




1 Institute of Bioinformatics, International Technology Park, 560066 Bengaluru, Karnataka, India
2 Manipal Academy of Higher Education (MAHE), 576104 Manipal, Karnataka, India
3 Somaiya Institute of Research and Consultancy (SIRAC), Somaiya Vidyavihar University (SVU), Vidyavihar, 400077 Mumbai, Maharashtra, India
Abstract
Eribulin, a synthetic marine based drug has received extensive attention recently due to its promising anticancer activities against a wide variety of cancer types as evidenced by preclinical and clinical data. Eribulin is predominantly shown to exhibit microtubule inhibitory activity, however recent reports indicate that it acts via multiple molecular mechanisms targeting both the cancer cells as well as the tumor microenvironment. In this review, a comprehensive account on various modes of action of eribulin on cancer cells is presented along with important clinical aspects in the management of cancer through a comprehensive literature review. We have also highlighted approaches including combination therapy to improve the efficacy of eribulin in cancer treatment. Currently, eribulin is used to treat heavily pretreated patients with metastatic breast cancer, for which it gained FDA approval a decade ago and more recently, it has been approved for treating anthracycline-pretreated patients with metastatic liposarcoma. Novel therapeutic strategies should aim at resolving the toxicity and resistance conferred due to eribulin treatment so that it could be integrated in the clinics as a first-line treatment approach.
Keywords
- Eribulin
- Cancer therapy
- Microtubule inhibition
- Reversal of EMT
- Review
Over the recent years, significant decrease in the mortality due to cancer has been observed globally [1]. One of the plausible reasons could be better understanding of molecular mechanisms underlying different types of cancers. Such knowledge has led to the current era of targeted therapies and precision medicine. Nevertheless, chemotherapy is still a powerful approach and various studies are carried out recently to improve the efficacy of the existing drugs and to identify potent new drugs [2]. Among the several classes of anticancer agents, microtubule inhibitors are considerably effective, since they interfere with significant cellular processes including mitosis and angiogenesis [3]. Interestingly, all the FDA approved microtubule-targeting agents are either isolated or derived from natural products of plant or marine origin [4, 5]. Plant based vinca alkaloids and taxanes are incredibly successful microtubule inhibitors till date which were approved about five decades ago for cancer therapy [3]. More recently a unique microtubule inhibitor, eribulin gained a lot of interest owing to its excellent anticancer properties.
Eribulin is a simplified synthetic analog of Halichondrin B (Fig. 1), which is
isolated from a rare marine sponge Halichondria okadai [6, 7, 8]. In spite
of potent anticancer properties on a wide variety of cancers [7], Halichondrin B
could not be developed as a therapeutic drug due to its limited availability and
complicated isolation process from the marine source. Among its several
simplified structural analogs, eribulin retains the remarkable anticancer
activity of the parent compound as evidenced through preclinical studies on
various cancer cell lines [6, 9]. A number of clinical trials were conducted
before the FDA approval of eribulin for heavily pretreated patients with
metastatic breast cancer [10] and patients with metastatic liposarcoma [11].
Clinical trials on eribulin monotherapy or combination therapy with other
anti-neoplastic agents, are still been conducted primarily for MBC and soft
tissue sarcoma (STS) in addition to several other cancer types also. Currently
eribulin is marketed in more than 50 countries with trade name of
Halaven
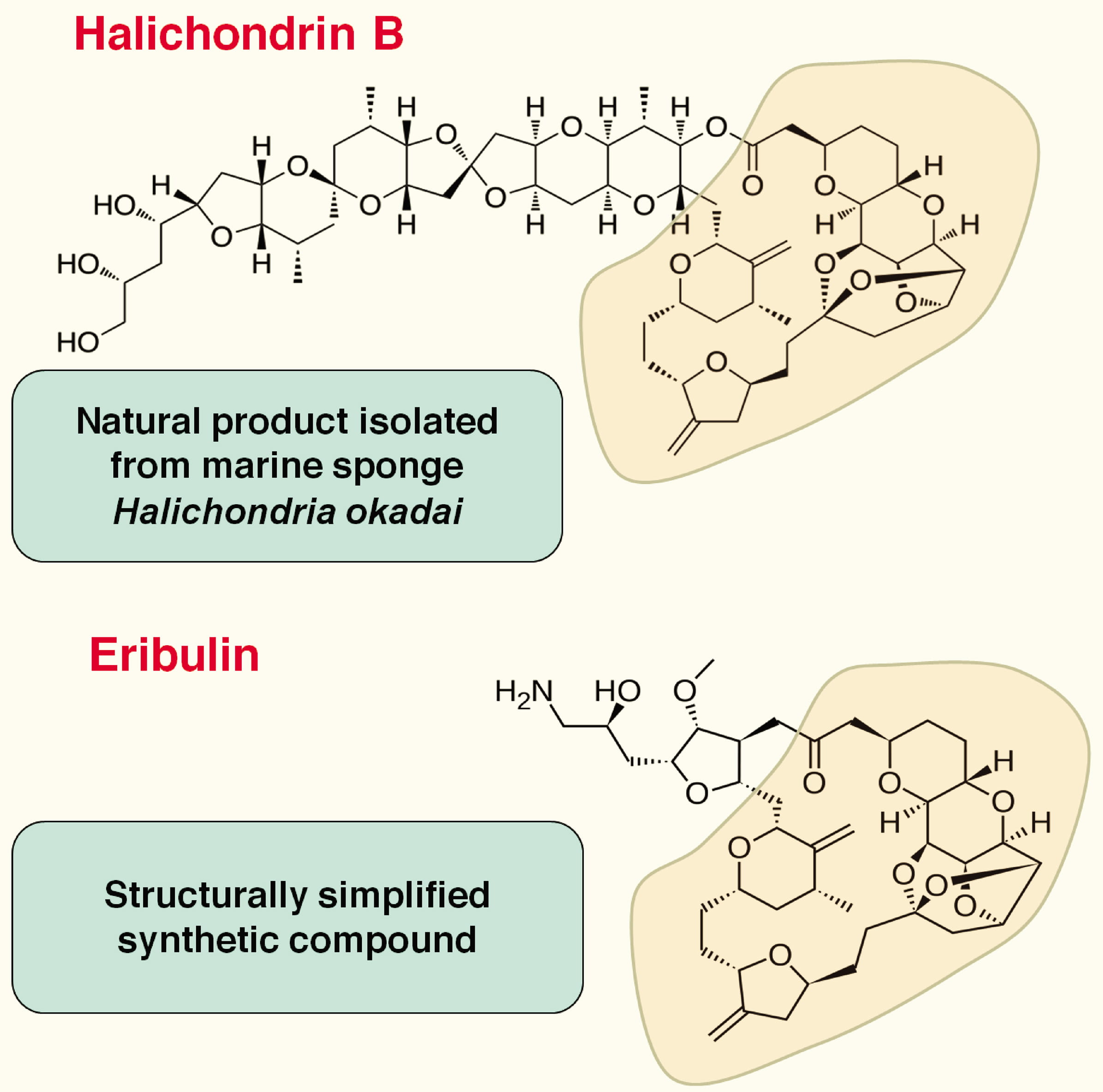 Fig. 1.
Fig. 1.Structures of Halichondrin B and Eribulin.
Eribulin exerts its anticancer property primarily through tubulin inhibition and mitosis [14, 15, 16, 17]. However, recent studies revealed that eribulin participates in various other molecular mechanisms including changes in the tumor microenvironment (TME) that impact significantly on tumor migration, invasion and metastasis. This section discusses the involvement of eribulin in various signaling pathways along with those that regulate mitosis.
The role of microtubules in cancer is well established and inhibitors of microtubule growth exhibit excellent anticancer properties [18, 19]. One of the earliest studies on the effects of eribulin on purified microtubules as well as in living cells showed that it inhibits microtubule growth by an end-poisoning mechanism [14]. This is in contrast to other anti-mitotic drugs such as vinblastine and paclitaxel that diminish the shortening and growth phases of microtubule dynamic instability. Eribulin does not cause the shortening of tubulin but it turns them into aggregates as observed through electron microscopy [14]. Eribulin significantly suppressed centromere dynamics in mitotic spindles at drug concentrations that arrested mitosis [15]. Results from binding studies on soluble tubulin suggested that eribulin had the ability to bind with high affinity to microtubule plus ends thereby it suppressed the dynamic instability [17]. Later studies involving X-ray crystallography eribulin was shown to bind to a site on beta-tubulin that is required for proto filament plus end elongation. In addition, single eribulin molecule was found to specifically interact with microtubule plus ends and is sufficient enough to either trigger a catastrophe or induce erratic microtubule growth [20]. Eribulin caused irreversible mitotic arrest unlike other microtubule inhibitors that leads to consistent inactivation of Bcl-2 and ended up with apoptosis [21, 22]. Molecular consequences of microtubule inhibition due to eribulin treatment are summarized in the Figure (Fig. 2).
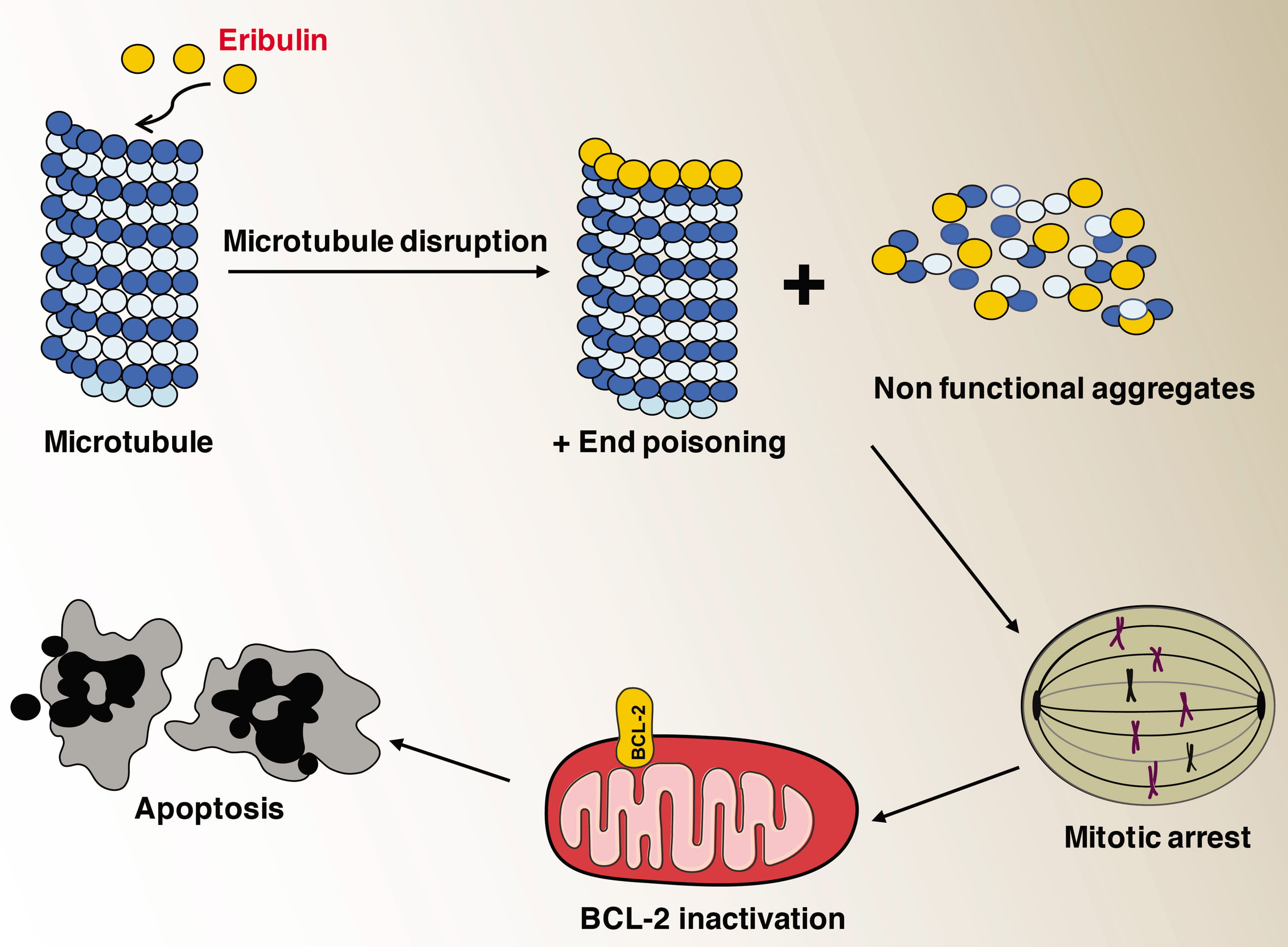 Fig. 2.
Fig. 2.Effects of microtubule inhibition due to eribulin treatment.
Recent evidences show that eribulin not only acts on the cancer cells but also shows its effects on the entire Tumor microenvironment (TME) [23, 24]. TME includes the entire cellular environment containing blood vessels, immune cells such as T cells, natural killer (NK) cells, activated fibroblasts and endothelial and mesenchymal cells along with the tumor cells [25, 26, 27]. The events orchestrated by all of these components of TME play a major role in tumor progression. Evidences indicate that effectiveness of the therapy depends on the capability of the anticancer agent to reduce the abnormalities in the TME [28]. Some of the alterations in the tumor environment due to eribulin treatment as observed until recently are discussed below. The different processes that are altered/ activated in the TME due to eribulin treatment is summarized in Fig. 3.
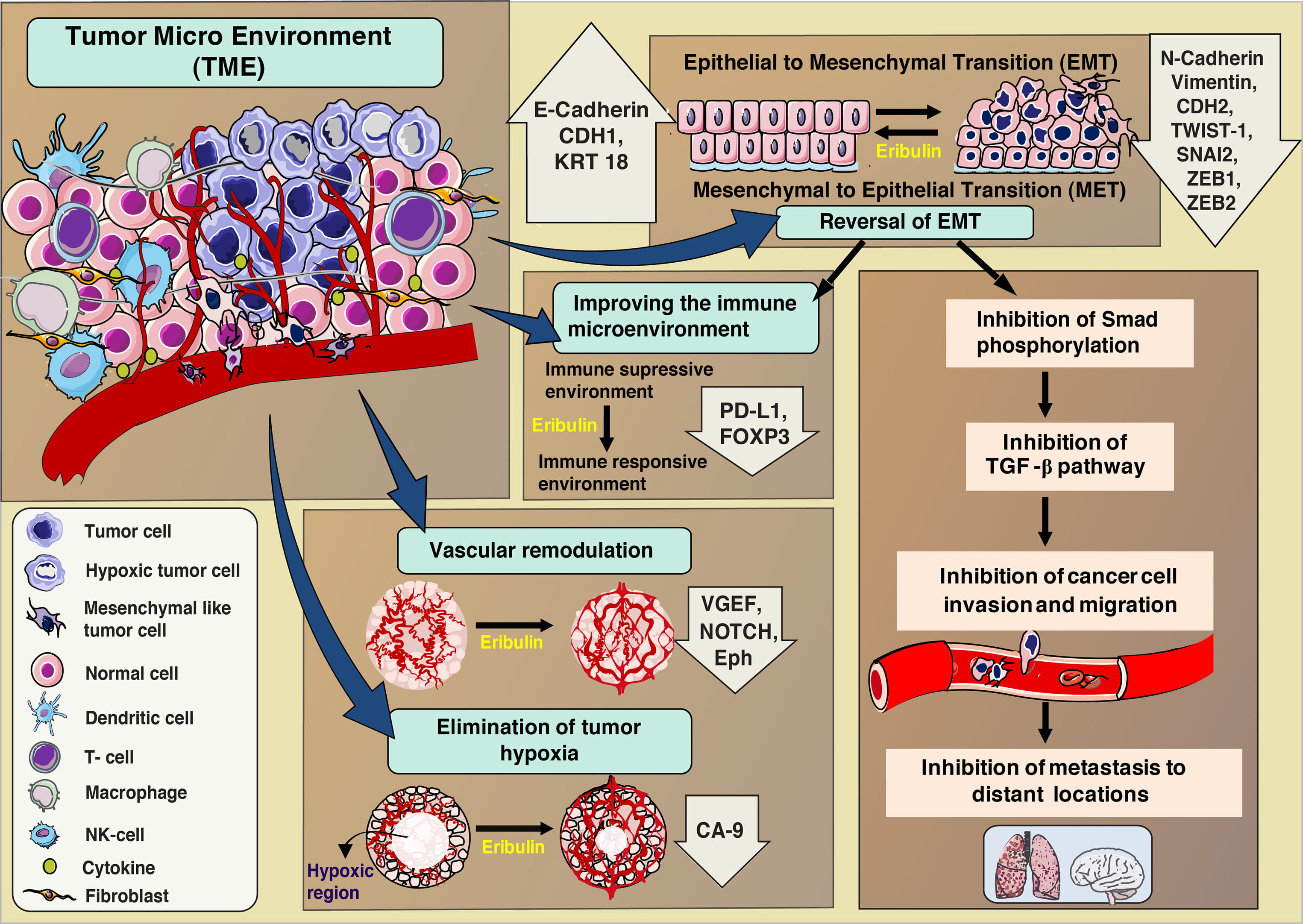 Fig. 3.
Fig. 3.Effects of eribulin treatment on Tumor microenvironment (TME).
Epithelial cells lose their flat and stationary nature and gain spindle like morphology with highly motile mesenchymal characteristics through epithelial mesenchymal transition (EMT) has been widely established in cancer [29, 30, 31]. This EMT process plays a major role in tumor progression, maintenance, drug resistance, and metastasis [32, 33, 34]. Tumor cells acquire mesenchymal properties by up regulation of EMT transcriptional factors such as TWIST1/2, SNAI2/SLUG, and ZEB1 [35, 36, 37]. Further, the expression profile of adhesion molecules (cadherins) and regulators such as Notch and Wnt ligands are analyzed to understand EMT process [38, 39]. Various preclinical evidences indicated that eribulin could reverse the EMT process to MET, while exerting its anticancer activity [23, 40, 41, 42]. In vitro studies on TNBC cell lines after treatment with eribulin for 7 days resulted in the reversal of spindle-like to epithelial-like flat morphology, up regulation of mRNA expression for epithelial markers (CDH1, KRT18) and down regulation of mesenchymal markers (CDH2, VIM, TWIST1, SNAI2, ZEB1, and ZEB2) [23]. Through IHC studies on xenograft models, eribulin treatment was shown to induce increased tumor expression level of epithelial markers (E-cadherin) in parallel to decreased mesenchymal markers (N-cadherin, ZEB1) [23]. One of the studies analyzed the differently altered genes between eribulin and paclitaxel in different cancer cell line panels. The results showed that the EMT pathway was activated with significantly altered expression between the drugs for breast and endometrial cancers, but not for ovarian cancer [43]. In human breast cancer xenograft models, 13 EMT-related mesenchymal marker genes, including Snai1, Snai2, Tgfb1, Tgfb2 and Vim, showed decreased expression after eribulin treatment as observed through gene profiling studies [24]. Similar pattern of gene regulation was observed in Oral Squamous Carcinoma cell line (OSCC), and in addition, cells with mesenchymal phenotype were found to be about 100 times more sensitive to eribulin compared to other cell lines [41]. The gene expression profiles of genes involved in EMT were altered between sensitive and resistant cell lines and were predictive of eribulin sensitivity in the breast and endometrial cancer panels [43].
EMT also plays a key role in immunosuppression in cancers and is shown to be involved in the activation of different immune checkpoint molecules, including PD-L1 [44, 45, 46]. EMT reversal due to eribulin treatment altered the immune suppressive to an immune responsive environment as indicated by reduced expression of the immunosuppressive drivers (PD-L1 and FOXP3) in metastatic breast cancer patients [47].
TGF-beta signaling pathway plays a major role for the induction of EMT and Smad proteins are crucial mediators of this process [48, 49]. In the absence of TGF- beta, Smad proteins tend to bind with microtubules [50]. Upon TGF-beta stimulation Smad proteins dissociate from microtubule and in involve in phosphorylation to Smad2, Smad3 and eventually activate transcription [50]. In a TGF-beta induced model of EMT, eribulin initiated the reversal of EMT by decreasing the phosphorylation of Smad2 and Smad3 [23]. There exists a correlation between microtubule inhibition by eribulin and EMT reversal. In fact, microtubule inhibition leads to interrelated mechanistic connection between other pathways. Eribulin binds to growing microtubule ends as discussed already. This stabilizes the association between Smad proteins and microtubules, and as a result inhibit Smad phosphorylation and TGF-beta signaling there by reverse the EMT. Recently it was hypothesized that the ability of eribulin to reverse EMT in cancer cells, xenograft models and CTCs in patients could be due to a prompt microtubule depolymerisation-mediated inhibition of TGF-beta signaling [51]. Inhibition of tubulin polymerization led to the blocking of Smad2/3 transport to the nucleus, which in turn inhibits expression of Snail in breast cancer [51].
Through the ability to alter the TME and reverse the EMT process, eribulin could influence the metastasis and tumor progression even if it loses its efficacy towards cancer cells. This can be correlated with the prolonged OS in eribulin treated patients observed during clinical trial [52].
In tumors, vascular networks are altered with complex branched patterns leading to decreased blood flow [53]. Two types of anti-vascular activities of tubulin inhibitors are reported namely, anti-angiogenic activity and vascular disruptive activity. In contrast, eribulin exhibits a unique anti vascular activity through vascular remodeling (increased numbers of smaller functional micro vessels) which reduces abnormalities in the TME in preclinical human breast cancer models and STS [24, 42]. Eribulin significantly enhanced tumor blood perfusion in STS xenografts and induced morphological changes and up-regulation of differentiation marker genes such as MYLK, C/EBPbeta, and KIF23 [42]. In a functional endothelial-pericyte co-culture assay, in contrast to paclitaxel, eribulin showed a strong efficacy as a potent anti-vascular agent that affected pericyte driven in vitro angiogenesis [54]. In xenograft models, the expression of mouse genes associated with the VEGF, Notch and Eph signaling pathways are reduced by eribulin [24]. In vitro studies established that the expression of Dll4, Notch4 and Efnb2 are decreased due to eribulin treatment, indicating that in vascular cells it regulated the angiogenesis signaling pathways [24].
During aggressive cell proliferation, the ability to develop a network of blood
vessels decreases, as a result, the tumor loses access to oxygen and the hypoxic
regions are created [55]. In such hypoxic environment cancer cells acquire a high
metastatic potential [55, 56]. Eribulin treatment (0.3 and 1.0 mg/kg)
significantly decreased mouse VEGF protein expression in xenograft models. CA9
protein, a marker for hypoxia that is regulated by hypoxia-inducible factor, was
also observed to have a lower expression when treated with 1.0 mg/kg eribulin in
the MDA-MB-231 model. These observations suggest that eribulin treatment resulted
in vascular remodeling [24] leading to an increase in the intra tumoral oxygen
supply and decrease in tumor aggressiveness. Immunohistochemical studies on
tissues of patients with MBC indicated that the expression of EMT markers and
cellular hypoxia (E-cadherin, N-cadherin, vimentin, and carbonic anhydrase 9
(CA9)) were suppressed by eribulin treatment [40]. Autoradiographic, ex
vivo and histological studies of the human tumor xenograft models (MDA-MB435),
demonstrated that after eribulin treatment, the
EMT results in the mobilization and spread of primary tumor cells to distant locations due to the morphology and mobility of the mesenchymal cells [32]. In a preclinical study, it was demonstrated that loss of the tubulin polymerase ch-TOG from the growing plus ends of microtubules is caused by eribulin, which is causative to variations in microtubule dynamics [58]. During metastasis, tumor cells gain the ability to invade the tissue beyond their normal boundaries and enter the circulation, and seed new tumors at other locations [59]. Watanabe et al. [52] observed concentration dependent effects on metastatic osteosarcoma cell line- LM8 due to eribulin treatment. Cell cycle arrest and apoptosis was instigated due to high doses of eribulin whereas, low doses led to changes in the cell morphology, decreased LM8 migration and reduced the directionality during migration. In a murine osteosarcoma metastasis model, lung metastasis and primary tumor growth were inhibited sue to increased doses of eribulin administered on a standard schedule [52]. TNBC metastasis is shown to be associated with Wnt/beta catenin signaling pathway [60]. In the non-basal-like cells, Wnt3a expression considerably, leading to the inhibition of Wnt/beta catenin signaling cascade and induced cancer cell apoptosis [61]. Eribulin might be a candidate for Small bowel adenocarcinoma (SBS) treatment due to its inhibitory effect on Wnt/beta catenin signaling [62].
Besides the different mechanisms mentioned already, eribulin is shown to have specific inhibitory activity against TERT-RdRP as well [63]. TERT-RdRP is responsible for mitotic progression through the elevation of heterochromatin assembly and the maintenance of stem-cell property [64]. Eribulin-sensitive ovarian cancer cell lines expressed high levels of human TERT, and inhibition of the TERT expression reduced the eribulin sensitivity. Thus, suppression of the cancer cell growth by eribulin treatment was shown to be dependent on the TERT status [63]. The human TERT is shown to localize into mitotic spindles and centromeres during mitosis [65], Therefore it has been hypothesized that eribulin inhibited human TERT functions by interfering with the interaction between TERT and microtubules [63]. Subcutaneous U87MG brain tumor xenografts treated with eribulin was observed to have a decreased TERT-RdRP activity in a dose-dependent manner [66].
Several in vitro studies have accelerated the efforts to undertake the
clinical use of Eribulin (Table 1, Ref. [6, 14, 21, 67, 68]). Eribulin treatment
with wide varieties of human cancer cell lines such as breast, prostate,
melanoma, colon, lung, ovaries, histiocytic lymphoma, pharyngeal squamous cell
carcinoma and uterine sarcoma cell lines resulted in effective inhibition of cell
growth even at very low concentrations (IC
| Cell line | Origin | IC50 | Aim of the study | Conclusions | References |
|---|---|---|---|---|---|
| COLO 205 DLD-1, HL-60, U937, LNCaP DU 145, MDA-MB-435 | Colon cancer, promyelocytic leukemia, histiocytic lymphoma, prostate cancer, breast cancer | 0.4 |
To analyse the growth inhibitory activities of Eribulin and its mode of action | Induced G2/M cell arrest | [6] |
| MCF7 | Breast cancer | 1.5 nM | To analyze the mechanism of action of Eribulin | Inhibition of microtubule growth | [14] |
| U937, Jurkat, HL-60, HeLa | Lymphoma, T cell leukemia, cervical cancer | 10 nM | To analyze mitotic block reversibility/irreversibility pharmacodyanamics | compound-specific reversibility characteristics of antimitotic agents contribute to interactions between cell-based pharmacodynamics | [21] |
| Calu-1, A549 | Non-small cell lung carcinoma (NSCLC) | 0.5 pM | To analyze the anticancer activity | Anticancer activity achieved | [67] |
| U87MG, U251MG, U118MG, LN229 | Glioblastoma | Below 1 nM | To analyze the growth inhibition efficiency of Eribulin in glioblastoma | Inhibition of growth a subnanomolar concentrations | [68] |
Eribulin treatment with human tumor xenograft models originated from breast, ovary, colon, lung, melanoma, pancreatic, and fibrosarcoma resulted in increased survival rates, tumor growth delays and reductions in size at dose levels below the maximum tolerated dose [6]. Eribulin showed in vivo anticancer efficacy in MDA-MB-435, COLO 205, and LOX xenograft models at much lower doses when compared with paclitaxel [6]. It has been proven that intermittent dosing was less toxic and more effective than daily dosing in preclinical models. Dose scheduling studies with eribulin on MDA-MB-435 breast, HT-1080 fibrosarcoma, U251, glioblastoma, SR-475 head and neck cancer, SK-LMS-1 leiomyosarcoma, NCI-H322M and NCI-H522 NSCLC, PANC-1 pancreatic cancer, and NCI-H82 small cell lung cancer (SCLC) models showed that maximal efficacy and minimal toxicity is achieved with moderate intermittent dosing [21]. Results from Pediatric preclinical testing program in childhood cancer models of different cancer histologies showed that eribulin exerted higher antineoplastic activity in ES xenografts than VCR [70, 71]. A latest report showed that eribulin penetrated the brain tumor tissue and prolonged the survival of mice by inhibiting the growth of glioblastoma cells transplanted subcutaneously or intracerebrally into mice [66].
Phase-1 trials are intended to identify the maximum tolerated dose,
pharmacokinetics, toxicity profile and dose-limiting toxicities. First phase-1
trial of eribulin was conducted in patients with advanced solid tumors [72].
During such phase-1 trials, different dosing schedules were checked.
Pharmacokinetic data showed linear, dose-proportional kinetics with a rapid
distribution phase and a slower elimination phase [73]. As of now, 25 phase-1
studies are completed [74] in patients with different cancer types including
advanced solid tumors, breast, ovary, head and neck, colon and bladder cancers.
Eribulin has been well tolerated and has demonstrated good clinical response in
breast, NSCLC, urothelial/bladder, and thyroid cancers. Febrile neutropenia,
fatigue, anorexia, and peripheral neuropathy are some of the most common doses
limiting toxicities reported [72, 73, 75, 76, 77, 78, 79]. The pharmacokinetic profile of
eribulin showed an extensive volume of distribution, slow-to-moderate clearance,
and slow elimination. Area under the plasma concentration versus time curve (AUC)
and maximum plasma concentration (C max) has been reported to be directly
proportional to the dose of eribulin (0.25–4 mg/m
Reassuring tumor response data from the phase-1 trials led to the initiation of
phase-2 studies in breast cancer, as well as in other solid tumor types. Phase-2
clinical trials gathered preliminary data about eribulin effectiveness on cancer
patients compared to similar participants receiving a different treatment, Safety
and adverse events were evaluated. As of now, 39 phase-2 studies on eribulin
treatment are completed [74] in patients with cancer types including breast
cancer, NSCLC, sarcoma, prostate, head and neck, urothelial, ovarian and
pancreatic cancers [80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102]. In order to address the eribulin intolerance, a
phase II, non-randomized, prospective study was conducted recently, which
reportedthat a bi-weekly eribulin schedule (1.4 mg/m
Phase-3 trials of eribulin involved more number of participants, different populations and different dosages of the drug in combination with other drugs were tested to ensure safety and effectiveness. As of now, 6 phase-3 studies are completed [74] in patients with breast cancer, STS and NSCLC [10, 11, 104, 105, 106]. After getting FDA approval for marketing, two phase-4 trials (sponsored by Eisai) have been completed as of now, to obtain additional information on the safety, efficacy, and optimal use of eribulin by the study sponsor [107]. Very recent results favoring eribulin as a potent candidate for first-line treatment of patients with HER2-negative advanced breast cancer are obtained in a multicenter, prospective, post-marketing, observational study from Japan [108].
Clinical trials that gained approval for eribulin as a drug for cancer treatment are given in the table (Table 2, Ref. [10, 11, 93, 102, 104]). US Food and Drug Administration (FDA) approved eribulin in the year 2010 for the treatment of patients with metastatic breast cancer who were already pretreated with anthracycline and a taxane regimen. One of the pivotal phase-3 studies on eribulin treatment was Study 305/EMBRACE (Eisai Metastatic Breast Cancer Study Assessing Physician’s Choice vs E7389 Study). It was the first mono-therapy study of a cytotoxic agent to show increase of survival in patients with heavily pretreated metastatic breast cancer. Overall survival of eribulin (OS) was observed as 13.1 months versus treatment of physician’s choice (TPC) where the OS was 10.6 months. Survival benefit of 2.5 months was considered statistically significant. Neutropenia was the most common clinical grade 3 or 4 adverse event with eribulin (grade 3: 106 [21%] of 503 patients; grade 4: 121 [24%] of 503 patients) [10].
| Trial design | Cancer type | Eligibility criteria of the patients | Treatment | Number of patients | Efficacy | References | ||
| ORR | Median PFS | Median OS | ||||||
| (%) | (months) | (months) | ||||||
| Phase-2 | MBC | Women with MBC pre-treated with anthra- | eribulin mono therapy | 291 | 9.3 | 2.6 | 10.4 | [93] |
| Single arm | cyclin, taxane and capecitabine | (eribulin mesylate (1.4 mg/m |
||||||
| Phase-3 | Locally recurrent or MBC | Women with MBC who had received 2 to 5 prior anthracycline- and taxane-based therapy | eribulin vs Treatment of Physicians Choice (TPC) | 762 (total) | 12 vs 5 | 3.7 vs 2.2 | 13.1 vs 10.6 | [10] |
| Study 305 (EMBRACE trial) | 501 (eribulin) | |||||||
| Open label, randomized (2:1), parallel, multi-centred (North America, Western Europe, Eastern Europe, Latin America, South Africa, Asia) | (eribulin mesylate (1.4 mg/m |
254 (TPC) | ||||||
| Phase-3 | MBC | Women with MBC who had received prior | eribulin vs capecitabine | 1102 (total) | 11 vs 11.5 | 4.1 vs 4.2 | 15.9 vs 14.5 | [104] |
| Study 301 | anthracycline- and taxane-based therapy | 554 | ||||||
| Open label, randomized (1:1), | (eribulin mesylate (1.4 mg/m |
(eribulin) | ||||||
| multi-centered (North America, Western Europe, Eastern Europe, Latin America, South Africa, Asia) | on days 1 and 8 versus capecitabine 1.25 g/m |
548 (capecitabine) | ||||||
| Phase-2 | Four independent strata of patients with mesenchymal tumours: | Patients with progressive or high-grade STS who had received not more than one previous combination chemotherapy or up to two single drugs for advanced disease and have no pre-existing neuropathy higher than grade 2 | eribulin mono therapy | 128 (total) | Not reported | 2·6 (adipocytic sarcoma) | Not reported | [102] |
| Non randomized, multi-centered (14 sites in five European countries) | (i) Adypocytic sarcoma | Following sarcomas were defined as ineligible: embryonal rhabdomyosarcoma, chondro-sarcoma, osteosarcoma, Ewing family of tu- | (eribulin mesylate (1.4 mg/m |
37 (Adipocytic sarcoma) | 2·9 (leiomyosarcoma) | |||
| (EORTC -trial 62052) | (ii) Leiomyosarcoma | mours, gastro-intestinal stromal tumours, dermato fibrosarcoma protuberans, and inflamma- | 40 (Leiomyosarcoma) | 2·6 (synovial sarcoma) | ||||
| (iii) Synovial sarcoma | tory myofibroblastic tumours. | 19 (Synovial sarcoma) | 2·1 (Other types of STS) | |||||
| (iv) other STS | 32 (Other STS) | |||||||
| Phase-3 | Advanced or metastatic STS | Patients with intermediate-grade or high-grade advanced liposarcoma or leiomyosarcoma who had received at least two previous systemic regimens for advanced disease (including an anthracycline) | eribulin vs dacarbazine | 452 (total) | 5 vs 6 | 2.6 vs 2.6 | 13.5 vs 11.5 | [11] |
| Open-label, randomized, multi-centered (across 110 study sites in 22 countries) | eribulin mesylate (1.4 mg/m |
228 (eribulin) | ||||||
| 224 (dacarbazine) | ||||||||
| MBC, metastatic breast cancer; STS, Soft tissue sarcoma; ORR, Objective response
rate; OS, Overall survival; PFS, Progression free survival. TPC | ||||||||
In another phase-3 trial (study 301), eribulin showed almost same efficacy as
capecitabine while considering overall survival (OS) or progression free survival
(PFS). The treatment had manageable safety profiles consistent with the known
adverse effects; most adverse events were grade 1 or 2 [104]. As per the request
from European Medicines Agency (EMA), pooled analysis of the above two phase-3
trials was carried out, to assess whether specific patient sub-groups, previously
treated with an anthracycline and a taxane, benefited from eribulin. In this
pooled analysis, serious adverse events were presented in similar magnitudes in
the two groups (eribulin 21.1%; control 22.6%). Conclusively, 10.5% of
patients in the eribulin group and 12% in the control group had an adverse event
that led to discontinuation of the treatment and [109]. A subgroup analysis of
392 patients with human epidermal growth factor receptor 2 (HER2)-negative MBC
showed a longer OS with eribulin (16.1 months) versus capecitabine (13.5 months)
in this subgroup. The most frequent treatment-emergent adverse effect in the
eribulin group of this study were neutropenia (53.3%), alopecia (34.8%),
leukopenia (31.0%), peripheral neuropathy (pooled term, 23.9%), and anemia
(21.2%) grade 3 or 4 neutropenia occurred in 43.5% of eribulin treated patients
[110]. Very recent Post hoc, pooled exploratory subgroup analysis of EMBRACE and
Study 301 revealed that eribulin provided a median OS benefit of 2.1 months over
TPC/capecitabine in patients who received fewer prior regimens (
Phase-3 study by Schoffski et al. [11] showed that better improvement of OS in the liposarcoma group compared to all sarcoma subtypes. The PFS was similar in both the study arms. Similar results were also observed in the MBC trial (study 301) with improvement in OS but without notable difference in the PFS (Table 2). The benefit of eribulin monotherapy based on improvement in OS led to approval by the FDA and EMA in 2016 for patients with unresectable (inoperable) or metastatic liposarcoma who had already undergone treatment with anthracyclines.
Combination therapy is a treatment modality in which rational combinations of different anticancer agents are employed [112]. Synergistic effect of eribulin and other chemotherapeutic drugs are carried out to minimize the toxicities and to improve the drug sensitivity. Eribulin makes effective chemotherapeutic combinations with several cytotoxic and targeted inhibitors from widely differing mechanistic classes. The combination of S-1 (5-FU) and eribulin exerts a synergistic effect for TNBC cell lines through MET-induction by eribulin. Such combination therapy could be a potential treatment option for TNBC [113]. Several investigations were conducted in which, eribulin was combined with either one of the cytotoxic agents (capecitabine, carboplatin, cisplatin, doxorubicin, gemcitabine) or targeted agents (bevacizumab, BKM-120, E7449, erlotinib, everolimus, lenvatinib, palbociclib) in tumor xenograft models of breast cancer, melanoma, non-small cell lung cancer (NSCLC), and ovarian cancer. Eribulin showed positive combination activity with most of the agents with more efficacy than the individual agents [114]. Synergistic effects of eribulin on CDK inhibition [115], PIK3 inhibition [116, 117] and mTOR inhibition [118, 119] in TNBC and pAKT inhibition in STS [120] were carried out with corresponding inhibitors. In all these studies, positive synergistic effects were noticed. It is indeed a powerful strategy to increase the therapeutic index of the drug combination to improve drug efficiency even at the lowest doses on the aggressive or resistant cancer types. Recently several phase-1and phase-2 clinical trials were conducted with eribulin in combination with other chemotherapeutic agents to evaluate and improve the therapeutic efficiency [76, 77, 78, 79, 80, 82, 84, 100, 101, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132].
Resistance to chemotherapeutic drugs is a major obstacle to therapeutic success [133]. A common mechanism of drug resistance in various tumors is identified as over expression of a drug efflux pump such as P-glycoprotein (Pgp)/(ABCB1) [134]. Eribulin increased the mRNA and protein expression of P-glycoprotein in LS174T cells and MDR1 promoter activity in human breast cancer MCF7 cells thereby the drug efflux transporter P-glycoprotein is activated [135].
In a study involving 7 different eribulin resistant cell lines, ATP-binding cassette subfamily B member 1 (ABCB1) and subfamily C member 11 (ABCC11) were observed to be overexpressed and had an increased gene expression as well [136]. Both were responsible for the development of eribulin resistance in breast cancer cells regardless of the molecular subtype. Thus, it was suggested that ABCB1 and ABCC11 could be used as a biomarker for predicting the eribulin response in patients with breast cancer [136]. Studies have proven that therapeutic targeting of EMT may be beneficial in treatment-resistant cancer subtypes. Vimentin and claudin-1 are important markers of EMT pathway activation which were significantly up regulated in eribulin resistant cell lines [43]. Eribulin-resistant leiomyosarcoma cell lines were investigated to understand the mechanism of resistance [137]. Eribulin resistant cell lines express higher TUBB3 levels and grow more promptly than the parent cell line. Knockdown of TUBB3 by siRNA resulted in the inhibition of cell growth. Therefore, it could be predicted that if over expression of TUBB3 is observed for a type of cancer, eribulin would be suggestively be the drug of choice for treatment and alternative treatment approaches should be considered.
Further mechanistic understanding of Pgp activation, ABCB1 and ABCC11 activation, TUBB3 over expression might lead to novel therapeutic regimen to bridle eribulin resistance in the future.
EMT plays a crucial role in cancer by the generation of Circulating Tumor Cells (CTCs) that exist in peripheral blood as single cells or as clusters of tumor cells along with platelets and lymphocytes and involved in tumor metastasis [138]. Enumerating the CTCs is an advanced liquid biopsy approach and an emerging tool for predicting patient outcomes and treatment effects [139, 140, 141]. The effectiveness of eribulin treatment is evaluated by enumerating CTCs thereby predicting the efficiency of the drug. Preliminary studies have indicated that assessment of both mesenchymal and epithelial CTCs might be important for predicting eribulin responsiveness [142, 143].
Detection of serum miRNAs has emerged recently as potent tool to predict the prognosis and response to cancer therapy [144]. A serum micro-RNA (miRNA)-based prediction model has been established for the emergence of new distant metastases after eribulin treatment [145]. Evidences indicate that serum miRNA profiling may serve as a biomarker for the responsiveness to eribulin and for predicting the development of new distant metastases in metastatic breast cancer [145]. An exploratory clinical trial was aimed at identification of putative miRNA biomarkers that associate with eribulin sensitivity or resistance in STS. Among the 756 miRNAs assessed, the expression of 26 individual miRNAs differed significantly between non-responders and responders [146].
Primary cultures moderately imitate the in situ microenvironment of the diseased condition and also preserve the crosstalk between malignant and healthy constituents. Such features are to be involved in the different responses to therapies and in all stages of the natural history of malignant tumors, i.e., carcinogenesis, migration, invasion and metastatic dissemination. Therefore, ex vivo models permit a more faithful reproduction of tumors and are a valid strategy especially for clinical and preclinical analyses [147]. Recently, efforts have been made to develop methods for tumor cell culture from biopsies and expansion of patient-derived circulating tumor cells CTCs) for individualized drug susceptibility [140, 141, 148]. A recent study by De et al. [149] had established an ex vivo 3D system for culturing CTCs derived from either primary or metastatic tumors that recapitulates their physiological characteristics. This scaffold imitates the architecture and stiffness of breast tumors and allows improved cell-matrix interactions. Patient-derived organoid models are also established as they offer spatial and temporal insights into cancer biology. These models conserve the heterogeneity of the primary tumor, which makes them more appropriate for identifying therapeutic targets and corroborating drug response [150]. The anticancer activity of eribulin on the patient-derived primary culture of a liposarcoma patient was assessed its antiproliferative effect by the arrest of cell motility and initiation of apoptosis [151, 152].
Even though eribulin is an approved clinical drug, sustained investigation of its antitumor characteristics may contribute enormously towards improved cancer management. Deeper understanding of molecular pathways would provide a better strategy of using eribulin in combination with other therapeutic regimens. As our knowledge on resistance mechanisms improves, specific drugs and drug combinations that would work on specific cancer type could be identified. Emerging techniques involving CTCs and serum miRNA profiling as prognostic methods for cancer patients are to be explored widely and employed in the future studies. Novel molecular and therapeutic strategies should aim at combating the disadvantages associated with the toxicity and resistance, which would enable eribulin therapy to be deliberated as a first-line treatment option.
PK conceptualized the draft and reviewed the manuscript. PS was involved in writing the manuscript and preparation of the figures. BD was involved in the revision and rebuttal of the manuscript.
Not applicable.
Not applicable.
This study received no external funding.
The authors declare no conflict of interest.
MBC, Metastatic breast cancer; STS, Soft tissue sarcoma; NSCLC, Non small cell lung cancer; TNBC, Triple negative breast cancer; FDA, Food and drug administration; TME, Tumor microenvironment; EMT, Epithelial mesenchymal transition; TGF- beta, Tumor Growth factor-beta; OS, Overall survival; PFS, Progression free survival; CTC, Circulating Tumor cells.