



1 Department of Cardiology, Second Affiliated Hospital of Shantou University Medical College, 515000 Shantou, Guangdong, China
2 Shantou University Medical College, 515000 Shantou, Guangdong, China
†These authors contributed equally.
Abstract
Atherosclerosis (AS) is a chronic inflammatory vascular disease that begins with
endothelial activation followed by a series of inflammatory responses, plaque
formation, and finally rupture. An early event in endothelial dysfunction is
activation of the nuclear factor-
Keywords
- TLR2
- TLR4
- NF-
B - atherosclerosis
Based on ample experimental and clinical evidence, atherosclerosis (AS) is now recognized as a chronic inflammatory vascular disease [1, 2]. The first step in AS pathology is endothelial activation, followed by inflammatory responses, plaque formation, and eventually rupture [3]. These disease stages lead to vascular stenosis, or the blockage of blood flow [4]. It is now clear that endothelial cell (EC) dysfunction is pivotal in the initiation and progression of AS lesions [3, 5].
The pathological process of AS broadly involves inflammation and immunity.
Toll-like receptors (TLRs) and nuclear factor-
MicroRNAs (miRNAs) are a class of small, single-stranded noncoding RNA that act
as negative post-transcriptional regulators of gene expression [8]. Circular RNAs
(circRNAs) are a novel class of endogenous non-coding RNAs with covalently
closed-loop structures [9]. Both miRNA and circRNA play essential roles in
coordinating cellular functions such as inflammation, differentiation, and
apoptosis [10]. Recent studies have also focused on the involvement of miRNA and
circRNA regulating TLR2/4 and NF-
In this summary, we present the current state of knowledge with regard to TLR2/4
and NF-
ECs act as sensors and effectors that release biological agonists and antagonists [3]. They form a functional cell layer that lines the entire circulatory system and helps to maintain multi-organ health and homeostasis [11]. The functions of healthy endothelium include serving as a semi-permeable barrier, maintaining vascular tone, angiogenesis, vascular repair, regulating hemostasis, mediating mechano-transduction, and regulating metabolism [12]. ECs synthesize and release endothelial-derived relaxing factors such as nitrous oxide (NO) and endothelium-derived hyperpolarization factor (EDHF), as well as metabolites of arachidonic acid and peptides (endothelin, reactive oxygen species [ROS], etc.) that regulate vasoconstriction and vasodilation [13]. NO is produced by the endothelial isoform of NO synthase (eNOS) and also affects the behavior of other cell types in the vascular system. For example, NO reduces the contractility of adjacent vascular smooth muscle cells (VSMCs), inhibits platelet activation and adhesion, and reduces leukocyte adhesion [5]. The single layer of ECs in the aortic intima of the normal aorta is a complete structure that encloses the elastic plate. Without a complete EC structure, or in the event of uneven hyperplasia within the EC layer, the inner elastic plates break and compress, thus allowing the accumulation of foam cells, lipid plaques, and VSMCs [14]. These processes begin due to endothelial dysfunction, which is defined as a variety of non-adaptive alterations in normal functional phenotypes [5]. ECs can switch from a quiescent phenotype to a host defense phenotype during the response to proinflammatory agents, such as endotoxins, modified lipoproteins, advanced glycosylation end products (AGE), and disturbed flow-derived IL-1 and TNF [15]. Such sustained maladaptive changes lead to endothelial dysfunction characterized by reduced eNOS activity and resulting in decreased bioavailability of NO. Other pathologic changes are subsequently triggered, including lipid-laden inflammation and oxidative stress [12]. Endothelial dysfunction occurs during sepsis [16], cardiovascular diseases such as atherosclerosis and hypertension [17, 18], pulmonary hypertension [19], metabolic disorders such as type-2-diabetes and obesity [20], and autoimmune diseases such as autoimmune hepatitis [21] and rheumatoid arthritis [22].
While endothelial dysfunction is the initial step in AS pathology, an early
event in the inflammatory response is activation of the NF-
TLRs are one of the vital pattern recognition receptors (PRRs) of the innate immune system. They are responsible for recognizing pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) during embryonic development [24]. TLRs are type I transmembrane glycoproteins that recognize PAMPs or DAMPs through their extracellular leucine-rich repeat (LRR) domain. PAMPs and DAMPs activate downstream signaling pathways through their cytoplasmic Toll/interleukin (IL)-1 receptor (TIR) domain [25]. TLR-dependent signaling leads to the activation of transcription factors and inflammatory responses [26].
To date, 13 TLRs have been characterized in mammals, of which TLRs 1-10 are functional in humans [27]. TLRs in mammals are synthesized in the endoplasmic reticulum (ER), potentially undergo sequential glycosylation in the Golgi complex, and are then transported to the membranes of plasma or intracellular vesicles, such as the endoplasmic reticulum and endosomes [28]. TLRs located in the plasma membrane include TLR1, TLR2, TLR4, TLR5, and TLR6, while those located in intracellular vesicle membranes include TLR3, TLR7, TLR8, and TLR9 [26]. Although endosomal TLRs require trafficking factors, TLRs localized in the plasma membrane are transported to the plasma membrane through conventional secretory pathways [28].
Activation of specific TLR signaling pathways in mammals is dependent upon both
the agonist and the cell type [26]. For example, during the response to the
prototypical ligand for TLR4, LPS, ECs primarily express IL-6, IL-8, CSF2, CSF4,
ICAM-1, and SELE, but little TNF-
TLRs are expressed in all innate immune cells, including monocytes, macrophages, dendritic cells (DCs), neutrophils and natural killer cells, as well as vascular cells like VSMCs and fibroblasts. However, the expression of specific TLR types varies between different cell types [30], as well as mediation of the innate immune response [24]. For example, ECs express TLR2, 4, 6, 11, 12 and 13, whereas macrophages express TLR5 in addition to these TLRs [30]. VSMCs have also been reported to express TLR2 and 4 [31, 32].
Because of different ligand-binding structures [25], TLRs can recognize a variety of ligands including PAMPs and DAMPs. In response to PAMPs, TLR2 dimerizes with either TLR1 or TLR6. The resulting structure is able to detect lipoproteins. Similarly, TLR3 detects double stranded and ribosomal RNA, while TLR4 can detect LPS when accompanied by the co-receptor MD-2. Moreover, TLR5 detects flagellin, while TLR7 and TLR8 bind ssRNA, and TLR9 detects DNA containing cytosine-phosphate-guanine (CpG) motifs [28]. In addition, TLR2 and TLR4 recognize intracellular DAMPs such as HMGB1, histones, heat shock proteins, and extracellular DAMPs such as short fragment hyaluronan and extracellular matrix [33]. Finally, TLR2 and TLR4 recognize oxidized lipoproteins within endogenous lifestyle-associated molecular patterns (LAMPs) [33], which is critical in the formation of atherosclerotic plaques.
TLR signal conduction is a three-step process [28]. First, agonistic ligands
bind and mediate the dimerization of TLRs. Through interactions between ligand
and TLR LRR domains, the TLR extracellular domain induces the formation of
homodimers such as TLR3, 4, 5, 7, 8 and 9, or heterodimers such as TLR2/1 and
TLR2/6, both of which possess a typical “m”-shaped structure [34]. Second, TLR
dimers mediate assembly of the supramolecular organizing center (SMOC), which is
widely considered as the subcellular site of signal initiation. Dimerization of
the extracellular LRR domains results in dimerization of the intracellular TIR
domains, which subsequently activates cell-extrinsic, cell-intrinsic, and
cell-type-specific responses and TLR signaling pathways [28]. TIR domains are
detected by the receptor-proximal membrane protein TIRAP/MAL, and are necessary
for the initiation of TLR2, 4, 7, and 9 signaling. Alternatively, this action can
occur through the peripheral membrane protein TRIF (TIR domain-containing
adaptor-inducing IFN-beta)-related adaptor molecule (TRAM), which detects the
dimerization of TLR4 and TLR3 and further stimulates the assembly of SMOC. SMOCs
are essential in multiple innate immune pathways, including putative ribosomes
that govern TLR signaling. Third, SMOCs mediate the activation of kinases, which
then primarily drive transcription and glycolysis. The myddosome, an oligomeric
complex formed by the interaction between myeloid differentiation primary
response protein 88 (MyD88) and IL-1R-associated kinase (IRAK) family
serine-threonine kinases, subsequently auto-phosphorylates and recruits the E3
ubiquitin ligase tumor necrosis factor receptor-associated factor 6 (TRAF6) [28].
TRAF6 then activates the kinase transforming growth factor b (TGF-b)-activated
kinase 1 (TAK1) to stimulate transcription factors including IkB kinase, which
activates NF-
TLR signaling pathways are generally classified as being MyD88-dependent. These are initiated by TIRAP/MAL and are governed by the myddosome. This signaling axis results mostly in the production of proinflammatory cytokines. Signaling by TRIF is initiated by TRAM and governed by the triffosome. This signaling axis is primarily responsible for the production of type I interferons [28].
TLR2 and TLR4 are both located on the plasma membrane and transmit signals
through the MyD88-dependent axis to activate NF-
TLRs in ECs serve as sensors for the innate immune system and initiate inflammatory responses that subsequently activate the adaptive immune system [26]. With the initiation and progression of AS, TLR2 and TLR4 expression increases within the endothelium, atherosclerotic plaques, and circulating blood cells [38]. Using laser scanning confocal microscopy (LSCM), Mullick et al. [39] found that vascular perturbations associated with a high fat diet (HFD) increased the expression of EC TLR2. Qu et al. [40] found that abnormal fluid flow increased the expression of TLR4 in ECs within the inner curvature of the aorta in vivo. This result was mimicked in vitro by HUVECs. Symptomatic plaques that are unstable and display vascular stenosis or blood flow blockage show higher expression of TLR4 in ECs than asymptomatic plaques [41]. Inhibition of TLR2 and TLR4 inhibits inflammatory response and AS progression. In Mullick et al. [39] study, they also found that TLR2-/- mice showed reduced intimal leukocyte accumulation within the endothelial layer compared to wildtype mice, and exhibited less intimal lipid foam cell accumulation in AS lesions.
NF-
NF-
In unstimulated conditions, NF-
The non-canonical NF-
The TLR family are essential members of innate PRRs. Their signals activate the
canonical NF-
The TLR2,4/NF-
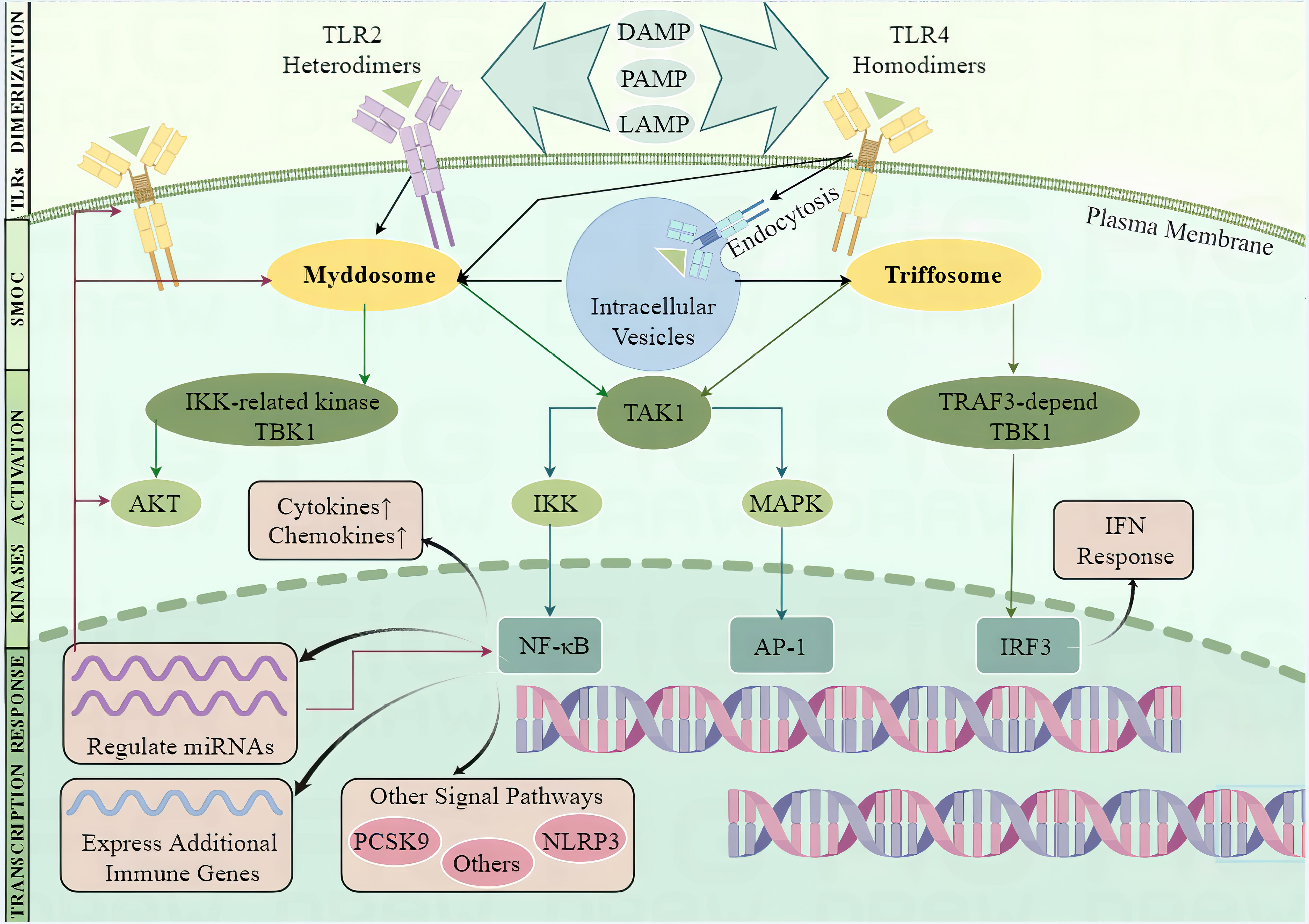 Fig. 1.
Fig. 1.TLR2 and TLR4 signaling in endothelial cells and regulation of
miRNAs. The transmission of toll-like receptor (TLR) signaling follows three
steps. First, agonistic ligands mediate the formation of TLR2 heterodimers and
TLR4 homodimers in the plasma membrane. These include pathogen-associated
molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs) and
lifestyle-associated molecular patterns (LAMPs). The TLR4 homodimers subsequently
translocate from the cell surface to intracellular vesicles via endocytosis.
Second, TLR2 heterodimers and TLR4 homodimers mediate assembly of the
supramolecular organizing center (SMOC), including the Myddosome which governs
MyD88-dependent signaling, and the Triffosome which governs TRIF-dependent
signaling. TLR2 and TLR4 mediate assembly of the Myddosome, while TLR4 also
mediates assembly of the Triffosome (black arrows). Third, SMOCs mediate the
activation of kinases, which then activate the transcriptional response. The
Myddosome activates the inhibitor of kappa B kinase (IKK)-related kinase Tumor necrosis factor receptor-associated factor (TRAF)
family member associated Nuclear
factor-
HFD that is rich for example in cholesterol and trans-fatty acids will increase the levels of plasma cholesterol and serum triglycerides. This induces ECs to switch to a maladaptive phenotype characterized by excessive inflammatory responses, insulin resistance, ROS production, and impaired vasorelaxation. Bhaskar et al. [52] reported that a hypercholesterolemic diet in mice enhanced the inflammatory response. This was indicated by elevated levels of serum C-reactive protein (CRP), cyclooxygenase (COX), 5-lipoxygenase (5-LOX), NOS and myeloperoxidase (MPO), as well as upregulating the mRNA level of IL-6. Kramer et al. [57] fed mice a long-term western diet characterized by a high fat and high carbohydrate content. This increased the levels of fasting serum triglycerides and serum non-esterified free fatty acid (NEFA), resulting in glucose intolerance, insulin resistance, and increased arterial blood pressure. Kondo et al. [58] reported that 4 weeks of a high trans-fatty acid (TFA) diet led to the accumulation of plasma lipids and delayed catabolism of circulating TFA-containing lipids, while also triggering prothrombogenic activity in ECs.
Lipid accumulation upregulates the expression of TLR2 and TLR4, as well as
downstream signaling components such as MyD88, TRAF6, and NF-
Apart from ox-LDL, saturated and trans-fatty acids have also gained considerable
attention for their ability to induce an inflammatory response and endothelial
dysfunction through TLR signaling in ECs. Palmitate (PA) is the most abundant
saturated fatty acid in the human bloodstream. Kim et al. [62] found
that PA increased the expression of numerous proinflammatory genes in HAECs,
including E-selectin, TNF
Unlike ox-LDL which directly binds TLR2 or TLR4 dimers, PA induces an
inflammatory response by altering cell metabolism, gene expression, lipid
metabolism, and membrane lipid composition through TLR signaling [64]. Ren
et al. [50] found that TLR activation induced by PA differs from
canonical TLR agonists in several ways. First, it takes longer for PA-dependent
E-selectin expression to plateau, and for peak phosphorylation of JNK and
activation of NF-
A number of infectious agents have been implicated in atherosclerotic pathogenesis, including Chlamydia pneumoniae [66, 67], Porphyromonas gingivalis [68], Helicobacter pylori [68], cytomegalovirus (CMV), Epstein-Barr virus (EBV), human immunodeficiency virus (HIV), herpes simplex virus-1 (HSV-1), HSV-2, and hepatitis C virus (HCV) [30]. Microbial cell surface components are detected by TLRs through PAMP-PRR interaction. These include LPS, lipoprotein and flagellin, as well as dsRNA, ssRNA and CpG DNA of viral origin, and lipoproteins, CpG DNA, ssRNA and profilin from parasites [28]. After recognizing such pathologically-related factors and subsequently activating TLR signaling, ECs recruit immune cells through upregulation of the leucocyte adhesion molecules VCAM-1 and ICAM-1 [69], LOX-1 [54, 70], and CXC16 [53] following adhesion of the immune cell to ECs [69]. Benagiano et al. [66] observed that C. pneumoniae phospholipase D (CpPLD) increased the mRNA and protein expression of various chemokines and adhesion molecules in HUVECs, including CCL-4, CCL-2, CCL-20, CXCL9, ICAM-1 and sVCAM-1. Echavarria et al. [47] noted that exposure of HUVECs to LPS for 4 hr induced significant increases in leucocyte adhesion, adhesion molecule expression, and proinflammatory cytokine production, but did not affect the expression of anti-inflammatory cytokines such as IL4 and IL10.
LPS is a well characterized component of the outer membrane of most
gram-negative bacteria and is strongly recognized by the immune system during
inflammatory responses [71], endothelial dysfunction, and apoptosis [72]. LPS is
recognized by TLRs, of which TLR4 and myeloid differentiation factor 2 (MD2) are
the primary receptor and activator of subsequent signaling [25]. Zhao et al. [54] found that LPS increased the expression of TLR4 in ECs but reported no
change in TLR2 expression. The TLR4-MD2 heterodimer interacts with LPS at the
cell surface and promotes translocation to lipid rafts. TLR4-MD2 has a strong
affinity for TIRAP/MAL, thus promoting the assembly of myotomes and initiating
MyD88-dependent signaling [28]. CD14 mediates a process by which TLR4 undergoes
endocytosis to intracellular vesicles and subsequently engages with TRAM to
activate TRIF-dependent signaling [37]. Thus, activation of TLR4 initiates both
MyD88-dependent signaling at the cell surface and TRIF-dependent signaling in
intracellular vesicles, both of which lead to increased expression of
proinflammatory factors and subsequent inflammation. Qu et al. [40]
reported that LPS-induced TLR4 activation is mediated by a MyD88-dependent
mechanism, leading to phosphorylation of the NF-
The inflammation that occurs during AS is chronic and mimics the signal pathway
transmission and communication mechanisms observed in immune cells. The
inflammatory response induced by LPS varies according to the dose, duration, and
specific pathogen. Xiao et al. [53] noted that the viability of HUVECs
decreased significantly after treatment with LPS for 24 hr compared with lower
doses, and that viability rebounded within 24 hr when treated with 1 µg/mL
LPS. Zhao et al. [54] found that high doses of LPS inhibited EC
proliferation, and that LOX-1 expression increased significantly when the cells
were treated for a prolonged time or with a higher concentration of LPS.
Triantafilou et al. [68] observed that LPS from either H.
pylori or P. gingivalis activated ECs through TLR2/NF-
TLR signaling induced by LPS is interactive in nature. Lipid rafts are regularly
distributed in all cellular membranes and form unique microdomains that serve as
platforms for signal transduction and receptor activation [73]. Huang et al. [69] noted that LPS-induced TLR4 translocated to lipid rafts, whereas
nystatin, a lipid raft inhibitor, blocked the downstream upregulation of VCAM-1
and ICAM-1 expression. Other studies found that LPS from H. pylori and
P. gingivalis activated ECs through TLR2/NF-
Diabetes and AS share a close relationship and both diseases impair the
endothelium and cause endothelial dysfunction. Lu et al. [75] reported
that HFD-induced type 2 diabetes increased the size of AS lesions and intimal
lesions of AS plaques by increasing the accumulation of monocytes and
macrophages, the expression of IL-6 and MMP-9 expression in AS plaques, and the
collagen content of AS lesions. Using a streptozotocin-induced animal model of
diabetes, Leng et al. [49] reported that aortic rings showed impaired
vasodilation in response to acetylcholine (ACh), decreased levels of eNOS and NO,
increased levels of IL-6 and TNF-
Surprisingly, TLRs do not recognize glucose. However, TLR signaling participates
in hyperglycemia-induced endothelial dysfunction. Leng et al. [49] found
that the aortic rings obtained from diabetic mice had higher levels of TLR4 and
the NF-
The endothelium is located between circulating blood and tissue. It senses
changes in mechanical stress and in the concentration of metabolic factors and is
responsible for activating signal transduction in response to such changes [3].
High-risk factors including hypercholesterolemia, hypertension, diabetes
mellitus, and smoking [4] promote the formation of atherosclerotic lesions, which
tend to occur in regions with low wall shear stress (WSS). WSS tends to occur at
sites of disturbed flow (DF), such as vessel branch points and bifurcation [3].
ECs that are exposed to DF undergo changes in morphology and function, eventually
resulting in cell turnover and senescence [5]. DF also alters TLR gene
expression, and TLR signaling is activated by DAMPs such as fibronectin extra
domain A (EDA), hyaluronan fragments, HMGB1, and serum amyloid A. Tobias
et al. [2] found that murine ECs exposed to DF showed upregulation of
TLR2 expression. Furthermore, en-face laser scanning confocal microscopy
revealed that TLR2 was only observed in the DF area of murine aortic tissue,
while hyperlipidemia could increase TLR expression [39]. Qu et al. [40]
found that ECs resided in the human aortic arch. This area is constantly exposed
to DF, is activated by the TLR4/ MyD88/ NF-
Noncoding RNAs, including long noncoding RNA (lncRNA), microRNA (miRNA), long
intergenic noncoding RNA (lincRNA), and circular RNA (circRNA), play essential
roles in coordinating cellular functions such as inflammation, differentiation,
and apoptosis [10]. Noncoding RNAs are also associated with many
inflammation-related diseases, such as AS, diabetes mellitus, obesity, and
osteoarthritis [10]. The level of these noncoding RNAs is commonly dysregulated
in AS, and they participate in the stimulation of various signaling mechanisms
including the TLR/NF-
miRNAs are defined as being non-coding, single-stranded RNA oligonucleotides
with an average length of 22 nucleotide residues. Through binding to 3
Three different mechanisms have been described regarding the regulation of TLR
signaling by miRNAs [83]. First, miRNAs directly bind with a component of TLR
signaling. Chen et al. [61] found that TLR4 and TXNIP were direct
targets of miR20 due to the presence of an evolutionarily conserved miR-20a
binding site within their 3
| miR | Inducer | Expression level | Mechanism | Effects on ECs | Regulator | Ref. |
| miR-590 | ox-LDL | Inhibit TLR4/NF- |
attenuate atherosclerotic lesion, promote proliferation, block ox-LDL-induced apoptosis | [81] | ||
| miR-204-5p | ox-LDL | Inhibit TLR4 | increase cell viability and inhibit cell apoptosis, inflammatory response, and oxidative stress | [87] | ||
| miR-217 | ox-LDL | Inhibit TLR4 PI3K/Akt/ NF- |
increase the viability of atherosclerotic ECs, inhibit their apoptosis, inflammatory response, and EndMT | [89] | ||
| miR-20a | ox-LDL | Directly target TLR4 and TXNIP | inhibit TLR4-mediated inflammatory response and NLRP3 activation | [61] | ||
| miR-26a-5p | ox-LDL | Directly bind TLR4 and downregulate | inactivate TLR4/NF- |
Kaempferol | [85] | |
| miR-146a | LPS | Inhibit TLR4/NF- |
reduce LPS-induced CXCL16 | [53] | ||
| miR-146a, miR-155 | LPS | TLR4/NF- |
related to proinflammatory response potentially | Dapagliflozin | [88] | |
| miRNA-146a | LPS | Downregulate IRAK-1 and TRAF-6 | suppress the activation of NF- |
[86] | ||
| miRNA-126 | LPS | Downregulate VCAM-1 | suppress leukocyte adherence to endothelial cells | |||
| miR-146b-5p | LPS | / | Selectively reduce IRAK1 and TRAF6 expression | inhibits LPS-induced leucocyte adhesion, adhesion molecule expression, proinflammatory cytokine production, p38 and SAPK/JNK phosphorylation, and NF- |
Ang-1 | [47] |
| miR-21 | LPS | Directly target TLR4 | downregulate TLR4 expression | propofol | [84] | |
| miR-6835 | LPS | / | Interaction between TLR-4 and AdipoR1 in lipid rafts | enhance LPS-induced inflammation process | [90] |
Abbreviations: ox-LDL, oxidized low-density lipoprotein; LPS, lipopolysaccharide; TLR, Toll-like
receptor; NF-
Many molecules that regulate the inflammatory response modulate TLR signaling in
ECs through miRNA. Ang-1 was found to block TLR4 signaling by upregulating
miR-146-5p, which selectively attenuates the TLR4 signal transducers IRAK1 and
TRAF6 [47]. Propofol downregulated LPS-induced expression of TLR4 through the
upregulation of miR21 [84]. Dapagliflozin was reported to increase the expression
of anti-inflammatory miR-146a and to decrease the expression of pro-inflammatory
miR-155 [88]. Similarly, kaempferol inactivated TLR4/ NF-
Circular RNA (circRNA) is a form of endogenous noncoding RNA characterized by a
covalently closed loop structure. CircRNAs are formed through back-splicing,
whereby a covalent bond is formed between a downstream 5
There is some evidence to suggest that circRNAs play a role in the progression of AS, with several studies investigating the effects of different circRNAs on EC behavior, inflammation, and plaque destabilization. For example, circRNA-0006896 was found to be positively correlated with triglyceride, LDL-C and CRP levels in patients with unstable/vulnerable plaque AS, and negatively associated with albumin levels [96]. Another study found that knockdown of circRNA-PTPRA inhibited AS progression by repressing ox-LDL-induced EC injury by sponging miR-671-5p [97]. Additionally, circRNA RSF1 was found to regulate ox-LDL-induced EC inflammation in AS by modulating the miR-135b-5p/HDAC1 axis [98]. These studies suggest that circRNAs may regulate EC function and inflammation in AS.
To date, however, there is limited information on how circRNAs might affect the expression of genes related to EC dysfunction in AS. Some studies have shown that circRNAs can regulate autophagy, apoptosis, inflammation, oxidative stress, and proliferation of ECs and hence the development of AS. For example, circ_0068087 negatively regulated the level of miR-186-5p in HUVECs by interacting with this miRNA [99]. This promoted ox-LDL-induced injury in HUVECs by allowing upregulation of ROBO1 expression. Knockdown of hsa_circ_0005699 attenuated the inflammatory and apoptotic response induced by ox-LDL in HUVECs by regulating the miR-450b-5p/NFKB1 axis [100]. Taken together, these findings suggest that circRNAs may regulate gene expression associated with EC dysfunction in AS by enhancing the inflammatory response. However, more work is needed to fully understand the specific effects of circRNAs on the expression of genes associated with EC dysfunction in AS.
In conclusion, TLR2/4 and NF-
BY and XY: Conceptualization, Writing-original draft, Writing-editing. XC, XH, BX: Conceptualization, Writing-review. DL, JC, WL, YL: Conceptualization, Writing-review & editing. JY: Visualization. JL: Conceptualization, Writing-review & editing, Funding acquisition. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity. All authors read and approved the final manuscript. All authors contributed to editorial changes in the manuscript.
Not applicable.
Not applicable.
This research was funded by Special Fund Project for Science and Technology Innovation Strategy of Guangdong Province [2019]113-55(190826155555147) and Shantou Medical Health Science and Technology Plan [2020]66 (ST2020027).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.