





 , Krishnapriya M. Varier 1,2,4, Wenxue Li 1,2, Qin Liu 1,2,4, Qing Rao 1,2, Yubing Hang 1,2, Xiangchun Shen 1,2,4, Sheng Liu 1,2, Lei Huang 1,2,*
, Krishnapriya M. Varier 1,2,4, Wenxue Li 1,2, Qin Liu 1,2,4, Qing Rao 1,2, Yubing Hang 1,2, Xiangchun Shen 1,2,4, Sheng Liu 1,2, Lei Huang 1,2,* , Mei Xu 1,2,*, Yanmei Li 1,2,*
, Mei Xu 1,2,*, Yanmei Li 1,2,*1 State Key Laboratory for Functions and Applications of Medicinal Plants, Guizhou Medical University, 550014 Guiyang, Guizhou, China
2 Natural Products Research Center of Guizhou Province, 550014 Guiyang, Guizhou, China
3 School of Basic Medicine, Guizhou Medical University, 550025 Guiyang, Guizhou, China
4 School of Pharmaceutical Sciences, Guizhou Medical University, 550025 Guiyang, Guizhou, China
5 Department of Pharmacy, Guizhou Provincial People’s Hospital, 550002 Guiyang, Guizhou, China
†These authors contributed equally.
Abstract
Background: Glucocorticoids (GCs) are commonly used as the primary chemotherapy for lymphoid malignancies, including acute lymphoblastic leukemia (ALL). However, the development of GC resistance limits their prolonged use. Methods: In this study, we investigated the potential of a newly synthesized indole derivative called LWX-473, in combination with the classic GC Dexamethasone (DEX), to enhance the responsiveness of Jurkat cells to GC treatment. Results: Our findings demonstrate that LWX-473 alone or in combination with DEX significantly improves GC-induced cell apoptosis and arrests the cell cycle in the G1 phase. Notably, the combination of LWX-473 and DEX exhibits superior efficacy in killing Jurkat cells compared to LWX-473 alone. Importantly, this compound demonstrates reduced toxicity towards normal cells. Conclusions: Our study reveals that LWX-473 has the ability to restore the sensitivity of Jurkat cells to DEX by modulating the mitochondrial membrane potential, activating the expression of DEX-liganded glucocorticoid receptor (GR), and inhibiting key molecules in the JAK/STAT signaling pathway. These findings suggest that LWX-473 could be a potential therapeutic agent for overcoming GC resistance in lymphoid malignancies.
Keywords
- leukemia
- glucocorticoid resistance
- indole derivative
- Jurkat cells
- apoptosis
Glucocorticoids (GCs) play a pivotal role in the metabolism of cancer cells by binding to the glucocorticoid receptor (GR) in the cytoplasm. GCs can homodimerize, translocate into the nucleus, interact with the glucocorticoid response element (GRE) to induce gene transcription, change the expression of various oncogenes, and induce cell cycle arrest and apoptosis [1, 2]. The primary response of cancer patients to GCs corresponds to the overall clinical outcomes of therapy, but the development of initial or acquired drug resistance renders treatment ineffective or leads to chemoresistance [3].
Resistance to glucocorticoids is a complex process in many cancers, such as leukemia, lymphoma, and liver cancer [4]. It has been found that the level of the glucocorticoid receptor and its ligand determines the sensitivity of glucocorticoids in inducing leukemic cell apoptosis [5], thus affecting the effectiveness of GCs [6]. Researchers have also discovered that inhibiting the JAK/STAT pathway can overcome IL-7-induced resistance to glucocorticoids [7]. Additionally, the regulation of glucocorticoid resistance in pediatric T-cell acute lymphoblastic leukemia by increasing BIM expression with PI3K/mTOR inhibitors [8] can assist in tumor therapy. Despite continuous discoveries regarding the mechanism of glucocorticoid resistance, finding new treatments or drugs to overcome this resistance remains a scientific challenge.
Indole compounds play an important role in the development of anticancer drugs. For example, some natural or synthetic indole compounds, such as vincristine, vinorelbine, semaxanib, and sunitinib, have been widely used in various clinical anti-tumor treatments [9, 10]. Additionally, indole compounds have promising potential in addressing drug resistance. The indole alkaloid reserpine, which is used as an antihypertensive, increases tumor cell susceptibility to doxorubicin by inhibiting the efflux function of the P glycoprotein [11]. Newly synthesized indole compounds identified as double Aurora B/FLT3 inhibitors have nanomolar anti-leukemia activity and show good effects on sunitinib-resistant FLT3-ITD AML cells [12]. Therefore, the development of a new indole derivative to overcome glucocorticoid resistance in the treatment of leukemia holds great promise. In this study, a newly synthesized indole compound (LWX-473) was combined with GCs to evaluate its efficiency in overpowering chemoresistance in Jurkat cells. This was achieved by inhibiting the PI3K/AKT/JAK2/Stat3 pathways and inducing apoptosis of Jurkat cells through activation of the GR receptor and the mitochondrial pathway, as well as arresting the cell cycle at their G1 phase.
The human cell lines Jurkat (acute T cell leukemia; STCC10904G) and CEM/C1 (T
lymphoblast; STCC10910G) were purchased from Wuhan Servicebio Technology Co.,
Ltd, Wuhan, China . CEM-C7H2 (acute T lymphoblastic leukemia; GD-C60404492) and HL-7702 (normal
liver; GD-C60306615) were purchased from Shanghai Guandao Bioengineering Co.,
Ltd, Shanghai, China. All cell lines were validated by STR profiling and tested negative for
mycoplasma. All cells were cultured in RPMI 1640 medium (Gibco, Carlsbad, CA, USA) supplemented
with 5% FBS (Biological Industries, Beijing, China) in a 37 °C humidified incubator
with 5% CO
A solution of Methyl 3,5-dichloro-2-oxoindoline-3-carboxylate (1.0 g, 3.85 mmol)
and N-benzyl-2-(1H-indol-3-yl) ethan-1-amine (0.96 g, 3.85 mmol) in 10 mL of MeCN
was prepared. To this solution, DIPEA (2.01 mL, 11.54 mmol) was slowly added at 0
°C. The resulting mixture was then stirred at room temperature for 2
hours. Finally, the mixture was concentrated under reduced pressure. The crude
product was purified by flash column chromatography (DCM/MeOH = 300:1–150:1) to
obtain the product (1.15 g, yield 63%). Methyl 3-((2-(1H-indol-3-yl) ethyl)
(benzyl) amino)-5-chloro-2-oxoindoline-3-carboxylate: white solid, m. p:
153.2–154.5 °C
Cells (1.5
For apoptotic detection, cells (20
For the cell cycle analysis, Cells (20
The cells (2
The mitochondrial membrane potential (MMP) variations were detected using the
MMP assay kit with JC-1 (Beyotime, China) following the manufacturer’s protocol.
Briefly, the collected cells were incubated with JC-1 dye solution for 20 min.
Afterward, the cells were collected and washed (600 g, 4 °C) in JC-1 dye
buffer (1
Cells (5
All statistical analyses were performed using the ANOVA test, followed by
post-hoc analysis using GraphPad Prism9 Software (GraphPad Software, Inc., San
Diego, CA, USA). All experiments were conducted in triplicates with three
independent experiments. The data were expressed as mean
Natural indole compounds have shown promise in addressing drug resistance [18, 19]. However, these compounds are naturally produced in small amounts. To overcome this limitation, a new indole derivative, methyl 3-((2-(1H-indol-3-yl)ethyl)(benzyl)amino)-5-chloro-2-oxoindoline-3-carboxylate (LWX-473), was designed and synthesized using a previously reported method. The synthesis involved the reaction of methyl 3,5-dichloro-2-oxoindoline-3-carboxylate with N-benzyl-2-(1H-indol-3-yl)ethan-1-amine, as depicted in Fig. 1A–D.
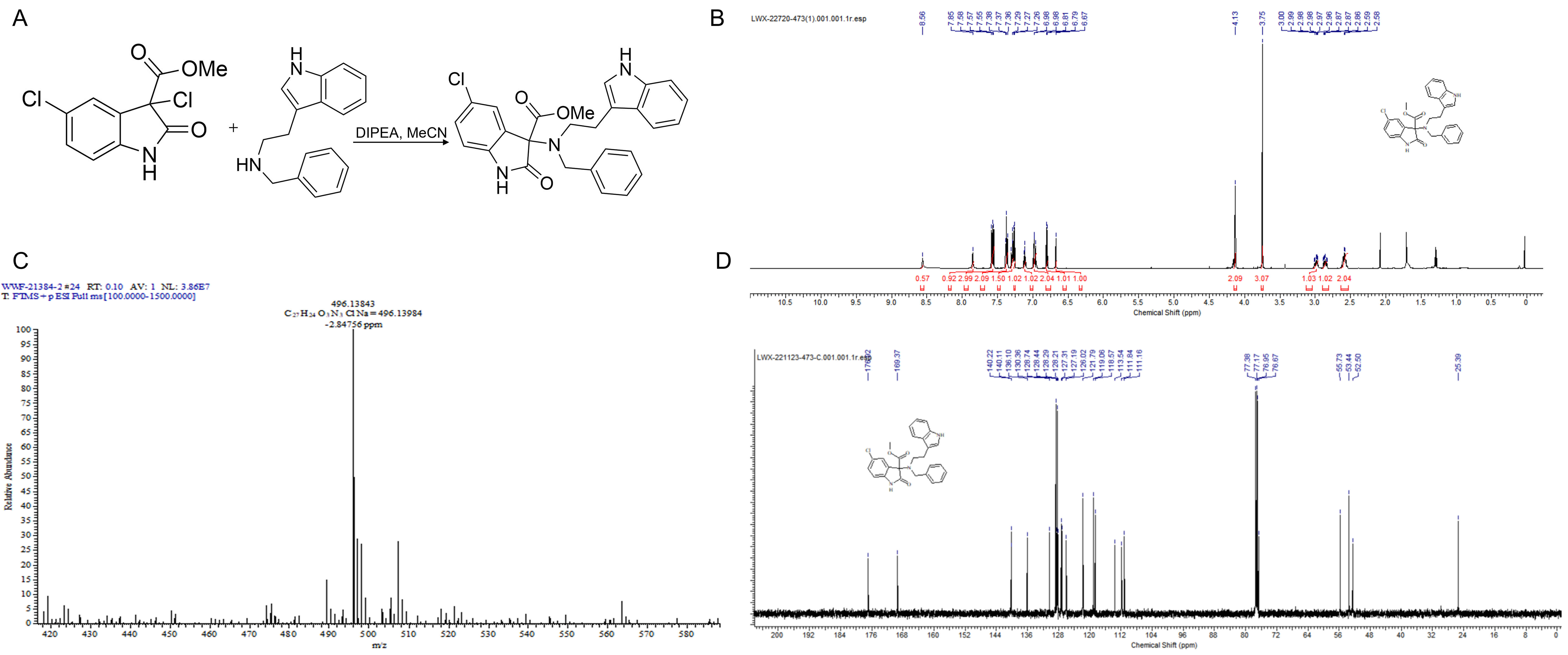 Fig. 1.
Fig. 1.Synthesis and characterization of LWX-473. (A) The synthesis of
compound LWX-473. (B) The
The T-cell leukemic Jurkat cells have been reported to exhibit resistance to glucocorticoids (GCs) due to qualitative (GR mutations) or quantitative defects in GR expression [20]. To investigate the resistance of Jurkat cells to DEX-induced cell death, we treated the cells with different doses of DEX for varying durations and observed their response. The results showed that even at a high dose of 100 µM, DEX was unable to induce cell death in Jurkat cells (Fig. 2A–C). To assess the sensitivity of the novel indole derivative LWX-473 to DEX, we treated Jurkat cells with LWX-473 alone, DEX alone, or a combination of LWX-473 and DEX for different durations (24, 48, and 72 h). The results demonstrated that LWX-473 inhibited the growth of Jurkat cells in a time- and dose-dependent manner. Notably, the co-treatment of LWX-473 with DEX exhibited a more pronounced growth inhibition compared to LWX-473 alone at 48 and 72 h. These findings suggested a synergistic growth inhibition effect of LWX-473 and DEX on Jurkat, CEM/C1, and CEM-C7H2 cells (Fig. 2D–F; Supplementary Fig. 1). Morphological changes such as cellular loss, membrane bubbling, and cell death, leading to a loss of cell viability, further contributed to the synergistic effect of LWX-473 and DEX in Jurkat cells (Fig. 2G). Importantly, LWX-473 displayed minimal toxicity on non-tumorous human normal liver cells HL7702, indicating its potential selectivity towards cancer cells (Fig. 2H).
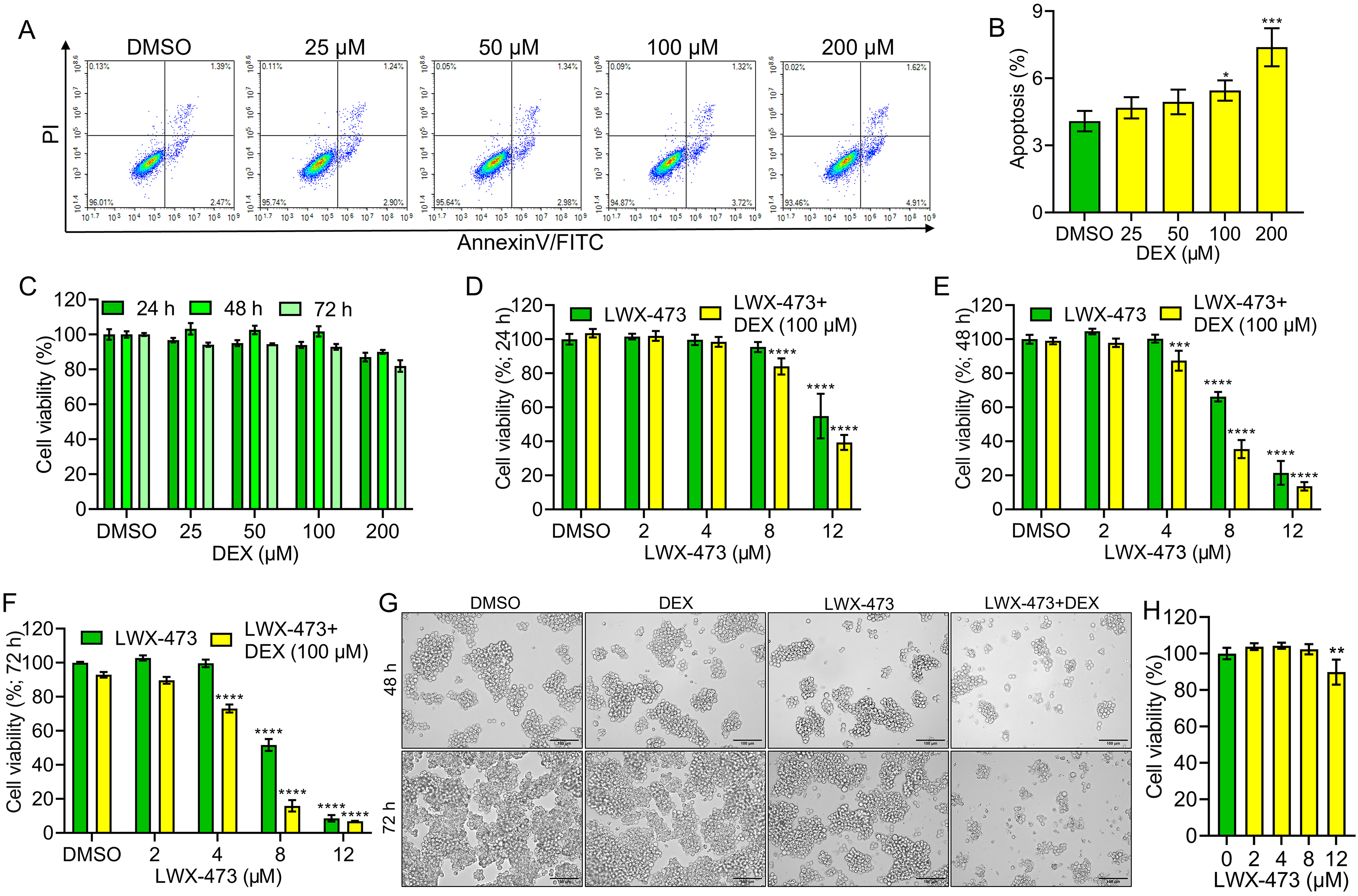 Fig. 2.
Fig. 2.Effect of LWX-473 combined with Dexamethasone on the
proliferation of Jurkat cells. (A) Flow cytometric analysis of indicated
Dexamethasone (DEX) concentrations on the proliferation of Jurkat cells. (B)
Densitometry analysis of apoptotic effects of different DEX concentrations on
Jurkat cells at 48 h. (C) Cell viability effect of different concentrations of
DEX on Jurkat cells at indicated times. (D–F) The effect of various
concentrations of LWX-473 on the proliferation of Jurkat cells in the presence of
DEX (100 µM) for 24 (D), 48 (E), and 72 h (F) of incubation. (G)
Microscopic images (20
Glucocorticoid resistance in leukemia cells allows them to evade normal apoptosis processes and survive. Therefore, we investigated whether LWX-473 could induce apoptosis in Jurkat cells. We observed that LWX-473 could induce apoptosis in a dose-dependent manner. Moreover, when combined with DEX, LWX-473 exhibited a more pronounced induction of apoptosis, overcoming the drug resistance effect of DEX (Fig. 3A,B). Hoechst staining further confirmed these findings, as treatment with DEX or LWX-473 alone did not significantly induce apoptosis, whereas the combination of DEX and LWX-473 resulted in significant apoptosis and cellular fragmentation, characterized by the presence of apoptotic bodies (Fig. 3C). Furthermore, we examined the levels of various apoptotic markers and found that the combination of DEX and LWX-473 significantly increased the levels of cleaved PARP, cleaved caspase3, cleaved caspase9, BIMEL, and BIMSL. Additionally, it significantly reduced the levels of NOTCH1, c-Myc, and Bcl-2, promoting the apoptotic effects, compared to the control (Fig. 3D,E). These results suggest that the combination of DEX and LWX-473 enhances apoptosis in Jurkat cells by modulating key apoptotic proteins.
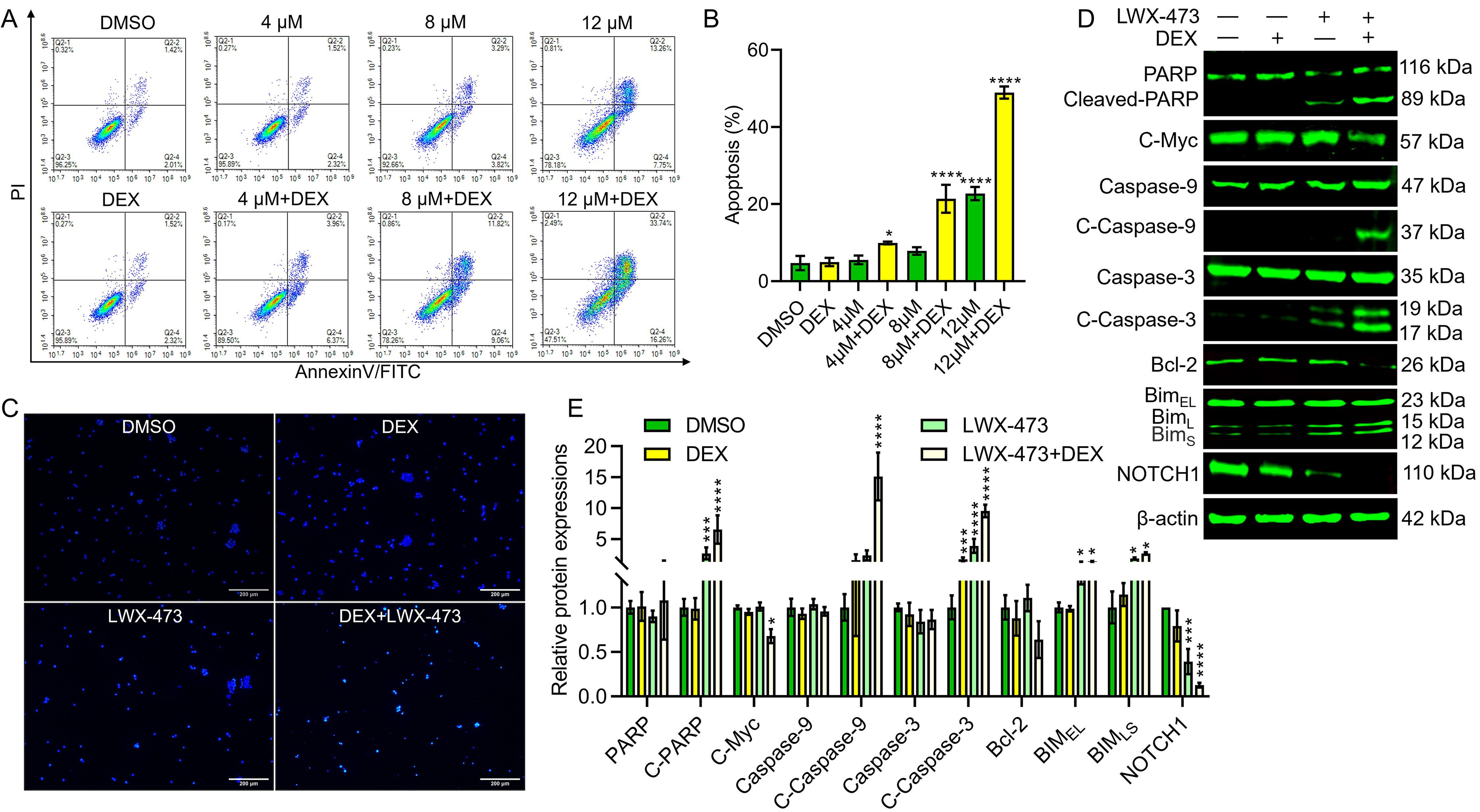 Fig. 3.
Fig. 3.LWX-473 combined with DEX induces apoptosis of Jurkat cells.
(A) Flow cytometric analysis to detect the effect of LWX-473 with or without DEX
on apoptosis of Jurkat cells. (B) Apoptotic effects of different combinational
concentrations on Jurkat cells at 48 h of incubation. (C) Jurkat cells treated
with various indicated combinations were stained with Hoechst (20
As an early event in apoptosis, there is a decrease in mitochondrial membrane potential (MMP) [21]. To assess the effect of the combination of LWX-473 and DEX on MMP, we used JC-1, a fluorescent probe to detect MMP. In Fig. 4A, compared to the other groups, the combination group exhibited stronger green fluorescence (JC-1 monomers) and weaker red fluorescence (JC-1 aggregates), indicating a significant decrease in MMP (Fig. 4B). Furthermore, it is known that DEX can induce G1 phase cell cycle arrest in cancer cells [22]. To investigate whether the co-administration of LWX-473 and DEX affects the cell cycle of Jurkat cells, we treated the cells with different concentrations of LWX-473 and DEX individually. The results demonstrated that LWX-473 could dose-dependently induce G1 phase cell cycle arrest in Jurkat cells (Fig. 4C,D). Additionally, we examined G1 phase-related proteins using western blot analysis. When LWX-473 was combined with DEX, the levels of CDK2 and cyclin E1 proteins were significantly downregulated (Fig. 4E,F) in Jurkat cells, indicating the blockade of the cell cycle in the G1 phase.
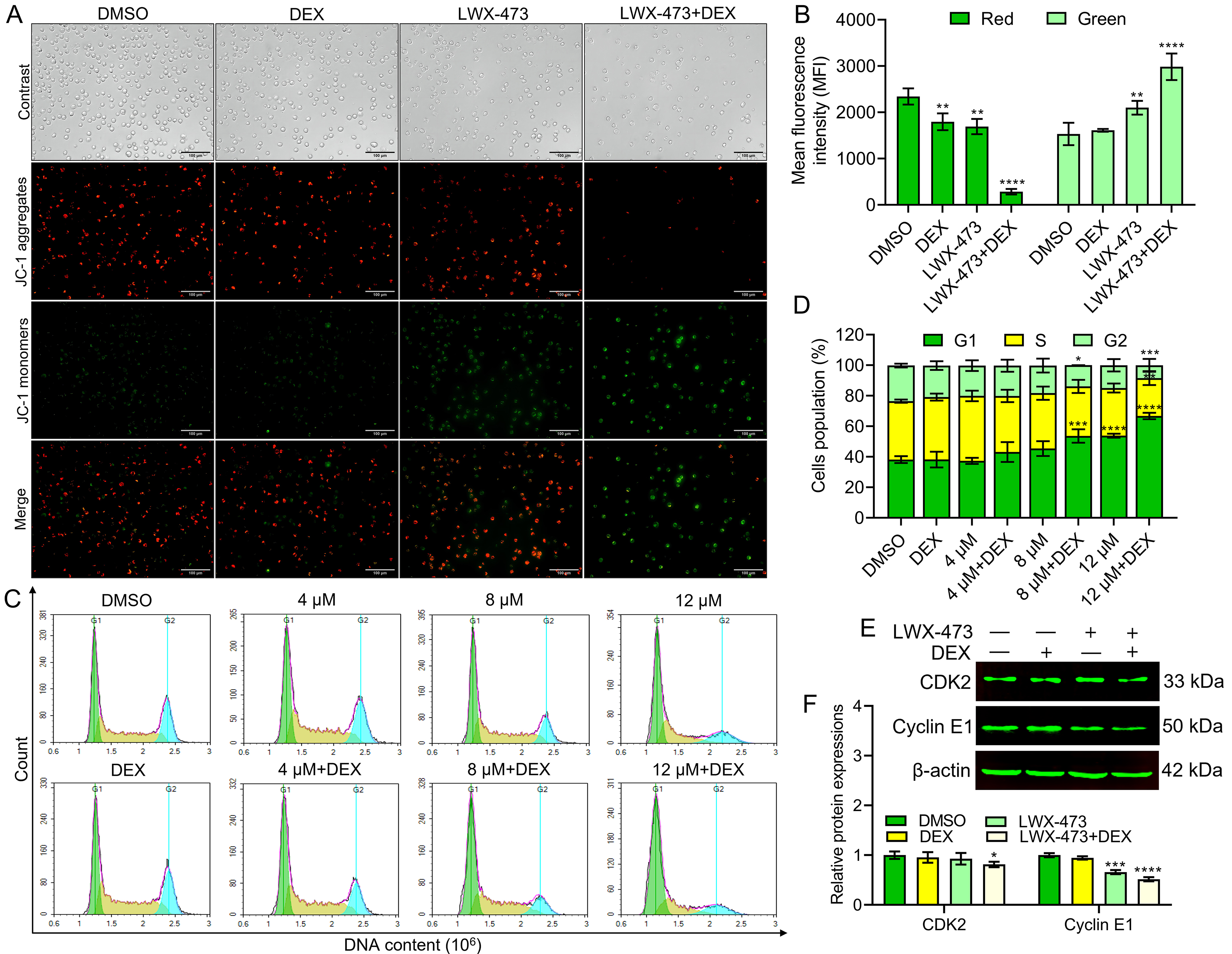 Fig. 4.
Fig. 4.LWX-473 combined with DEX induced mitochondrial membrane
potential (MMP) changes and G1 phase cell cycle arrest of Jurkat cells. (A)
Cells treated with indicated combinations for 48 h were stained with JC-1 and
analyzed by fluorescent microscope (40
Inhibition of the JAK/STAT pathway has been shown to enhance the sensitivity of DEX-induced apoptosis. To investigate the effect of LWX-473 and DEX on this pathway, we analyzed the protein expression of phosphorylated JAK2, STAT3, PI3K, and AKT. We observed that the combination of LWX-473 and DEX significantly reduced the phosphorylation of JAK2. Neither DEX nor LWX-473 alone had an impact on the expression of phosphorylated JAK2 protein compared to the control (Fig. 5A). Furthermore, we found that co-treatment with LWX-473 and DEX significantly reduced the phosphorylation of PI3K and AKT proteins, indicating their involvement in the regulation of this pathway (Fig. 5B). These findings suggest that the combinational treatment of LWX-473 and DEX can overcome glucocorticoid resistance in Jurkat cells through the JAK2/STAT3/PI3K/AKT signaling pathway. Additionally, during co-treatment, there was an increased expression of phosphorylated GR (Ser211), confirming the escape of cells from the chemoresistance of glucocorticoids in Jurkat cells.
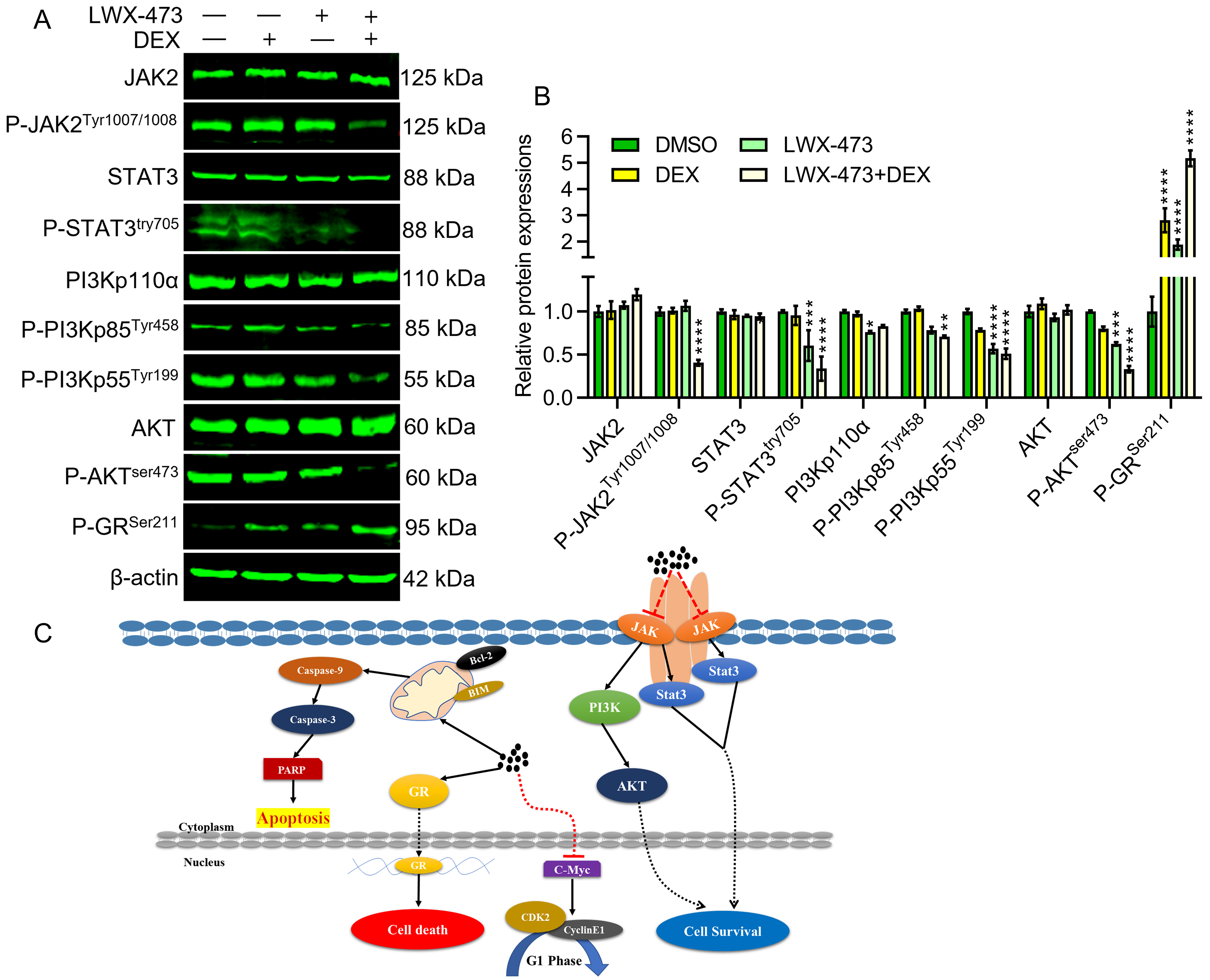 Fig. 5.
Fig. 5.LWX-473 enhances glucocorticoid DEX sensitivity of Jurkat cells
by inhibiting JAK2/STAT3/PI3K/AKT pathway. (A) Western blot images showing
variations in indicated protein levels after the intervention of different
indicated drug combinations,
GCs regulate cell proliferation, inflammatory response, and metabolism by binding to the glucocorticoid receptor (GR) [23]. Moreover, resistance to glucocorticoids forms a complex process in leukemia. Thus, therapeutic agents to overcome the resistance by enhancing apoptosis in leukemic cells are needed for anti-cancer therapy. This study investigated a novel indole compound (LWX-473), which was studied to enhance the apoptotic effect in the presence of a well-known GC, DEX.
The apoptosis, or programmed cell death, by employing cell cycle arrest contributes to the first line of chemotherapy. It can even lead to mitochondrial damage by increasing the level of cleavage of caspases and PARPs. In addition, the Bcl-2 family is related to glucocorticoid resistance. It has been reported that the increase in BIM could effectively overcome glucocorticoid resistance [24, 25]. Similarly, the combination of drugs, LWX-473 and DEX, could downregulate the anti-apoptotic Bcl-2 and upregulate the apoptosis-promoting BIM isomers. Similar effects were seen in the apoptosis and cell cycle analysis. In co-administration, we can decipher enhanced apoptosis by accelerating G1 cell cycle arrest. The level of BIM is closely related to the increase of glucocorticoid resistance, while the decrease of Bcl-2 can accelerate mitochondrial membrane potential damage and apoptosis in Jurkat cells.
The c-Myc gene is one of the essential members of the Myc gene family, and the activation of the c-Myc gene is closely related to the occurrence and development of many cancers. Targeted inhibition of the c-Myc pathway reduces tumor growth and restores antitumor immunity [24, 25]. In addition, many studies have shown that c-Myc can affect the process of the cell cycle and interact with cyclin to arrest the tumor cell cycle [26, 27]. The c-Myc gene is abnormally activated in a variety of cancers and can regulate the cell cycle of tumor cells to make them divide rapidly [28]. However, in our study, LWX-473 combined with DEX significantly reduced the C-Myc protein and arrested Jurkat cell proliferation at its G1 phase, causing a significant reduction in NOTCH1, CDK2, and cyclin E1. This suggests that LWX-473’s mechanism of action is potentially NOTCH1-dependent.
The abnormal activation of the Janus kinase/signal transducer and activator of the transcription (JAK/STAT) signaling pathway can cause many diseases. Mutations or overexpression of the JAK2/STAT3 pathway can be observed in children and adults with acute lymphoblastic leukemia [29, 30]. Moreover, it can also restore the sensitivity of leukemia cells to glucocorticoids in leukemia treatment [31]. Additionally, studies have shown that the PI3K pathway is involved in generating drug resistance, and the FDA has approved many PI3K inhibitors. Furthermore, studies have shown that the abnormal activation of JAK kinase can activate the PI3K pathway [32]. PI3K (phosphatidylinositol 3-kinase) participates in cell growth, survival, and metabolism. Increased activity of PI3K kinase is usually associated with various cancers [33]. Some studies have shown that the inhibition of PI3K and glucocorticoids cooperate in treating T-lymphoid leukemia [34, 35]. Moreover, glucocorticoids can activate GR receptors to regulate target genes, which is a classic way for glucocorticoids to induce leukemia cells and an effective way to overcome glucocorticoid resistance [36]. This study found that the phosphorylation of JAK2 and STAT3, as well as PI3K/AKT pathway proteins, were significantly reduced in the presence of both DEX and LWX-473. However, the individual presence of either DEX or LWX-473 did not offer significant inhibition.
In the absence of glucocorticoids, the glucocorticoid receptor (GR) is located in the cytoplasm in an inactive form, bound to regulatory chaperones such as FKBP52, HSP90, and HSP70. Upon binding to glucocorticoids, GR is released from the chaperone complex and translocated to the nucleus as a dimer to bind specific DNA sequences called glucocorticoid response elements (GRE), thereby enhancing or inhibiting the transcription of specific target genes [37]. It has been proven that GR-mediated transcriptional activation is regulated by phosphorylation, and phosphorylation of Ser211 is a biomarker for GR activation in vivo [38, 39]. Our experimental results show that LWX-473 enhances the sensitivity of Jurkat cells to DEX and improves apoptotic machinery in leukemia cells through the mitochondrial pathways and phosphorylation of GR (Ser221), inhibiting cellular proliferation.
When the synthesized indole compound LWX-473 was administered along with the glucocorticoid DEX, there was a significant enhancement in the apoptotic machinery of the chemotherapeutic drug. In other words, this co-administration was advantageous due to its efficiency in overcoming DEX resistance. It can enhance apoptosis by promoting cell cycle arrest and activating apoptotic molecules through the inhibition of anti-apoptotic mediators. Furthermore, the combinational therapy could enhance the inhibition of JAK2/STAT3/PI3K/AKT pathways by activating the phosphorylation of GR, contributing to the suppression of chemoresistance during anti-cancer LWX-473 treatments (Fig. 5C). These findings offer a novel arena of drug development with potential enrichment strategies to bypass the chemoresistance of glucocorticoids.
All data generated or analyzed during this study are included in this published article.
JS and KY designed the research study, performed the experiments, and wrote the original draft. KMV, WL, QL, QR, YH, XS, and SL performed formal analysis. XS, SL, LH, BG, MX, XH, and YL provided writing revisions. LH, BG, MX, XH, and YL conception of the work, supervised and contributed to writing, interpretation of data, funding acquisition, review, and editing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The authors thank to Yaacov Ben David, State Key Laboratory for Functions and Applications of Medicinal Plants, Guizhou Medical University who provide a platform to finish some experiments.
This research was funded by the grants from the National Natural Science Foundation of China (82360035, 81960546, 81872772, 82160808 & 82260040), The Science and Technology Department of Guizhou Province (QKHPTRC [2020]5008, QKHZC[2022]YB192).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.