




 , Evgeny I. Barabanshchikov 3,†
, Evgeny I. Barabanshchikov 3,†1 Laboratory of Deep Sea Research, A.V. Zhirmunsky National Scientific Center of Marine Biology, Far Eastern Branch, Russian Academy of Sciences (NSCMB FEB RAS), 690041 Vladivostok, Russia
2 Chair of Water Biological Resources and Aquaculture, Far Eastern State Technical Fisheries University, 690087 Vladivostok, Russia
3 Laboratory of Biological Resources Continental Reservoirs and Fishes Estuarine Systems, Pacific branch of Russian Federal Research Institute of Fisheries and Oceanography (TINRO), 690950 Vladivostok, Russia
†These authors contributed equally.
Abstract
Background: The mitochondrial genome is a powerful tool for exploring and confirming species identity and understanding evolutionary trajectories. The genus Cambaroides, which consists of freshwater crayfish, is recognized for its evolutionary and morphological complexities. However, comprehensive genetic and mitogenomic data on species within this genus, such as C. wladiwostokiensis, remain scarce, thereby necessitating an in-depth mitogenomic exploration to decipher its evolutionary position and validate its species identity. Methods: The mitochondrial genome of C. wladiwostokiensis was obtained through shallow Illumina paired-end sequencing of total DNA, followed by hybrid assembly using both de novo and reference-based techniques. Comparative analysis was performed using available Cambaroides mitochondrial genomes obtained from National Center for Biotechnology Information (NCBI). Additionally, phylogenetic analyses of 23 representatives from three families within the Astacidea infraorder were employed using the PhyloSuite platform for sequence management and phylogenetic preparation, to elucidate phylogenetic relationships via Bayesian Inference (BI), based on concatenated mitochondrial fragments. Results: The resulting genome, which spans 16,391 base pairs was investigated, revealing 13 protein-coding genes, rRNAs (12S and 16S), 19 tRNAs, and a putative control region. Comparative analysis together with five other Cambaroides mitogenomes retrieved from GenBank unveiled regions that remained unread due to challenges associated with the genome skimming technique. Protein-coding genes varied in size and typically exhibited common start (ATG) and stop (TAA) codons. However, exceptions were noted in ND5 (start codon: GTG) and ND1 (stop codon: TAG). Landscape analysis was used to explore sequence variation across the five available mitochondrial genomes of Cambaroides. Conclusions: Collectively, these findings reveal variable sites and contribute to a deeper understanding of the genetic diversity in this genus alongside the further development of species–specific primers for noninvasive monitoring techniques. The partitioned phylogenetic analysis of Astacidea revealed a paraphyletic origin of Asian cambarids, which confirms the data in recent studies based on both multilocus analyses and integrative approaches.
Keywords
- mitochondrial genome
- Cambaroides wladiwostokiensis
- genome skimming
- landscape variation
- phylogenetics
- partitioned analysis
Molecular genetic techniques, such as polymerase chain reaction (PCR), DNA barcoding, and genomic sequencing, have significantly impacted the processes of species identification and classification. These techniques allow researchers to directly examine the genetic material of organisms, thereby providing a precise method to differentiate between closely related species, understand their evolutionary relationships, and perform accurate classifications [1, 2, 3, 4, 5]. Furthermore, they have transformed our understanding of mitochondrial genomes, allowing us to investigate their intricate details with unparalleled precision [6, 7, 8, 9, 10]. Within this context, the genus Cambaroides (Decapoda: Astacidea) has emerged as a focal point in research. Researchers are keen to use genetic markers to decipher the complex web of species identities and their interrelationships, thereby positioning this genus as a distinct avenue of study. Until now, only seven species have been described within this genus: the “Daurian crayfish” C. dauricus (Pallas, 1773), the “Schrenck’s crayfish” C. schrenckii (Kessler, 1874), the “Korean crayfish” C. similis (Koelbel, 1892), the “Japanese crayfish” C. japonicus (De Haan, 1841), the “Sakhalin crayfish” C. sachalinensis (Birstein et Winogradow, 1934), the “Vladivostok crayfish” C. wladiwostokiensis (Birstein et Winogradow, 1934), and the “Kozhevnikov crayfish” C. koshewnikowi (Birstein et Winogradow, 1934). However, their range is limited in the north by the Amur River basin, in the east by Sakhalin Island and the northern part of the Japanese islands, in the west by the lower Selenga River basin (Lake Baikal basin), and in the south by the southern part of the Korean Peninsula [11, 12], in addition to personal observations. Notably, they also serve as intermediate hosts for the trematode Paragonimus westermani ichunensis, the causative agent of paragonimiasis—a severe parasitic disease [13].
These seven species of East Asian freshwater crayfish can be subdivided into three groups, each with unique ecological characteristics: The Daurian crayfish group (including Daurian, Japanese, Korean, and Vladivostok crayfish), which consists of stenobiotic rheophilic species that exclusively inhabit clean waters and can serve as indicators of a water body’s purity; the Schrenk crayfish group (Schrenk’s crayfish, Sakhalin crayfish), which are eurybiotic species, capable of inhabiting even polluted waters, small puddles, and swamps; Kozhevnikov’s crayfish are ecologically distinct, are found only in the lower part of the Amur River—the estuarine zone—and are a stenobiotic species. Within the Daurian crayfish group, the Vladivostok crayfish (Cambaroides wladiwostokiensis) is a species with a narrow niche, meaning it requires special attention for its conservation within its range. This is particularly relevant considering its highly stressed state due to water body pollution, and its reduced ecological capacity for survival. Its range includes water bodies of the Sea of Japan basin from the northern part of the Korean Peninsula to the Black and Kievka rivers, situated north of Cape Povorotny. It is erroneously indicated to be present in the basin of the Mulinhè River in the territory of the People’s Republic of China [11, 14] through personal observations. Considering the fragmentation of the ranges of individual species, a revision of their taxonomic status is required. For this purpose, genetic analysis of various groupings is necessary as an auxiliary tool.
The use of genetic markers to delineate species boundaries within this genus holds promise for determining the taxonomic status of individual species. Moreover, it can reveal broader patterns of genetic variation and the evolutionary history among closely related taxa [11, 15, 16]. Such investigations underscore both the benefits and challenges of harnessing genetic data to distinguish species identities in a group marked by complex evolutionary trajectories and morphological similarities.
Freshwater crayfish have been proposed to form a monophyletic group closely related to clawed lobsters and are found on every continent except Antarctica [17]. From a taxonomic perspective, freshwater crayfish are divided into two monophyletic superfamilies: the northern hemisphere’s Astacoidea and the southern hemisphere’s Parastacoidea [17]. A comprehensive phylogenetic analysis, which encompassed representatives from 44 extinct and 27 extant crayfish families, including Polychelida, Achelata, Glypheidea, and Astacidea, culminated in the identification of a new superfamily—Glaessnericarioidea [11]. Additionally, three new families were recognized: Glaessnericariidae, Neoglypheidae, and Litogastroidae [11]. In another pivotal study, the debated relationships of major clades of reptant decapods were elucidated using a combined analysis of 16S, 18S, and 28S rRNA sequences, paired with morphological data [18]. The resulting optimal tree demonstrated that Glypheidea is the sister group to Astacidea. This relationship, in conjunction with the monophyletic Astacidea, which encompasses both freshwater crustaceans (Astacida) and marine clawed lobsters (Homarida), aligns with the findings of most previous studies.
Prior research into mitochondrial genomes has significantly contributed to our understanding of the evolutionary pathways of various species within the infraorder Astacidea [16, 17, 18]. Notably, these analyses have both reaffirmed the existence of conserved genetic elements and shone a light on structural variations, including gene rearrangements, thereby offering a deeper understanding of genome evolution processes [19].
Building on this foundation, the present study endeavors to validate the species identity of C. wladiwostokiensis through meticulous analysis of mitochondrial genetic markers. This involves elucidating the wider landscape of genetic diversity and evolutionary history within the genus and its sister lineages by leveraging mitochondrial genome sequences. The results of the genetic analysis will help to determine the place of C. wladiwostokiensis within the group of both the Daurian crayfish and East Asian River crayfish. Moreover, beyond the immediate taxonomic implications, our findings also have the potential to pave the way for more accurate species identification, which can improve the management of parasitic diseases linked with some members of this genus. Furthermore, by examining the mitochondrial genomes, we are laying a basis that can be instrumental for future conservation and management strategies, and for the broader understanding of evolutionary processes in freshwater crayfish.
An individual C. wladiwostokiensis was captured in the area of Gerasimov Creek (Kievka River basin), Primorsky Krai, Russia, in May 2018. Species identification was conducted by leveraging descriptive data sourced from relevant literature [20, 21, 22, 23]. After capture, the specimen was completely fixed in 95% ethanol. DNA extraction from the prefixed chela muscle tissue was performed using the “K-Sorb” kit (LLC “Sintol”, Moscow, Russia). Total DNA sequencing was conducted on the Illumina NovaSeq 6000 platform (Novogen, Tianjin, China). Approximately 7.15 Gb of raw paired-end reads with a length of 150 bp were obtained. After using FastQC (version 0.12.0, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) [24] to assess read qualities, AdapterRemoval (version 2.2.2, https://adapterremoval.readthedocs.io/en/stable/) [25] was employed to trim standard Illumina adapters. Mitochondrial genome assembly was performed using SPAdes (version 3.15.5, Saint Petersburg State University, Saint Petersburg, Russia) [26] and NOVOPlasty (version 4.3.3, https://github.com/ndierckx/NOVOPlasty) [27] in parallel since the preliminary runs of these assemblers did not yield the full expected genome length of the cyclic form. Initially, contigs were de novo assembled in SPAdes with default parameters and a kmer length of 21. A reference database was formed using the available complete mitochondrial genome sequences of Cambaroides representatives (Table 1, Ref. [16, 19, 28, 29]) from GenBank (https://www.ncbi.nlm.nih.gov/). This reference was utilized to select the most homologous contigs from the SPAdes assembly to the target organism. Then, the selected contigs were used as seeds for the NOVOPlasty assembly, which can be considered reference-based. All contigs obtained in this manner, homologous to Cambaroides mitochondrial genomes, were aligned against C. similis and C. dauricus (considered to be the closest) using the MUSCLE algorithm [30] and implemented in MEGA (version 7, Mega Limited, Auckland, New Zealand) [31]. Manual curation was performed to form a consensus sequence.
| Family | Species | NCBI accession numbers | Capture localities | Source |
| Astacidae | Astacus astacus | MT862440 | - | GenBank |
| Austropotamobius pallipes | KP205430 | France: Lucelle, Alsace region | Grandjean et al., 2017 [16] | |
| A. torrentium | KX268734 | Germany: Kammel, Bavaria | Grandjean et al., 2017 [16] | |
| Pacifastacus leniusculus | KX268740 | United Kingdom: Greenwich Ecology Park, South London | Grandjean et al., 2017 [16] | |
| Cambaroididae | Cambaroides dauricus | OL542521 | Qingdingzi forest farm (Huinan County, Tonghua City, China) | Luo et al., 2023 [28] |
| C. japonicus | KX268736 | Japan: Bibai, Hokkaido | Grandjean et al., 2017 [16] | |
| C. schrenckii | KX268737 | Russia: Southeast Russia | Grandjean et al., 2017 [16] | |
| C. similis | JN991196 | ravine in the Gwanak Mountain in South Korea | Kim et al., 2012 [19] | |
| C. wladiwostokiensis | OR353741 | Russia: Primorsky Krai: Kievka River basin: Gerasimov Creek | Original data (this study) | |
| Cambaridae | Cambarus robustus | KX268738 | USA: Oberlin, Ohio | Grandjean et al., 2017 [16] |
| Orconectes rusticus | KU239994 | USA | GenBank | |
| O. limosus | KP205431 | France: Vonne, Poitou-Charentes region | Grandjean et al., 2017 [16] | |
| O. luteus | KX268739 | USA: Fouche Renault, Missouri | Grandjean et al., 2017 [16] | |
| O. punctimanus | KX119150 | Oriskany, Virginia, USA | GenBank | |
| O. sanbornii | KU239995 | USA | GenBank | |
| Procambarus acutus | KX268741 | USA: Prairie Fork Pond, Missouri | Grandjean et al., 2017 [16] | |
| P. alleni | KT074363 | See ref | Vogt et al., 2015 [29] | |
| P. clarkii | OL542520 | Yangtze River Fisheries Research Institute of Chinese Academy of Fishery Sciences | Luo et al., 2023 [28] | |
| P. clarkii | JN991197 | pet market in Incheon, South Korea | Kim et al., 2012 [19] | |
| P. fallax | KT074364 | See ref | Vogt et al., 2015 [29] | |
| Parastacidae | Cherax quinquecarinatus | HG799091 | Australia: Dunsborough, southwest Western Australia | GenBank |
| Engaeus cunicularius | HG942173 | Australia: Robbins Ck, South of Naracoopa, King Island, Tasmania | GenBank | |
| Geocharax gracilis | HG942174 | Australia: Yaloak Ck, East of Panmure, Victoria | Grandjean et al., 2017 [16] |
Annotation of the obtained sequence was carried out using the MITOS Web Server [32] with reference sequences being manually cross-referenced. The annotated sequence was deposited in GenBank under accession number: OR353741. To estimate assembly parameters and exclude possible artifacts, we mapped reads onto both the newly assembled genome and the C. dauricus (OL542521) genome using Bowtie 2 (version 2.3.4.1, https://bowtie-bio.sourceforge.net/bowtie2/manual.shtml) [33], and then sorted and indented the reads in SAMtools (version 1.7, https://github.com/samtools/samtools) [34]. Coverage and assembly quality were also assessed using SAMtools (depth and flagstat functions). Visualization of reads per reference was performed in the Tablet alignment viewer version 1.21.02.08 [35]. Genome map visualization (Fig. 1) was conducted using the web-implemented CGView program [36], with a sliding window width of 50 bases.
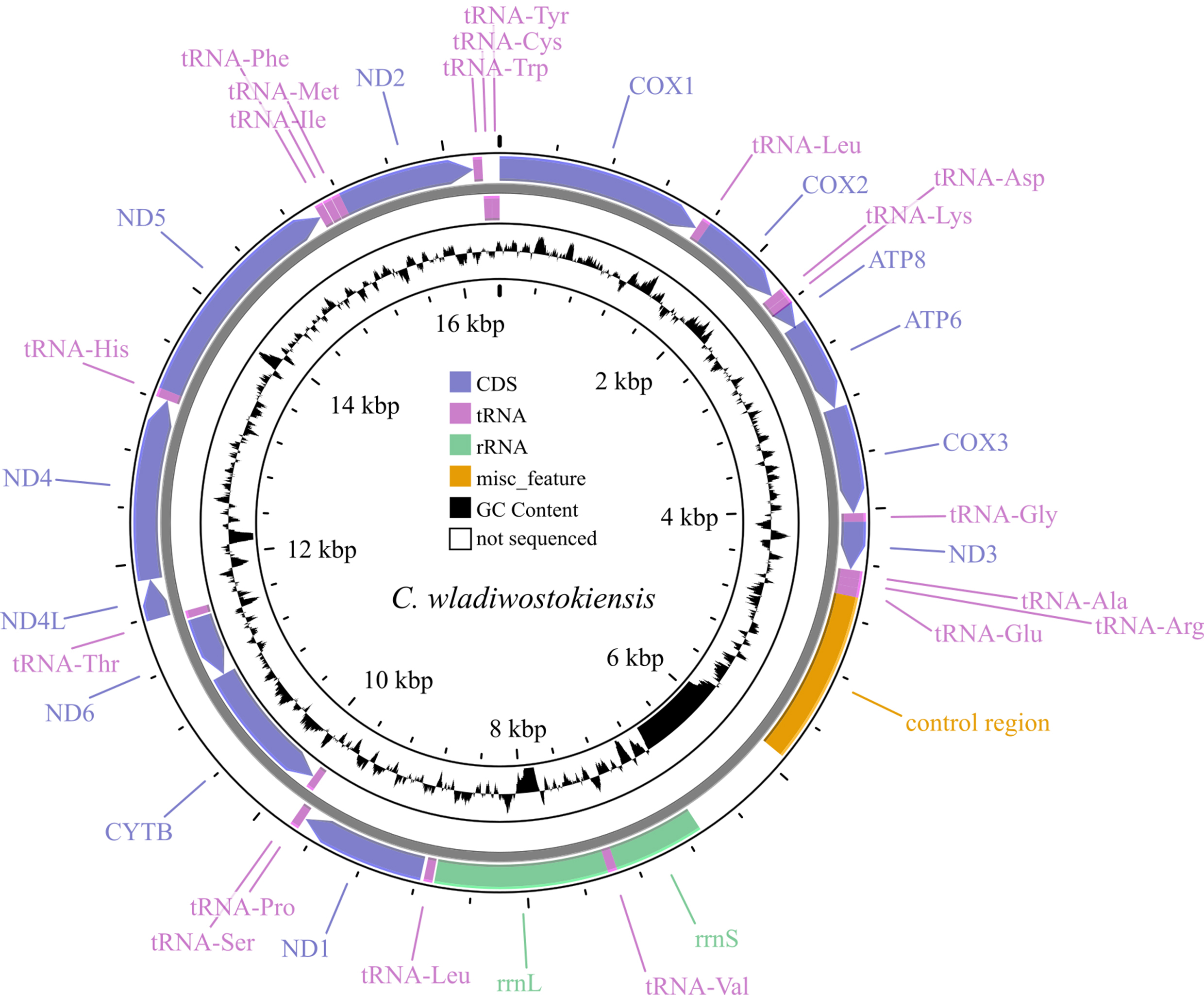 Fig. 1.
Fig. 1.Map of the C. wladiwostokiensis mitochondrial genome. Note that non-sequenced regions appear as “N” scaffolds, leading to GC view artifacts. Refer to Table 3 for details.
To confirm the species identity for the genetic material obtained from the specimen, an additional analysis was conducted on a 502 bp fragment of COX1 and a 524 bp fragment of 16S [12, 19] from Cambaroides representatives; A. astacus was used as an outgroup. We selected these fragments because they are the only ones that constitute a reliable reference, which follows the results of studies that used an integrative approach in the systematics of this genus. Sequences for comparison were downloaded from GenBank. Alignment was performed by MUSCLE [30], and genetic p-distance calculations (Table 2) were conducted for species groups using MEGA [31]. In addition, we performed distance-based NJ-phylogenetic sequence analyses using MEGA.
| Species names and GenBank accession numbers | C. wladiwostokiensis | C. dauricus OL542521, DQ666837 | C. similis JN991196, DQ666841 | C. schrenckii KX268737, DQ666835 | C. japonicus KX268736, DQ666839 | A. astacus KX268736 |
| C. wladiwostokiensis | - | 0.02 | 0.06 | 0.03 | 0.06 | 0.13 |
| C. dauricus AY820883, OL542521 | 0.08 | 0.01/0.02 | 0.07 | 0.04 | 0.06 | 0.13 |
| C. similis AY820880, JN991196 | 0.09 | 0.09 | 0/0.06 | 0.07 | 0.06 | 0.15 |
| C. schrenckii AY820882, KX268737 | 0.11 | 0.1 | 0.11 | 0.01/0 | 0.06 | 0.14 |
| C. japonicus AY820881, KX268736 | 0.11 | 0.11 | 0.11 | 0.11 | 0.03/0 | 0.14 |
| A. astacus MT862440 | 0.14 | 0.17 | 0.18 | 0.17 | 0.15 | - |
Along the diagonal, average intraspecific distances are presented for COX1/16S fragments. Lack of intraspecific sampling is indicated as “-”.
To identify variation landscapes of mitochondrial genome sequences of five Cambaroides representatives, a sliding window analysis was implemented in the Spider package (version 1.4-2, https://rdrr.io/rforge/spider/) [37]. The sequence matrix was concatenated from 15 fragments, totaling 13,288 bases. The control region was not included in the matrix due to its absence in C. similis. Unread regions in the sequenced fragments of C. wladiwostokiensis were substituted with “-”. The window width was set to 500 bases with a 1-base interval. For each window, average genetic p-distances were calculated using the dist.dna function in the APE package version 5.5 [38]. Then, the distances were visualized (Fig. 2) using base R tools version 4.1.0 (https://cran.r-project.org/bin/windows/base/old/4.1.0/).
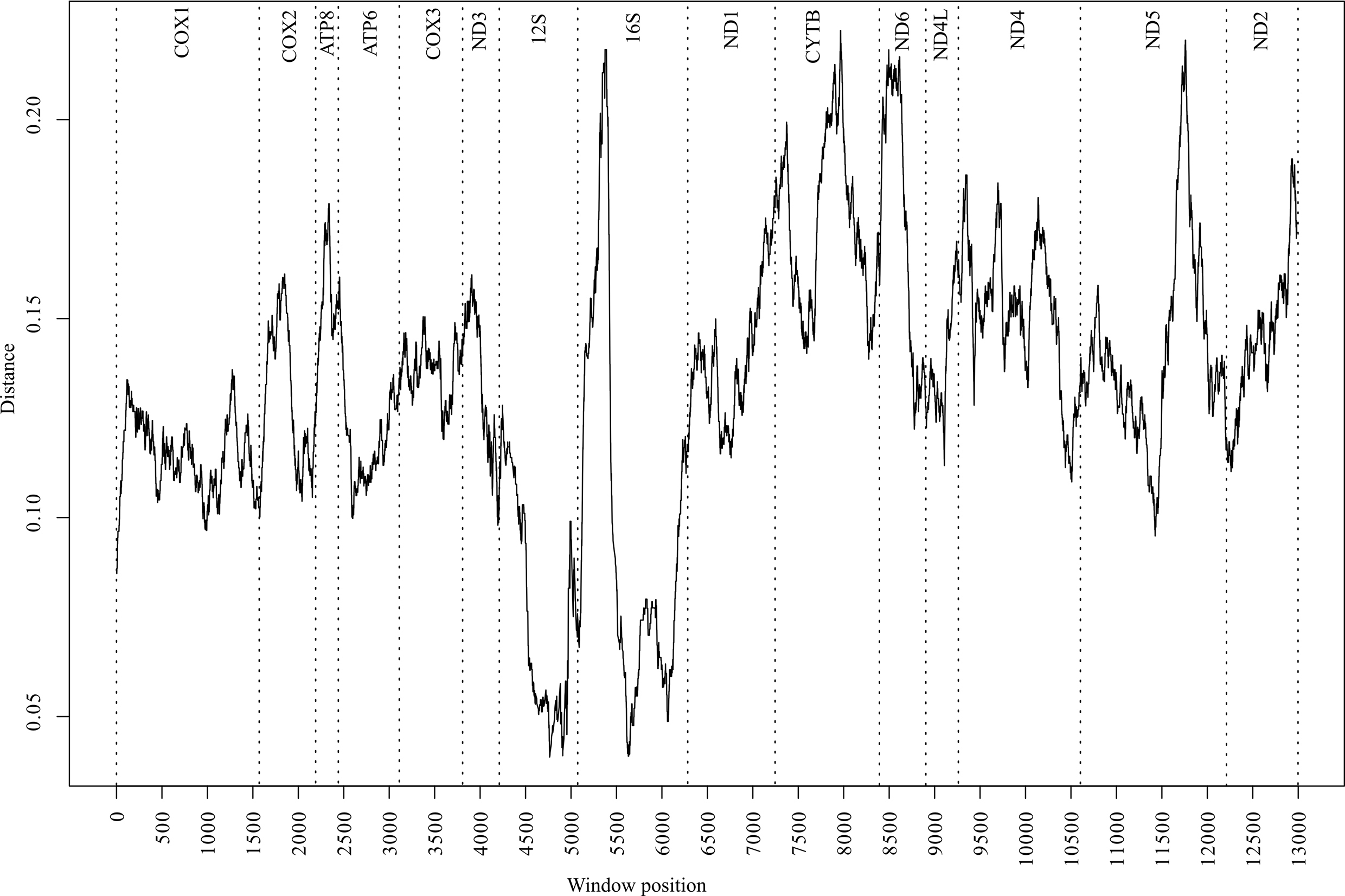 Fig. 2.
Fig. 2.Distribution of divergence values (p-distance) along the matrix of 15 mitochondrial genome fragments from five representatives in the genus Cambaroides. The analysis was conducted using the sliding window algorithm. Vertical dashed lines indicate fragment boundaries. Fragment names are provided at the top.
To define the position of C. wladiwostokiensis within the genus and of Cambaroides in the Astacidea infraorder, we conducted a phylogenetic analysis of 23 representatives spanning three families in this infraorder. Complete mitochondrial genome sequences for these representatives were sourced from GenBank (as detailed in Table 1). Parsing sequences, calculating basic statistics, aligning fragments, concatenating them into a supermatrix, and preparing them for phylogenetic analysis were carried out using the PhyloSuite platform version 1.1.14 (https://github.com/dongzhang0725/PhyloSuite/releases) [39, 40]. Alignment of all fragments was performed using MAFFT version 7.505 [41] with default parameters. The concatenated matrix consisted of 15 fragments and encompassed 13,612 bases. Simultaneous determination of partitions and the selection of optimal substitution models for them were performed using PartitionFinder version 2 [42], based on the Bayesian information criterion. Bayesian analysis (tree inference) using the previously determined scheme by PartitionFinder was conducted in MrBayes version 3.2.7 [43]. Tree topology searching and marginal posterior probability values were generated by two parallel runs of four Markov chains for 2,000,000 generations. The sampling frequency of topologies and parameters by the Metropolis-coupled algorithm was 1 per 1000 generations. The first 25% of trees corresponding to the burn-in step were discarded as non-optimal. A consensus tree was generated based on the remaining 3002 trees. Convergence indices (ESS, PSRF) indicated sufficient sampling across all parameters. The average standard deviation of split frequencies approached 0.000047 at the end of the run. Maximum likelihood analysis was performed in IQ-TREE [44] with simultaneous model selection for designated partitions and bootstrap support assessment using 50,000 ultrafast bootstrap replicates [45]. The Bayesian phylogeny was chosen as the basis for presenting the results of the phylogenetic analysis (Fig. 3).
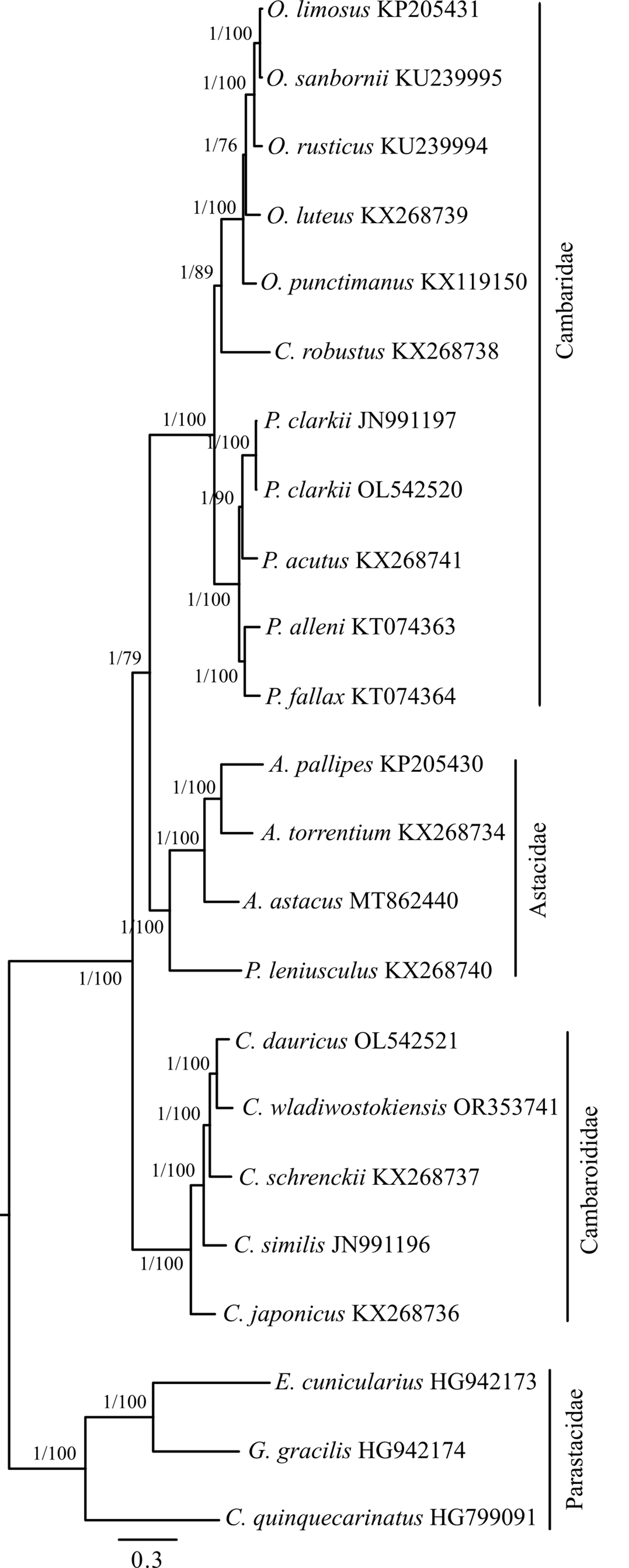 Fig. 3.
Fig. 3.Bayesian inference (BI) phylogenetic tree illustrating the relationships among representatives of the genus Cambaroides and the position of the genus among other groups within the Astacidea infraorder. Construction based on the concatenated matrix of 15 mitochondrial genome fragments (detailed in the Materials and Methods). The tree is rooted at the midpoint. Node support values are provided as Bayesian posterior probabilities (BI) and as percentages of 50,000 replicates in the ultrafast bootstrap test (ML). They are denoted as BI/ML.
The assembled mitochondrial genome of C. wladiwostokiensis comprises 16,391 base pairs. We identified a total of 13 protein-coding genes, 12S (rrnS) and 16S (rrnL) rRNAs, 19 tRNAs, and a putative control region (Tables 3,4 and Fig. 1). Through alignment with other available Camboroides mitogenomes, we discovered regions that were not successfully sequenced and remain unretrievable from the raw reads obtained. These regions include a 784 bp fragment between the control region and rrnS, where tRNAs Gln, Ser, and Asn might be located (see Table 3 and [28]), 18 bases between tRNA-Val and rrnL, 154 bases between rrnL and tRNA-Leu, and 107 bases within the ND4 gene. As a result, 1063 bases, or 6.5% of the genome remained unread. We did not detect any rearrangements in the genome. The non-sequenced regions did not have any reads to be covered with, and are not artifacts of assembly, as was proved by mapping the reads onto the genomes of C. wladiwostokiensis and C. dauricus (see Supplementary Figs. 1,2). The mean coverage was 26.9 reads per position. From the total 20,861,158 reads that were obtained from the sequencing run, 3152 reads (0.02%) were successfully mapped to the reference genome. Among the mapped reads, 2994 (0.01%) were properly paired, indicating the correct alignment of read pairs. Additionally, 114 reads were identified as singletons, where the mate did not map to the reference genome.
| Region | Strand | Position (bp) | Size (bp) | Start/Stop codons |
| COX1 | + | 1–1536 | 1536 | ACG/TAA |
| TRNA-Leu | + | 1538–1602 | 65 | - |
| COX2 | + | 1603–2290 | 688 | ATG/TAA* |
| TRNA-Lys | + | 2288–2351 | 64 | - |
| TRNA-Asp | + | 2353–2416 | 64 | - |
| ATP8 | + | 2417–2575 | 159 | ATG/TAA |
| ATP6 | + | 2569–3243 | 675 | ATG/TAA |
| COX3 | + | 3243–4031 | 789 | ATG/TAA |
| TRNA-Gly | + | 4030–4091 | 62 | - |
| ND3 | + | 4092–4445 | 354 | ATT/TAA |
| TRNA-Ala | + | 4447–4507 | 61 | - |
| TRNA-Arg | + | 4508–4568 | 61 | - |
| TRNA-Glu | + | 4569–4636 | 68 | - |
| Control region | + | 4637–5898 | 1262 | - |
| Non-sequenced region | n/a | 5899–6682 | 784 | - |
| rrnS | + | 6683–7343 | 661 | - |
| Non-sequenced region | n/a | 6893–6961 | 69 | - |
| TRNA-Val | + | 7344–7411 | 68 | - |
| Non-sequenced region | n/a | 7358–7375 | 18 | - |
| rrnL | + | 7412–8666 | 1255 | - |
| Non-sequenced region | n/a | 7812–7965 | 154 | - |
| TRNA-Leu | + | 8680–8744 | 65 | - |
| ND1 | + | 8769–9710 | 942 | ATA/TAG |
| TRNA-Pro | + | 9718–9782 | 65 | - |
| TRNA-Ser | - | 9786–9848 | 63 | - |
| CYTB | - | 9849–10983 | 1135 | ATG/TAA* |
| ND6 | - | 10983–11501 | 519 | ATT/TAA |
| TRNA-Thr | - | 11518–11580 | 63 | - |
| ND4L | + | 11583–11876 | 294 | ATG/TAA |
| ND4 | + | 11876–13216 | 1341 | ATG/TAA |
| Non-sequenced region | n/a | 12093–12199 | 107 | - |
| TRNA-His | + | 13216–13279 | 64 | - |
| ND5 | + | 13280–15010 | 1731 | GTG/TAA |
| TRNA-Phe | + | 15010–15070 | 61 | - |
| TRNA-Ile | + | 15077–15140 | 64 | - |
| TRNA-Met | + | 15144–15207 | 64 | - |
| ND2 | + | 15208–16200 | 993 | ATG/TAA |
| TRNA-Trp | + | 16200–16265 | 66 | - |
| TRNA-Cys | - | 16265–16328 | 64 | - |
| TRNA-Tyr | - | 16329–16391 | 63 | - |
The asterisk (*) indicates the exception where the TAA stop codon is completed by the addition of 3’ A residues to the mRNA. Non-sequenced regions are inferred based on alignment with the closest taxa. n/a, not applicable.
Protein-coding genes vary in size from 159 (ATP8) to 1731 (ND5) base pairs. ATG is the most common start codon, and TAA is the most common stop codon. Notably, the ND5 fragment contains a unique start codon (GTG), and ND1 contains a unique stop codon (TAG). A putative transcription exception was observed, where the TAA stop codon might be completed by the addition of 3’ A residues to the mRNA. This feature was detected in the COX2 and CYTB genes. Most protein-coding genes are located on the “+” strand, except for ND6 and CYTB. The large rRNA spans 1255 base pairs, while the small rRNA covers 661 base pairs, both on the “+” strand, although the small rRNA appears unfinished (Table 3). The tRNA fragment lengths vary from 61 (tRNA-Ala, tRNA-Arg, and tRNA-Phe) to 68 (tRNA-Glu and tRNA-Val) base pairs, with a common length of 64 bases. Only four tRNAs are located on the “-” strand: tRNA-Ser, tRNA-Thr, tRNA-Cys, and tRNA-Tyr. The control region, situated between tRNA-Glu and rrnS, spans 1262 base pairs. Features that are common to nucleotide content in the obtained genome are presented in Table 4.
| Regions | Strand | Size (bp) | GC (%) | AT skewness | GC skewness |
| Full genome | n/a | 16,391 | 26.0 | –0.079 | 0.200 |
| PCGs | all | 11,154 | 29.0 | –0.191 | 0.116 |
| PCGs | + | 9501 | 28.8 | –0.184 | 0.181 |
| PCGs | - | 1653 | 30.4 | –0.234 | –0.239 |
| tRNAs | all | 1215 | 25.1 | 0.026 | 0.204 |
| tRNAs | + | 962 | 25.5 | 0.007 | 0.208 |
| tRNAs | - | 253 | 23.3 | 0.093 | 0.186 |
| rRNAs | all | 1916 | 22.9 | 0.015 | 0.338 |
| rRNAs | + | 1916 | 22.9 | 0.015 | 0.338 |
n/a, not applicable; PCGs, protein coding genes.
Following alignment, the COX1 fragment matrix consisted of 502 bases. Among them, 132 sites were variable, including 90 parsimony informative sites and 42 singletons. The 16S matrix, aligned to 524 bases, had 105 variable sites, including 48 parsimony informative sites and 57 singleton sites. Intraspecific variation for the COX1 marker in Cambaroides representatives ranged from 0 (C. similis) to 0.03 (C. japonicus). Interspecific divergence varied from 0.08 (between C. wladiwostokiensis and C. dauricus) to 0.11 (between C. japonicus and all other representatives of the genus) (Table 2). The outgroup exhibited the highest differentiation between all species. The variability in the 16S marker showed greater heterogeneity. Intraspecific variability ranged from 0 (C. japonicus and C. schrenckii) to 0.06 (C. similis). No clear interspecific threshold was identified based on this marker. Divergence ranged from 0.02 (between C. wladiwostokiensis and C. dauricus) to 0.07 (C. similis–C. dauricus and C. similis–C. schrenckii). The outgroup was distant from all species, with the highest divergence values. The phylogenetic NJ-trees (see Supplementary Figs. 3,4) showed separate clusters for each species on the 16S tree, except for C. similis, and an individual branch for C. wladiwostokiensis, yet express low bootstrap support on interspecific nodes. The outgroup naturally forms a basal position on both trees.
A comprehensive analysis of the p-distance landscapes was conducted across the aligned mitochondrial genomes of five Cambaroides representatives (Fig. 2). The resulting profiles depicted local sequence variations between all genomes, with the most conserved regions identified within the 12S and 16S fragments. Conversely, the 16S fragment displayed the most uneven variability, with a peak in the first half. Among the protein-coding fragments, CYTB, ND6, and ND5 exhibited the highest variability, whereas the COX1 fragment did not show high variability, thereby limiting the divergence profile to 0.09–0.13.
The phylogenetic tree (Fig. 3) shows a strongly supported clade comprising
representatives of Astacidae and Cambaridae. The chosen external group,
Parastacidae, occupied a naturally separated position and was supported in both
algorithms. Within this designated macroclade, representatives of the genus
Cambaroides from the family Cambaridae occupied an external position,
with slightly less support following in the ML topology, a division into two
clades based on belonging to the Astacidae and Cambaridae subfamily Cambarinae.
The latter subfamily includes two additional clades, one containing the genus
Procambarus and the other containing Orconectes with
Cambarus robustus. Thus, representatives of the family
Cambaridae exhibited a paraphyletic position in this topology. Within the genus
Cambaroides, C. japonicus held an external position, followed
by a sequential branching of C. similis, C. schrenckii, and the
closest grouping of C. dauricus with C. wladiwostokiensis. The
support for this topology was absolute in both algorithms. Representatives of the
genus Procambarus displayed an additional bifurcation into P.
alleni + P. fallax and P. clarkii + P. acutus.
Notably, independently sequenced P. clarkii sequences clustered
together. In the adjacent clade, C. robustus held an external position,
followed by sequential bifurcations within F. punctimanus
This study presents, for the first time, data on the mitochondrial genome sequence of C. wladiwostokiensis crayfish and compares it with other available Cambaroides genomes. Various methods have been previously employed to obtain mitochondrial genomes in this genus, such as assembly from multiple fragments after Sanger sequencing, including the primer walking technique [19], and shallow sequencing of total DNA, known as genome skimming [16]. Although we employed a relatively shallow sequencing depth in our research, it was still nine times more comprehensive than the data garnered in a comparable study [10]. However, in our study, this approach did not allow for the complete reading of certain genome regions (in total 1063 bases) (Table 3). Genome skimming has also proven to be effective for obtaining complete mitochondrial genomes of fishes [46]; nevertheless, the development of other methods to reduce the costs of obtaining mitochondrial genomes continues [47].
A plausible explanation for the existence of unread regions (specifically, the missed tRNAs) in the crayfish genome in this study might stem from the contamination of the total DNA from the target organism with genetic material sourced from the microbiome. Such contamination is not uncommon and has been previously documented in both genomic (including metagenomics) and transcriptomic sequencing endeavors [48, 49]. However, higher sequencing depth usually mitigates these issues. In theory, labor-intensive approaches based on pulling the complete genome sequence by pieces via PCR should eliminate this disadvantage. Additionally, it is important to note that the copy number of mitochondrial DNA varies significantly across different tissues [50, 51], which might potentially substitute for enrichment procedures. Furthermore, this circumstance implies that when comparing genome skimming results, it is necessary to specify tissue types and, ideally, the number of mitochondrial DNA copies in the study methodology.
The genome structure (Fig. 1, Table 3) and nucleotide composition (Table 4) of C. wladiwostokiensis are quite similar to those of other Cambaroides representatives (see Supplementary Table 1). The main differences relate to nucleotides. Furthermore, single-nucleotide indels are present within the regions of transfer RNA compared to the nearest species, C. dauricus. According to the comparison data with reference sequences of COX1 and 16S [12, 19], the identity of C. wladiwostokiensis can be confirmed, albeit relatively. Genetic distances indicate that the specimen from which the genome was obtained does not correspond to any of the available reference species—C. dauricus, C. japonicus, C. schrenckii, or C. similis—differing from them by 0.08, which is comparable to interspecies differences within this genus. Another species in this genus, C. koshewnikowi Birstein and Vinogradov, 1934, is missing from the analysis. However, the assignment of our specimen to this species is doubtful due to the more northern range of C. koshewnikowi, as well as the fact that the species is extremely rare and only known from a few records in the lower part (estuarine zone) of the Amur River [11, 52, 53].
We examined landscape variation data to identify genome regions that could be used to develop future species-specific primers and probes. Typically, for the development of species-specific assays for crayfish, researchers use mitochondrial DNA [54, 55, 56] or a combination of nuclear and mitochondrial fragments [57, 58]. This is driven by the fact that mitochondrial DNA is present in significantly higher copy numbers in cells than nuclear DNA. Moreover, the ecological characteristics of mitochondrial DNA suggest that it is less prone to degradation [59]. In this case, we are limited by the available resources of the mitochondrial genome. Additionally, we need to exclude the control region (CR) from consideration since it is also unread in the species C. similis. Based on the landscape data (Fig. 2), the COX1 fragment is the most conserved among the mitochondrial protein-coding regions of Cabraroides, while also having the most uniform distribution in variability. The COX1 encodes one of the essential components in the complexes of the electron transport chain [60, 61]. Thus, strong purifying selection may define its conservative nature [62], forming an expected threshold for distinguishing between intraspecific and interspecific variability, thereby making this marker suitable for species delimitation of most multicellular organisms [2], including many known crustaceans [63]. The observed pattern in our case is likely to be a candidate for probe design; however, it may have limitations when searching for primers for haplotype-specific PCR. The 16S, CYTB, ND6, and ND5 fragments are more likely to be suitable for this purpose since they showed the highest peaks in variability.
Based on an independent partitioned phylogenetic analysis of 15 mitochondrial fragments in the Astacidea infraorder, we have shown that the family Cambaridae is not monophyletic, yet more precisely, it exhibits properties of paraphyly (Fig. 3) when considered alongside representatives in Astacidae. Accordingly, our results support the view that East Asian cambarids (Cambaroididae) occupy a basal position relative to Astacidae and North American Cambaridae [16], thus, representing a naturally monophyletic group. This view is inconsistent with data based on an integrative approach [64], although it had a limited sample from the group under discussion. Topologically, similar results were obtained when analyzing new mitochondrial genomes of P. clarkii and C. dauricus from China [28], where East Asian cambarids also form a group external to the others. There is another view according to which “the genus Cambaroides continues to fall outside the Cambaridae, but clusters with different taxa depending on the data set used” [14]. Our results also indicate that C. wladiwostokiensis is the closest to C. dauricus, and together they form the cluster most recently diverged from all other Cambaroides.
This study introduces the mitochondrial genome of C. wladiwostokiensis crayfish and compares it with other Cambaroides genomes. Shallow sequencing revealed certain unread regions, possibly due to contamination or tissue-specific DNA copy variations, coupled with insufficient sequencing depth. Despite these gaps, we show that the structure and composition of the genome resemble other Cambaroides. While the genetic analysis affirmed the identity of C. wladiwostokiensis, it also underscored its distinctiveness from previously known species. Landscape variation data identified potential regions for species-specific primer development, excluding the unread control region. Partitioned phylogenetic analysis showed the paraphyletic origin of the Cambaridae family. The basal position of East Asian cambarids (Cambaroididae) supports its monophyletic status and is consistent with previous studies. This contributes to discussions about Cambaroides taxonomic placement and enriches insights into crayfish evolutionary relationships. Overall, this research sheds light on the genetic characteristics of C. wladiwostokiensis and its Cambaroides relatives, providing a foundation for future studies in crayfish genomics and contributing to a broader understanding of the evolutionary history of this diverse group. Further exploration and integration of additional data could refine the conclusions drawn from this study.
Raw data that support the findings of this study have been deposited in NCBI SRA archive with the identifier SRR26399148.
EB performed the collection, identification and conceptualization of the taxonomic position and ecology of C. wladiwostokiensis. ST designed the research, performed genetic analysis and analyzed the data. ST wrote the manuscript. Both authors contributed to editorial changes in the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity.
The experiments on crayfish in this study are consistent with the current Russian and international standards of law and regulations for research involving animals and were approved by the Commission of biomedical ethics of A.V. Zhirmunsky National Scientific Center of Marine Biology of the Far Eastern Branch of the Russian Academy of Sciences (the record # 1-080923 from the Meeting # 2, September 6, 2023).
The authors are grateful to Dr Alexey Boyko for the help in working on the “Umnik” computing cluster at the NSCMB FEB RAS.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.