
Diabetes, obesity and increased body mass index are associated with changes in metabolism that lead to an inadequate reservoir or use of ATP in the heart and susceptibility to arrhythmia. Lack of availability of ATP and abnormal levels of metabolic end products can cause gene reprogramming and electrical remodelling that make myfibers susceptible to arrhythmia. Understanding the metabolic aberrations that lead to arrhythmia require better understanding of cardiac metabolism. Here, I discuss metabolic genes, enzymes and reducing equivalents and functional aspects of metabolic-induced arrhythmia with a special focus on atrial induced arrhythmia. It appears that normalisation of altered Kv1.5 channel, an oxygen sensing ion channel and fulfillment of oxygen demand by myocardium might offer a new strategy for preventing alterations of repolarisation that cause arrhythmia.
Metabolism is a set of life-sustaining chemical reactions in organisms that provide energy and other substrates through distinct metabolic pathways. Metabolism provides the cellular ATP which is required in the regulation of normal physiological processes. The life-sustaining chemical reactions become abnormal in aberrant metabolism. This would consequently lead to inadequate generation of metabolic end products, and the cellular ATP, which the mammalian heart depends on to maintain its workload. On a wide range of workloads, the scale of the absolute cardiac ATP pool can vary minimally; however, the ATP turnover can vary significantly. As a result, moderate changes in cardiac metabolism can produce significant effects on cardiac function. Earliest studies of cardiac metabolism showed that reduced myocardial glucose and increased fatty acid oxidation in diabetic patients may be harmful to cardiac myocytes (1, 2). Abnormal signalling transduction, discordant heart rates and misconfiguration of action potential (AP) morphology may be linked to uncoupled respiration (3). Cardiac functions are therefore influenced by metabolism and signalling transduction, meaning that there is vicious feedback of numerous mechanisms that ensure effective response of the heart to its environment and the cardiac metabolism (Figure 1). In fact, abnormality in the series of chemical reactions, termed metabolic aberrance, that occur during glycolysis, Krebs cycle and oxidative phosphorylation in the heart can lead to aberrant gene and ion channel expression, which in turn constitute electrical remodelling and arrhythmogenesis. The impact of this is profound.
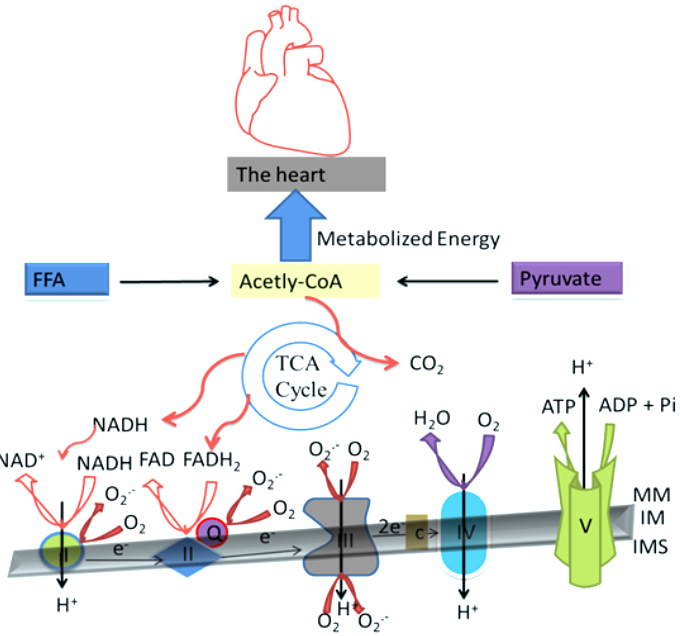 Figure 1
Figure 1Cardiac Oxidative Metabolism: Cardiac Oxidative Metabolism: The Krebs cycle aconitase produces NADH AND FADH2 as substrates for electron transport chain. The cardiac oxidative phosphorylation starts with entrance of the electrons into the electron-transport chain. The electrons pass through four complexes I–IV in the electron-transport chain (the small faint black arrows down) with the aid of the electron carriers, the cytochromes, ubiquinone (Q), and iron-sulphur proteins. The transfer of the electrons creates membrane potential (mV) that pump protons out of the mitochondrial matrix. This generates electrochemical energy in the form of a proton-motive force in the inner mitochondria space (IMS). This force then enables the production of ATP as protons flow passively back into the mitochondrial matrix through a proton pore that is associated with ATP synthase. MM is mitochondria membrane and IM is the inner mitochondria.
For instance, cardiovascular complications and diseases such as atrial fibrillation, emanating from metabolic disorders are the leading cause of sudden cardiac deaths in the developed countries (4-6). Combatting metabolic-induced cardiovascular complicates and diseases, including arrhythmia remains an unmet need. Tremendous efforts at designing therapeutic approaches for the treatment of the complications and diseases have been ongoing, though without much attention to cardiac arrhythmia. There was reduced mortality rate in patients with myocardial infarction who upon admission received glucose and potassium orally and insulin subcutaneously (7). Ranolazine, an inhibitor of β-oxidation of fatty acid, at therapeutic concentrations partially reversed cardiomyocyte hypertrophic-related cellular alterations via late sodium current (INa) inhibition (8). The revision of glucose-insulin-potassium in acute coronary syndrome and in energy-depleted heart re-enforces classically and historically this approach and its benefits in alleviating metabolic disorders in the heart diseases (9).
Despite these approaches and others including the use of neurohumoral antagonist and devises, first, benign worrisome cardiovascular related mortality induced by abnormal metabolism persists, reaching millions in the developed nations. Second, the efforts have been hampered because complete understanding of the functional mechanisms of metabolic induced cardiac complications and diseases is currently lacking. Therefore, further studies are needed to better understand the mechanisms to improve the treatment strategies. In this direction, it became necessary to evaluate metabolism in the mechanisms of arrhythmia. The discussion of the aberrant metabolic pathways is not the scope of this review. The goal of which is to review functional aspects (figure 2) of metabolic-induced arrhythmia in addressing the unmet need. Specific atrial mechanisms were specifically highlighted with the aim to provoke further studies and revolutionize the current therapeutic approach in the combat of arrhythmias.
Metabolic consequences are adverse effects of abnormal metabolism and energy imbalance. Patients with diabetes, obesity and abnormal body mass index are at increased risk of cardiovascular complications and diseases (10-14). Diabetes and obesity increased incidence of AF (10). Arrhythmia is defined as irregular heartbeat. It is common, complex, challenging and a major cause of mortality, globally. Studies are advancing our knowledge on the cellular and molecular mechanisms of arrhythmia. They demonstrate perturbation in metabolic pathways such as, glycolysis, Krebs cycle and oxidative phosphorylation as a major cause of arrhythmias. However, the cellular and molecular mechanisms of metabolic-induced arrhythmia are not yet clearly understood. Several hypotheses including alteration of structural proteins and interstitial fibrosis (15), autonomic system (16,17), Ca2+-handling (18-20) and electrical remodeling (8,21,22) have been put forward to explain metabolic mechanisms of cardiac remodelling (Figure 2), but further insights into the hypothesis are needed to better understand the mechanisms and improve strategies for therapy.
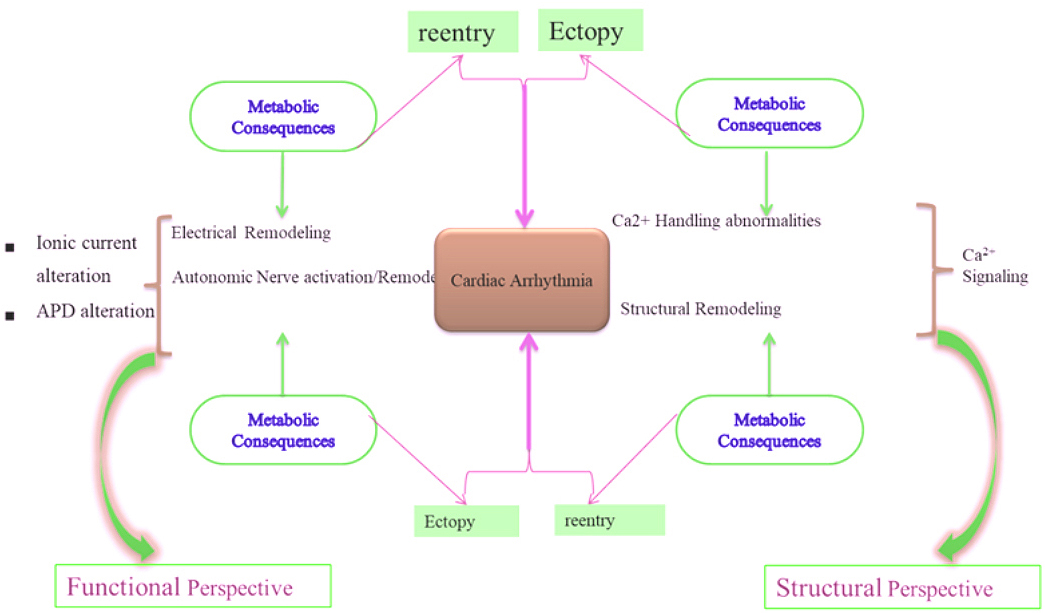 Figure 2
Figure 2Hypothetical effects of metabolic consequences on cardiac remodeling: Metabolic consequences induce functional and structural remodeling. Structural remodeling is characterized by tissue enlargement and fibrosis. Reentry substrate requires abbreviated refractoriness of the APD and/or conduction abnormalities. Ectopic firing occurs due to early after depolarisation, delay after depolarisation and action potential prolongation. These together produce cardiac arrhythmia.
The heart is a pump organ. It operates by mechanical action and therefore uses energy. Energy is the ability to do work and the mechanical actions of the heart that involve the use of energy include: excitation and contraction coupling, wall stress mechanics and heart rate. Oxygen is engaged in energy use. The heart as an aerobic organ engages oxygen in mechanical actions. Bing et al in 1949 suggested that mechanical efficiency of the heart is the ratio of functional energy performed to oxygen consumed. Adenosine triphosphate (ATP), an energy-rich phosphate ester, as described several years ago is highly important to the ratio (24).
ATP yield or turnover varies significantly according to substrate availability, oxygen supply, and metabolic demands of the body. The variability influences pathophysiology of the heart. Acute increase in afterload was characterised by switches in the utilisation of the major energy substrates and in the activities of metabolic enzymes (25) that regulate the energy substrates. More so, diabetes can be a function of metabolic reverse, energy status or ATP turn over, especially in occasional rise in blood glucose levels. Metabolic reverse is the unused potential energy available in response to an increase in cardiac workload according to demand. This is maintained by the heart’s complex metabolic energy machineries that breakdown carbon-based fuels or source for ATP and ensure that ATP concentration (ATP) remains constant regardless of changes in ATP turnover. The prevailing response of the heart to the vicious feedback process between cardiac metabolic demand and substrate availability could be detrimental, especially in chronicity.
Such a complex scheme of interlocking mutual response, characterised by insufficient availability of ATP, as result of aberrant metabolism, can produce electrical remodelling overtime. Electrical remodeling is the changes in normal working modality of a plethora of ion channels that generate and maintain AP. Changes in the expression of genes that carry ion channels and/or in channel current density cause electrical remodeling. Major principal culprits that inundate remodeling are glucotoxicity, lipotoxicity and glucolipotoxicity (12). Energy storage, intake or expenditure as critical components of energy imbalance produces the culprits. When fatty acid availability is more than fatty acid oxidation, intra-myocardial lipids accumulates, leading to lipotoxicity. When energy expenditure exceeds energy intake, the consequence is decrease in body mass, while when intake exceeds expenditure, the consequence is increase in body mass.
Hyperglycemia, lipidemia and hyperglycelipidemia are in fact pathological and affect the electrical remodelling of the heart and ionic homeostasis. Hyperglycemia can produce advanced glycation end-product (AGE)-associated reactive oxygen species (ROS) (26) and ROS alter electrical properties and cause remodeling (27). Advanced glycation end-product (28), hyperlipidemia associated with increased expression of the acyl transferase (29) and hyperglycemia (30) through reduced nitric oxide (NO) (31) decreased Na+/K+-ATPase activity, which is one of the most energy-demanding subcellular processes that maintains cellular gradient for Na+ and K+. Suppression of increased ROS production through application of superoxide dismutase and increase in the bioavailability and synthesis of NO through application of L-arginine reversed the decrease in the activity of Na+/K+-ATPase (31). Together, the findings suggest that metabolic ROS can alter ion channels both molecularly and genetically. The alteration is arrhythmogenic.
For instance, increased Na+ influx and Na+ overload in diabetes type 2 may contribute to arrhythmogenesis (32). How metabolic intracellular Na+ accumulation causes arrhythmia is very complex and multifactorial. In addition to initiating Ca2+ loading (33), it can trigger net loss in K+ due to lack of electro neutrality (33). Furthermore, increased Ca2+ influx following Na+ accumulation, when there is lack of oxygen supply, can be explained by pro-arrhythmic enhanced late INa, (34, 35). Intracellular Na+ accumulation and insufficient oxygen supply resulted in calcium overload that was attenuated by ranolazine inhibition of INa in ischemia (35). Consistently, in 191 patients under Monotherapy Assessment of Ranolazine In Stable Angina trial at 52 investigational sites from U.S., Czech Republic, Poland, and Canada, ranolazine offered a metabolic approach of treating ischemia by delaying and preventing imbalance in oxygen demand that characterize ischemia, glucose and fatty acid cardiac metabolic maladaptation (36).
Taken together, metabolic consequences influence cardiac function by changing the working modality of numerous cardiac ion channels and ionic components. How this occurs is not known completely. Whereas ventricular mechanisms have been extensively studied (27,33), atrial specific mechanisms remain largely known and advancement in the subject has not provided insight into underpinnings, correlations and into metabolic reducing equivalents such as flavin adenine dinucleotide (FADH2), that are molecular moieties, that transfer electron analogous in oxidation-reduction reactions in the tricarboxylic acid cycle (TCA) within the mitochondria electron transport chain (ETC) that might lead to arrhythmias. This in part highlights the importance of this study. With an interest in the atria, I start with succinct discussion on some ion channels that contribute in generating the electrical properties of the heart.
IKACh is a member of the K+ channel families. K+ channels form the largest ion channel family. IKACh as a transmembrane ion channel is directly modulated by G-proteins in atrial myocytes and neurons. IKACh regulates the parasympathetic activity of the heart (37, 16). The modulation involves acetylcholine binding from vagal nerve endings to M2-muscarinic receptors localized primarily in atrial cardiomyocytes. The binding triggers disassociation of G-proteins into Gαi2 and Gβγ subunits. The latter subunit activates the G protein–coupled inward rectifying K+ channel (GIRK1)2/(GIRK4)2 to produce acetylcholine-activated K+ current (IKACh). The IKACh is primarily localized in atrial (38, 39). To discuss IKACh and metabolism, I articulated the literature according to the available evidence and reflected on how the chemical energy insufficiency might remodel IKACh, leading atrial fibrillation.
Intracellular Na+ as well as Gβγ, the activating subunit of the G-proteins gates IKACh (40, 41). The gating mechanism could be dependent on the level of hydrolysis of intracellular ATP. Hence, blockade or reduction of phosphatidylinositol 4,5-bisphosphate (PIP2), a transducer protein that mimics perturbation of the intracellular ATP hydrolysis decreased the Gβϒ subunit and the intracellular Na+ dependent activation of IKACh (42). Conversely, ATP and PIP2 liposomes application enhanced IKACh (42). Strong evidences, in part, implicate ATP in IKACh functional activity (43, 44), which may be important in vagal tone remodelling and chemical energy insufficiency. In general, substrate switch and metabolic flexibility as features of physiological function are lost in maladapted heart. In maladaptation, the heart prefers to use fatty acid oxidation as the source of metabolic fuel instead of glucose oxidation. The reliance on fatty acid oxidation is less efficient to the heart as it uses more oxygen and the ATP. The fatty acid oxidation dependence remodels the parasympathetic system, as observed in patients with diabetic autonomic neuropathy, which developed arrhythmias (16) and subsequent sudden cardiac death (59).
Accordingly, an association has been described between sterol regulatory element binding protein-1 (SREBP-1), a lipid transcriptional factor implicated in atrial arrhythmia susceptibility and the parasympathetic function in diabetes. In Akita (diabetes type 1) mice, direct stimulation of the intrinsic parasympathetic signaling pathway with carbamylcholine was reduced compared to the control heart (46). The reduction in the intrinsic downstream signaling pathway showed that the level of expression of GIRK1& 4 and its conducting current might be reduced. Hence, insulin treatment conversely restored the parasympathetic dysfunction and increased GIRK expression (46). Accordingly, either insulin treatment or adenoviral expression of SREBP-1 conversely, corrected energy depletion, reversed parasympathetic dysfunction, increased GIRK expression and IKACh (46,47). Suggesting that in energy insufficiency GIRK activity might be reduced as well as, IKACh activation; decrease in GIRK activity has also been reported as a mechanism of permanent AF (48). In fact, metabolic derangement remodels IKACh.
Another member of the K+ channel families is Kv1.5. Kv1.5 channel underlines cardiac IKur and activates very rapidly. Its biophysical properties makes it difficult to separate from other K+ currents including Ito, which it overlaps at certain voltage ranges. The IKur α-subunit localizes largely in the atrial myocytes and to a very less extent in the ventricular myocytes (49), indicating that the expression and properties of Kv1.5 may be more predominant in the atrial. Consistently, Kv1.5 is not expressed in human ventricle myocytes and purkinje fibers, arguing that IKur may be a selective source for targeting atrial arrhythmias without constituting ventricular proarrhythmia (50).
The Kv1.5 channel is a known oxygen sensing ion channel (51,52). Metabolic alterations and associated ROS remodels Kv1.5. ROS are products of oxidative respiration. Cardiac Kv1.5 channel is regulated by ROS (53,54). In hypoxia, ischemia, and diabetes, Kv1.5 channel is reduced by high levels of ROS (53,55). The findings indicate that increase in ROS remodels IKur. IKur remodelling due to altered metabolism and associated ROS is arrhythmogenic. Studies demonstrating the role of Kv1.5 channels in metabolic arrhythmias are very scares. Morrow et al report appears to be among the very few works at present that has describe alteration of the Kv1.5 in metabolism in the context of cardiac specific genetic upregulation of peroxisome proliferator–activated receptor γ1 (PPARγ1) that recapitulates diabetes and obese-induced ventricular arrhythmia. In respect to lipids per se, the alteration of Kv1.5 in the Morrow et al report is reinforced by the hypothesis that Kv1.5 remodelling was followed by membrane depletion of cholesterol (56).
Considering the Kv1.5 sensitivity to oxygen availability and selective presence in the atria, protection from aberrant levels of metabolic ROS would preserve cardiac IKur and reverse alteration of repolarization, implying that normalisation of altered Kv1.5 might in turn regulate oxygen insufficiency in metabolic arrhythmias. Inhibition or knockout of Kv1.5 can correct alteration of repolarisation reserve and prevent arrhythmogenesis. Together, normalisation of altered Kv1.5 channel, an oxygen sensing ion channel and fulfillment of oxygen supply for the myocardium might offer a new strategy for preventing alterations of repolarisation that cause arrhythmia.
Structural intercalated disks join cardiac myocytes. Adjoining the disks is gap junctions formed by Cx. Cx transmit impulses from cell-to-cell in electrical propagation in neurons and cardiac myocytes. Cx ensure low membrane resistance and passage of the electrical impulse, second messengers and metabolites within apposing cells. This junctional communication can be selective because the junctions have 2-3 nm pore diameter that appose cells from each other within the plasma membrane. Gap junctions are expressed in non-excitable and excitable tissues and have huge functional significance.
They have different unique biophysical properties, which underline their different phenotypes in different cells types they are expressed (57). Cardiac myocytes express different connexin types (58). While the ventricular myocytes express Cx43 and Cx45 (59), the atrial myocytes express Cx40 in addition (59,60). The differential expression may have suggested that specific Cx perform impulse transmission within specific heart chambers. For instance, ventricular conduction velocity was reduced in mice lacking Cx43 without AF, but not in the atria (59). Conduction velocity in human atrial was principally regulated by Cx40 (60). However, atrial specific genetic deletion of Cx43 in a known healthy young female apparently resulted in AF (61).
Nonetheless, while alteration in Cx expression constitutes reentrant arrhythmia, normalisation of abnormal Cx expression to some extent is effective in preventing AF. Either Cx40 or Cx43 gene transfer abolished the development of AF and reentrant mechanism due to gap junction protein alterations (62). Furthermore, Gap junction modulator, rotigaptide reduced AF vulnerability in a canine mitral incompetence model of AF (63). Put the Guerra et al, Igarashi et al and Thibodeau et al reports together, it is comprehensible in their contexts that targeting Cx43 and 40 isotypes in AF rather than just Cx40, the principal atrial isotype would be rather a better approach in treatment of Cx abnormal conduction velocity. These reports highlight great functional roles of Cx43 in the atrial conduction velocity in contract to Thomas et al (59) and Dupont et al (60) reports. Together, it advocates further works on atrial Cx43 to critically evaluate their extent of localisation and roles in the pathogenesis of AF.
The functions of Cx have been suggested in metabolic homeostasis, cell differentiation (64), cell development (65), and in growth (64,66). In cancer cells, Cx act as conditional modulators of cell proliferation, adhesion and migration. Alterations in intercellular Cxs electrical coupling due to energy levels are critical for reentrant arrhythmias. It can slow the cardiac myocytes conduction velocity. Weak synchronous electrical coupling, underpinned by changes in the levels of Cx proteins, increased susceptibility to AF (60,62,63,67). Given that Cx hemichannels release chemical energy (68), a parsimonious explanation for the weak electrotonic coupling and reentrant arrhythmia, might be associated underlying ATP and oxygen use. For instance, increased dephosphorylated Cx43 in ventricular myocytes constituted the development of lethal arrhythmia in accumulation of toxic metabolites (69), due to depletion of the metabolic ATP and glucose (70). Obesity (13) and diabetes (71) can cause arrhythmogenesis through energy imbalance-induced Cx43 alteration, as shift in energy balance, which alters metabolic regulation is a feature of diabetes and obesity. Consistently, levels of ATP corresponded to astrocytes gap junction coupling (72). These observations are further supported by the illustration that energy depletion in ventricular arrhythmias is associated with Cx43 conduction slowing (73). These studies have highlighted that alteration of Cx43 due inadequate chemical energy can lead to the formation of ventricular arrhythmia. Patchy evidence exists in the atria and represents a significant gap.
Cardiac energetics is contended in substrate utilisation. The principal factors which determine substrate utilisation are mechanical load, oxygen demand or requirement and signaling transduction pathways. These factors are at times limiting in the setting of cardiac disturbances. The substrate-providing metabolites such as fatty acid which yield about 90% and glucose which yield about 10% (74,75) act as sensors to the expression of gene encoding proteins that regulate the transport of the substrate-providing products and their metabolism (76,12). Accordingly, fatty acid induced mRNA upregulation of malonyl-CoA decarboxylase (MCD), fatty acid-handling protein acyl-coenzyme A synthetase (ACS), muscle-type carnitine palmitoyltransferase-1 (MCPT-1), long-chain acyl-coenzyme A dehydrogenase (LCAD) and the uncoupling proteins 2 and 3 (UCP-2/-3)) at transcriptional levels (77). A family of 3 transcriptional factors, SREBPs-1, that regulate expression of genes involved in lipid and glucose metabolism is modulated by glucose hormone (78). More so, Meox2 and Tcf15 haplodeficiency disabled FA uptake in cardiac endothelial cells and reduced contractility and FA transport to cardiomyocytes (79). The studies indicate that alteration of the genes encoding the substrate-providing metabolites (Table 1), can affect cardiac performance.
| Genes | Roles | References |
|---|---|---|
| Meox2 and Tcf15 | FA uptake in cardiac endothelial cells | 79 |
| Xanthine oxidase | Breakdown of purine nucleotide | 91 |
| Glycerol-3-phosphate dehydrogenase | triglyceride synthesis | 75 |
| ACS | breakdown of some essential amino acids and beta-oxidation of fatty acids | 77 |
| MCPT-1 | Transports long chain fatty acyl-COA into mitochondria matrix | 77 |
| UCP-2, UCP-3 | Regulation of mitochondria oxidative phosphorylation | 77 |
| MCD | Inhibitor of mCPTI Activation of fatty acids by removal of malonyl-CoA | 85; 86 |
| SREBPs-1 | Lipids and glucose metabolism | 78 |
| LCAD | Mitochondrial ß-oxidation of long-chain fatty acids. | 92 |
In a complex scheme of continual interaction between metabolic consequences and the functional myocardium through the life process, temporary and chronic alteration in expression of the metabolic genes can cause pathogenesis. For instance, failing adult hearts retrogress to a fetal pattern of energy substrate metabolism and metabolic gene expression (80) that can cause arrhythmias (81,82). Furthermore, a spectrum of cardiomyopathpies such as hypertrophy and diabetes are associated with chronic alteration in energy metabolism and gene expression (75,83,84). Conversely, improved homeostatic regulation can normalise the expression of the genes and reverse the alterations. Thus, long and short-term deletion of cardiac malonyl coenzyme A decarboxylase MCD gene in mice dramatically increased glucose oxidation and improved functional recovery of the heart after ischemia by correcting metabolic shift in energy utilisation between fatty acid and glucose (85,86). Taken together, the influence of alteration of the metabolic genes, which are discrete genes that directly govern metabolic pathways (Table 1) on substrate availability, can converge as metabolic consequences, which are squealers for cardiac ion channel remodelling. Consistently, SREBPs-1 is a critical regulator of GIRK1/4 which generates IKACh of the parasympathetic system, and in diabetes there is IKACh dysregulation of the parasympathetic system (46). Despite this knowledge, the literature is not yet at the stage to give a better account on cardiac ion channels regulation, metabolic genes and cardiac energy insufficiency.
Intermediary metabolism represents the intracellular processes by which metabolic substrates are enzymatically degraded into cellular usable products. Mitochondria tricarboxylic acid cycle (TCA) synthesis metabolic reducing products, known as high energy biomolecule. NADH and FADH2 as metabolic reducing equivalents are called high energy biomolecules because they have stores of chemical energy mobilised from initial enzymatic degradation of glucose and fatty acid molecules. NADH and FADH2 are reduced form of NAD and FAD respectively. Their redox ability is utilised by electron transport chain (ETC) for bulk of ATP production. Proton (H+) gradient that enables ATP synthase to produce ATP from ADP is generated from the NADH and FADH2 in a complex series of electron transfer involving the mitochondria enzyme complexes through ubiquinone.
FAD is specifically a prosthetic group of protein, but both FAD and NAD are coenzymes. As coenzymes they facilitate electron coupling in anaerobic and aerobic respiration. This notwithstanding, current discussions (20, 87,88) have authoritatively favoured NAD+/NADH, whereas FADH2 has more positive reducing potential than NADH (which is equivalent to semiquinone or FADH) (27, 33). This represents an important gap in knowledge. For instance, the importance of the reducing equivalents on cardiac function, disease diagnosis and therapeutic targets are completely unknown. Understanding the roles of FAD/FADH2 is therefore crucial. FAD holds a lot of promise in optogenetics for disease monitoring. In vivo native fluorescence of FAD and NADH may be relevant in the molecular imaging of electroanatomical substrate, which is currently a major challenge in understanding the science and treatment of AF. Native fluorescence FAD and NADH varied in normal tissue and oral submucous fibrosis as early signs of invasive oral cancer (89).
Furthermore, FAD-dependent genes and enzymes that fine-tune metabolism include: acetyl-CoA-dehydrogenases in the breakdown of some essential amino acids (lysine, valine isoleucine and leucine) not synthesized de novo, and in beta-oxidation of fatty acids, xanthine oxidase in purine nucleotide, glycerol-3-phosphate dehydrogenase in triglyceride synthesis, and the TCA succinate dehydrogenase that oxidizes succinate to fumarate. These functions are important to note since the roles of FAD in energy substrate metabolism is currently underrepresented. It also noteworthy that during electron coupling process, mitochondria ROS can be generated through NAD and FAD as well. This has been appreciated for NAD+/NADH but not for FAD/FADH2, whereas the mechanisms may be similar.
For instance, in diabetes, fatty acid substrate utilisation is preferred over glucose, resulting in increased expression of the fatty-handling metabolic genes, through perhaps a proportionately high level of the FAD-dependent genes expression and enzymes activity. This can in turn increase FADH/FAD ratio to elevate ROS production. It follows that ROS production in the mitochondria is elevated by increased NADH/NAD+ ratio (90). The coenzymes as well as metabolic enzymes are crucial elements in arrhythmogenesis, as their metabolic products control cardiac ion channel (88).
For about five decades now, efforts at combating metabolic-induced arrhythmia have not yielded a complete result as sudden cardiac death emanating from benign worrisome cardiac metabolic-induced arrhythmia persists at significant proportion in the population. Being a critical unmet need, targeting all but a combinatorial based therapeutic approach (Figure 3) obtained from different levels of the systems organisation would be more helpful. This study reviewed a broad spectrum of observations in the pathogenesis of metabolic-induced arrhythmia. Cardiac metabolism is complex, and a better understanding of cardiac metabolism requires a model of cardiac metabolism (Figure 4). Clearly, the literature is lacking on the subject. Thus, the persistence of a significant proportion of metabolic-induced arrhythmia in the population and lack of adequate therapy. It is hoped that this study will increase the understanding, stimulate further discussions and revolutionize the current strategy for targeting arrhythmias associated with abnormal metabolism. It is stated that a better picture of metabolic-induced cardiac complications and disease, including therapeutic management would emerge from conditions examining to what degree of effect does exclusive cardiac oxidative metabolism cause to cardiac structure and functions (Figure 5). For the future, the question should be to what extent do abnormal intermediary metabolism, redox state, Krebs cycle and oxidative phosphorylation exclusively alter: signaling transducer molecules and sensors, enzymes, metabolic genes and reducing products, cardiac ion channels and their ionic currents. Therefore, an era that will text necessary caveats on the fidelity of the connections between enzymes, metabolic reducing products, metabolic genes and cardiac ion channels in metabolic-induced cardiac arrhythmia is eagerly waited.
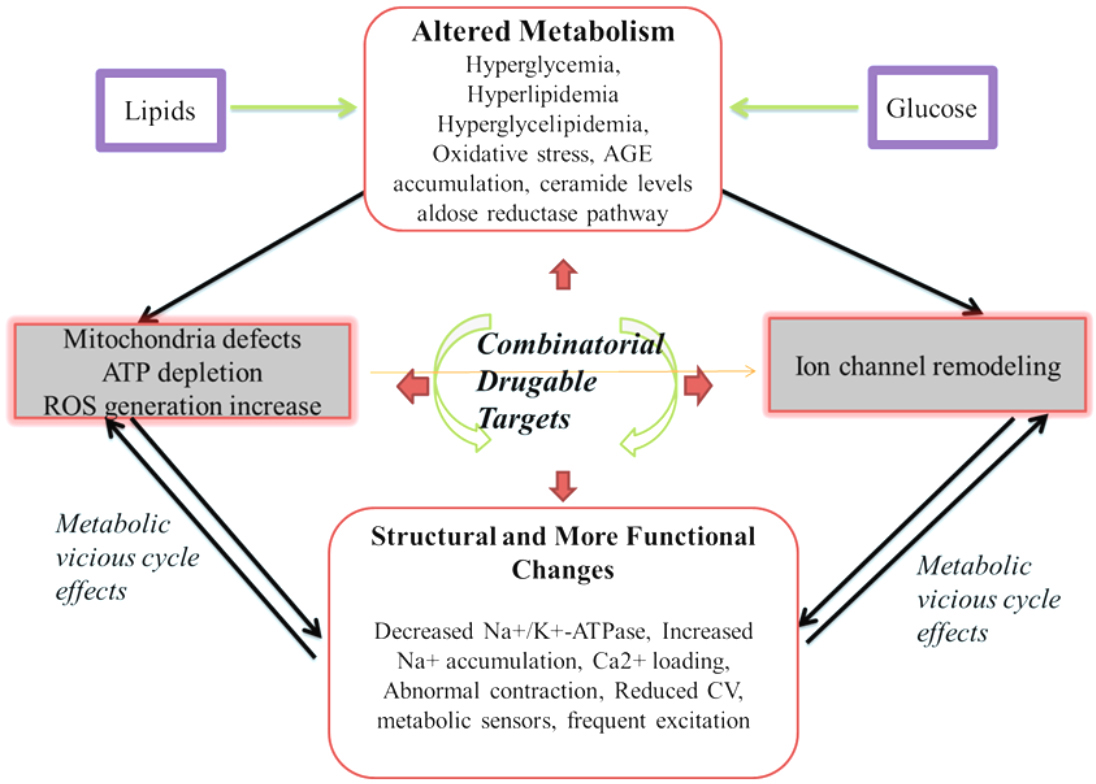 Figure 3
Figure 3Schematics of consequences and cellular effects: Abnormal glucose, lipid and fatty acid metabolism leads to hyperglycemia, hyperlipidemia hyperglycelipidemia, oxidative stress, advanced glycation-end products accumulation, changes in ceramide levels and in aldose reductase pathway. These phenotypes in turn effect normal working modality of ion channels, cause mitochondria defects, ATP depletion and increase in ROS production. Mitochondria defects, ATP depletion and increase in ROS production remodel ion channels (the central arrow). With a vicious cycle of effects, in between ion channel remodeling, and the mitochondria defects, ATP depletion and increase in ROS production are metabolic ionic imbalances. A combinatorial drugable approach would target all the phenotypes.
 Figure 4
Figure 4Model of cardiac metabolism: The average contribution of each component to the energy currency of the cell is highly critical.
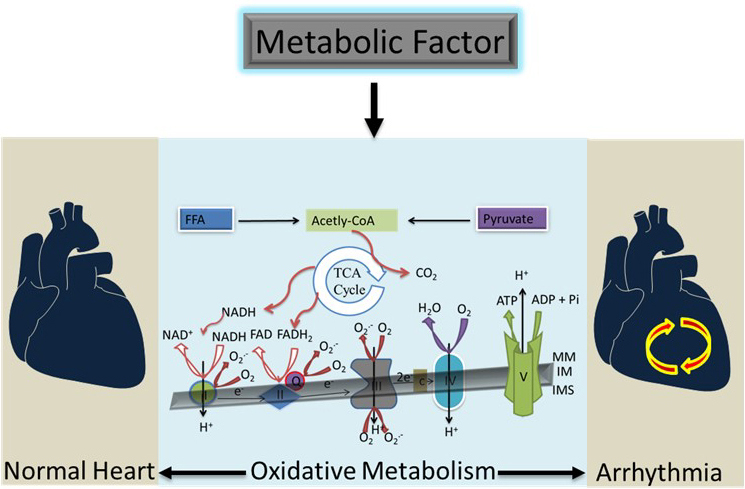 Figure 5
Figure 5The heart as the pump organ of the body requires oxygen which depends almost entirely on the aerobic oxidation of substrates for energy production. This consequently couples with cardiac performance, heart rate and wall stress. However, the exclusive impact of cardiac oxidative metabolism in the development of cardiac arrhythmia is not known and developing a model of cardiac oxidative metabolism to delineate the impact is necessary.
ADP
Adenosine diphosphate
Action Potential
Action Potential Duration
Adenosine monophosphate
Adenosine monophosphate-activated protein kinase
Adenosine triphosphate
Atrial Fibrillation
Connexins
Early after depolarisations
Excitation-contraction
Electron transport chain
Heart failure
Inward Ca2+ current
Inward Na+ Current
L-Type Ca2+ Channels
Malonyl-CoA decarboxylase
Sodium-calcium exchanger
Peroxisome proliferator-activated receptor γ1
Phosphatidylinositol-4,5-bisphosphate 3-kinase
Reactive Oxygen Specie
Tricarboxylic acid cycle
Transient outward potassium current
Two-pore-domain potassium
Small conductance Ca2+-activated K+ channels
Sterol regulatory element binding protein-1