [66].
According to the early findings, indicators linked to better mitochondrial
dynamics and function may be successfully modulated by both exercise and calorie
restriction. This was in connection with enhanced regulation of mitochondrial
oxidative capacity and reduced generation of reactive oxygen species in
individuals suffering from type 2 diabetes or related metabolic disorders. It
also was shown in a study [67] that endurance exercise improved mitochondrial
function to effectively use glucose oxidation by raising mitochondrial capacity
in skeletal muscle cells, mitochondrial oxidase activity, and regulating
mitochondrial lipid content. Additionally, it has been demonstrated that exercise
increases the expression of PGC1-
T2DM is the most common endocrine disease in humans and currently affects an
enormous proportion of the public. Thus, understanding the mechanisms involved at
each stage in the development and complications of T2DM is critical to
preventing, controlling, treating, and modifying the pathophysiology of T2DM
complications. Considering the main role of mitochondrial dysfunction in the
development of a number of metabolic disorders, new therapeutic strategies have
been developed in recent years to regulate mitochondrial function and biogenesis.
These approaches may be useful in restoring insulin action and pancreatic
AO, AB and VS designed the review plan. AM, LN and TK provided help and advice as scientific consultants. AB and LN wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity.
Not applicable.
Not applicable.
This work was supported by the Russian Science Foundation (Grant #23-45-00031).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.