
- Academic Editor
Metastasis remains a leading cause of mortality for patients with solid tumors. An expanding body of literature suggests interplay between the host, gut, and tumoral microbiomes may play a role in cancer initiation and distant dissemination. These associations have been particularly well-studied in colorectal cancer, where gut dysbiosis and an endotoxin-induced inflammatory milieu foster premalignant polyp formation, setting the stage for carcinogenesis. Subsequent violation of the gut vascular barrier enables dissemination of bacterial agents to sites such as the liver, where they contribute to establishment of pre-metastatic niches, which promote tumor cell extravasation and metastatic outgrowth. Intriguingly, breakdown of this vascular barrier has been shown to be aided by the presence of tumoral bacteria. The presence of similar species, including Fusobacterium nucleatum and Escherichia Coli, in both primary and metastatic colorectal tumors, supports this hypothesis and their presence is associated with chemotherapy resistance and an overall poor prognosis. Specific gut microbial populations are also associated with differential response to immunotherapy, which has a growing role in microsatellite unstable colorectal cancers. Recent work suggests that modulation of gut microbiome using dietary modification, targeted antibiotics, or fecal microbiota transplantation may improve response to immunotherapy and oncologic outcomes. Elucidation of the precise mechanistic links between the microbiome and cancer dissemination will open the doors to additional therapeutic possibilities.
Approximately 90% of cancer-related mortality can be attributed to metastatic disease [1, 2]. The metastatic cascade is a complex, multifaceted process that involves interplay between the primary tumor, tumor microenvironment, host immune system, and target organ. The microbiome, comprised of the collective bacteria, viruses, and fungi in a person’s body, plays an underappreciated role in this process. The average human hosts over 38 trillion bacteria and up to ten times as many viral particles, far exceeding the number of human cells [3, 4, 5]. With this colossal biomass, it is no surprise that microbial agents have been implicated in up to 20% of malignancies [6]. Advances in next generation sequencing technology and data processing algorithms have enabled an increasingly thorough characterization of the cancer-associated microbiome. Given the extensive study of the impact of the gut and tumoral microbiome on carcinogenesis, dissemination, and metastatic outgrowth in colorectal cancer (CRC), this will be our focus of review.
The colon and rectum contain the highest microbial density and diversity of all human organ systems. While the precise components of a “healthy” colorectal microbiome have not been defined, both culture-based methods and 16S rRNA gene sequencing have identified Bacteroides, Proteobacteria, Firmicutes, Verrucomicrobia, and Actinobacteria as the most common phyla in the normal human gastrointestinal tract [7, 8, 9]. Importantly, these gut microbial populations are dynamic, with numerous environmental factors shaping the gut microbiome including diet, antibiotic use, exposure to chemicals or toxins, and social interactions [10, 11, 12, 13, 14]. Imbalance in gut microbiome composition and function, termed dysbiosis, has been linked to the pathogenesis of multiple diseases, including colorectal cancer [15, 16, 17].
The relative overabundance of specific bacterial taxa in a dysbiotic colon have been linked to colorectal tumor initiation (Table 1, Ref. [18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38]). Escherichia coli was one of the earliest microorganisms linked to CRC. In 1998, Swidsinski and colleagues [18] using 16S rRNA sequencing in combination with a gentamicin protection assay, identified intracellular E. coli in colonic adenomas not present in healthy colorectal epithelium. This finding was replicated in colorectal tissues from patients with Crohn’s disease and CRC by Dejea et al. [19] using dithiothreitol mucolysis followed by bacterial culture to show E. coli’s ability to penetrate the mucus layer and adhere to the underlying colonic epithelium. They also treated tumor cells with gentamycin to deplete the extracellular microbiota and identified intracellular E. coli in Crohn’s and CRC epithelium that was not present in healthy control tissue. Moreover, the phenotype of E. coli isolated from Crohn’s and CRC patients was distinct, exhibiting upregulation of hemagglutinins, corresponding to increased mucosal adherence [19]. E. coli strains expressing polyketide synthases (pks), an enzymatic complex that synthesizes the genotoxin Colibactin, have particularly potent carcinogenic potential. Deletion of pks from these strains results in decreased tumor multiplicity and invasion in azoxymethane treated Il-10 deficient mice. Pks+E. coli have been found in colorectal tissues from a 40% of IBD patients and 67% of CRC patients [20]. As such, this and continued work studying Pks+E. coli have begun to demonstrate the importance of bacterial function as well as taxal associations in carcinogenesis.
| Study | Subject of study | N (human patients) | Findings |
| Eschericia coli | |||
| Swidsinski, et al. 1998 [18] | Patients | 65 healthy controls | Intracellular E. coli identified in patients with colonic adenomas and CRC but not healthy controls. |
| 29 with adenomas | |||
| 31 with CRC | |||
| Martin, et al. 2004 [33] | Patients, cell lines | 14 with Crohn’s disease | Mucosa-associated and intramucosal bacteria more commonly cultured from CRC and Crohn’s disease, of which E. coli accounts for majority of isolates. |
| 21 with ulcerative colitis | |||
| 24 healthy controls | |||
| 21 with CRC | |||
| Arthur, et al. 2012 [20] | Patients, mouse model | 21 with CRC | Colonization with pks+E. coli promotes CRC in colitis-susceptible mice. Mucosa-associated pks+E. coli enriched in patients with CRC and IBD. |
| 35 with IBD | |||
| 24 healthy controls | |||
| Dejea, et al. 2018 [19] | Patients, mouse model | 25 with FAP | E. coli-derived colibactin enriched in colonic mucosa of FAP patients. Mice co-colonized with pks+E. coli and ETBF exhibit faster tumor growth. |
| 23 healthy controls | |||
| Bacteroides fragilis | |||
| Wu, et al. 1998 [22] | Mouse model, cell line | N/A | B. fragilis toxin cleaves E-cadherin, producing morphologic changes dependent on target-cell ATP. |
| Wu, et al. 2009 [21] | Mouse model | N/A | ETBF, but not NTBF, triggers colitis and tumor growth in mice through Stat3 activation and mediated by a T |
| Purcell, et al. 2017 [34] | Patients | 150 undergoing colonoscopy | ETBF associated with pre-cancerous colonic lesions and more common in descending colon biopsies. |
| Dejea, et al. 2018 [19] | Patients, mouse model | 25 with FAP | ETBF toxin enriched in colonic mucosa of FAP patients. Mice co-colonized with pks+E. coli and ETBF exhibited faster tumor growth. |
| 23 healthy controls | |||
| Chung, et al. 2018 [35] | Mouse model, cell line | N/A | IL-17, NF- |
| Fusobacterium nucleatum | |||
| Kostic, et al. 2013 [24] | Patients, mouse model | 20 healthy controls | Fusobacterium spp. are enriched in adenomas and stool from adenoma and CRC patients. F. nucleatum increases tumor growth through myeloid cell recruitment and creation of a proinflammatory microenvironment. |
| 29 with adenomas | |||
| 27 with CRC | |||
| Rubinstein, et al. 2013 [23] | Patients, mouse model, cell lines | 14 healthy controls | The FadA adhesin produced by F. nucleatum binds E-cadherin and activates oncogenic signaling via |
| 16 with adenomas | |||
| 19 with CRC | |||
| Bullman, et al. 2017 [32] | Patients, mouse model, cell lines | 833 with CRC from multiple cohorts | F. nucleatum and associated taxa (Bacteroides, Selenomonas, and Prevotella) are maintained in CRC metastases. Metronidazole reduces Fusobacterium load and tumor growth in mice. |
| Yu, et al. 2017 [30] | Patients, mouse model, cell lines | 16 with recurrent CRC | F. nucleatum is associated with recurrence post-chemotherapy and promotes chemotherapy resistance through activation of autophagy. |
| 15 with non-recurrent CRC | |||
| Yang, et al. 2017 [36] | Patients, mouse model, cell lines | 105 with CRC | F. nucleatum increases CRC proliferation and invasion in vitro. F. nucleatum-induced TLR4 signaling leads to increased miR21. High levels of F. nucleatum DNA and miR21 associated with shorter OS in patients. |
| Serna, et al. 2020 [37] | Patients | 143 with rectal cancer | F. nucleatum persistence after neoadjuvant chemoradiation associated with decreased CD8 |
| Chen, et al. 2022 [25] | Patients, mouse model, cell lines | 380 with CRC | F. nucleatum activates YAP signaling, reducing METTL3 expression and increasing KIF26B expression. High KIF26B expression associated with shorter OS in CRC patients. |
| Jiang, et al. 2023 [31] | Patients, mouse models, cell lines | 42 with CRC | F. nucleatum metabolite succinic acid reduces CD8 |
| Akkermansia muciniphila | |||
| Wang, et al. 2020 [28] | Patients, mouse model, cell lines | 72 healthy controls | A. muciniphila was reduced in IBD patients and mice with colitis or CRC. A. muciniphila associated with cytotoxic T lymphocyte activation and decreased tumorigenesis. |
| 58 with ulcerative colitis | |||
| 18 with adenomas | |||
| 22 with CRC | |||
| Jiang, et al. 2023 [29] | Mouse model, cell lines | N/A | Acetyltransferase of A. muciniphila promote a cytotoxic T cell response and blunts tumorigenesis in mice. |
| Peptostreptococus anaerobius | |||
| Nakatsu, et al. 2015 [38] | Patients | 61 healthy controls | Metagenomic profiling linked multiple taxa, including Peptostreptococcus and Parvimonas, to CRC development and progression. |
| 47 with adenomas | |||
| 52 with CRC | |||
| Tsoi, et al. 2017 [26] | Patients, mouse model, cell lines | 49 healthy controls | P. anaerobius enriched in CRC patient stool and biopsy specimens. P. anaerobius exposure promoted cholesterol biosynthesis and cell proliferation in vitro and intestinal dysplasia in mice. |
| 45 with adenomas | |||
| 50 with CRC | |||
| Long, et al. 2019 [27] | Mouse model, cell lines | N/A | P. anaerobius adheres CRC cell integrins, activating the PI3K-Akt pathway and stimulating proliferation and myeloid cell recruitment. |
Abbreviations: PCR, polymerase chain reaction; CRC, colorectal cancer; qPCR, quantitative PCR; pks, polyketide synthase; IBD, inflammatory bowel disease; FISH, fluorescence in situ hybridization; ETBF, enterotoxic Bacteroides fragilis; FAP, familial adenomatous polyposis; N/A, not applicable; ATP, adenosine triphosphate; NTBF, non-toxigenic Bacteroides fragilis; TLR4, toll-like receptor 4; miR21, microRNA21; OS, overall survival; MDSC, myeloid-derived suppressor cell; KIF26B, kinesin family member 26B; ICAM1, intercellular adhesion molecule 1.
Similarly, a subgroup of the ubiquitous Bacteroides fragilis
species, enterotoxic B. fragilis (ETBF) is also strongly linked to colorectal
carcinogenesis. ETBF may present with asymptomatic colonization but can also
cause acute diarrheal illness. A study of ETBF in Apc-deficient multiple
intestinal neoplasia (Min) mice found that ETBF colonization leads to Stat3
activation and a T
Perhaps more than any other species, Fusobacterium nucleatum has been closely associated with CRC at every stage of disease. F. nucleatum is an oral commensal bacterium that reaches the colon through the digestive tract and by hematogenous spread [41]. Indeed, a study evaluating paired saliva and CRC samples found identical F. nucleatum strains at both sites [42]. In an evaluation of the CRC-associated gut microbiome, Ahn and colleagues found that feces from patients with CRC were enriched in F. nucleatum and Porphyromonas mRNA when compared to healthy controls [43]. Flanagan and colleagues similarly performed a qPCR-based evaluation of resected colorectal tumors and benign biopsy specimens and found higher levels of F. nucleatum in CRC patients. They further demonstrated an inverse correlation between F. nucleatum level and overall survival (OS). Importantly, a subgroup analysis of patients with pre-cancerous adenomas demonstrated F. nucleatum enrichment in specimens with high-grade dysplasia, suggesting that the carcinogenic impact of F. nucleatum occurs early in the adenoma-carcinoma progression [44]. Mima et al. [45] confirmed that Fusobacterium level was independently associated with shortened OS (Hazard ratio (HR) 1.58 [95% confidence interval (CI) 1.04–2.39]) and additionally noted that BRAF mutant tumors were enriched in F. nucleatum, linking the bacteria to a more aggressive phenotype.
Multiple mechanisms connecting F. nucleatum to CRC initiation have been
studied. The Fusobacterium virulence factor, FadA, has been shown to act
by binding to E-cadherin and activating Wnt/
Other genera including Peptostreptococcus, Parvimonas, Akkermansia, and Desulfovibrio have been linked to CRC initiation [26, 27, 28, 29, 46, 47]. However, dysbiosis-related CRC is not always driven by a single bacterial taxon. A generalized decrease in microbial diversity has also been linked to CRC in multiple studies. Wong et al. [48] found that oral gavage of fecal samples from CRC patients, but not healthy controls, promoted colonic polyp formation in germ-free and conventional mice treated with azoxymethane. The resultant gut microbiome in mice treated with CRC-derived stool was characterized by lower Fisher and Shannon-Weaver alpha diversity [48]. A fecal metagenomic comparison of patients with CRC and healthy controls similarly demonstrated reduced gene richness and alpha diversity in patients with CRC. Interestingly, control-enriched microbial genes occurred at a higher frequency and abundance than CRC-enriched genes, suggesting that CRC carcinogenesis is more commonly driven by an imbalanced gut microbiome rather than a dominant pathobiont [46].
The association between the microbiota and CRC extends beyond development of the primary tumor. Sun et al. [49] collected fecal samples from 30 patients with early-stage CRC and 30 with metastatic CRC and found consistent, generalized differences in fecal microbiome composition at both the genus and species levels. Bullman and colleagues [32] further demonstrated that Fusobacterium, and co-occurring anaerobes, were present in both primary colon tumors and matched liver metastases, suggesting that these agents may co-migrate with tumor cells to the metastatic target organ. The mechanisms by which the microbiome affects metastasis are multifactorial and have only begun to be understood but appear to involve both the tumor and gut microbiome. Current research links the microbiome to almost every stage of the metastatic cascade, including gut barrier penetration, pre-metastatic niche formation, epithelial-mesenchymal transition, intravasation, extravasation, and outgrowth (Fig. 1).
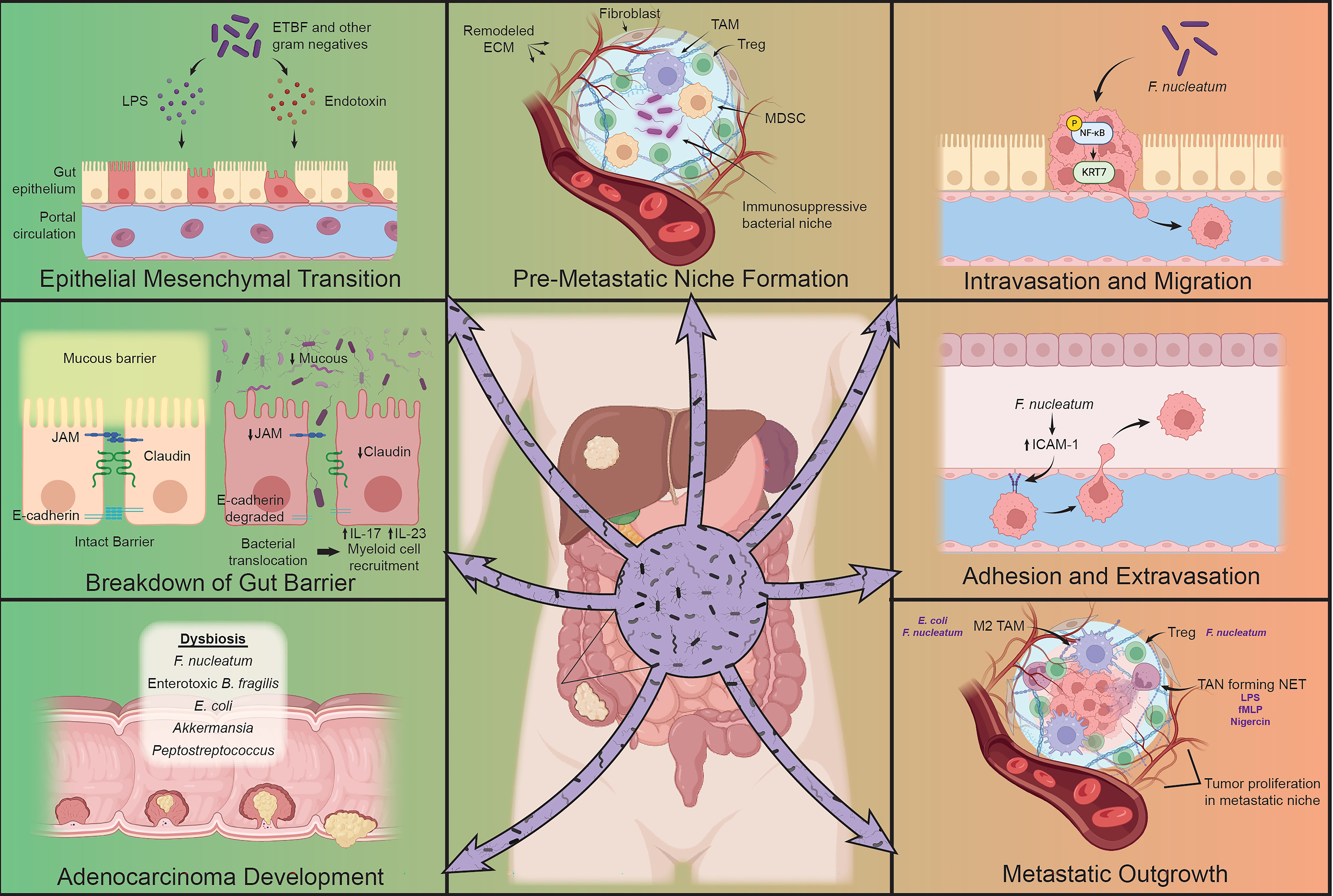 Fig. 1.
Fig. 1.The microbiome and colorectal cancer metastasis. The gut and
tumoral microbiomes modulate colorectal cancer metastasis at multiple levels,
including primary tumor development, breakdown of the gut vascular barrier,
epithelial mesenchymal transition, pre-metastatic niche formation, intravasation
and migration, adhesion and extravasation, and metastatic outgrowth. LPS, lipopolysaccharide; ETBF, enterotoxic bacteroides fragilis; JAM, junctional adhesion molecule; ECM, extracellular matrix; TAM, tumor associated macrophage; MDSC, myeloid derived suppressor cell; NF-
The gut vascular barrier is a complex system that acts as both a physical and
chemical defense, preventing harmful intestinal microbes and antigens from
entering the host circulation [50]. Penetration of the gut vascular barrier is a
critical step in CRC invasion and metastasis and is mediated, in part, by the gut
microbiome. Wu and colleagues [39] found the metalloproteinase enterotoxin of
ETBF cleaves the extracellular domain of E-cadherin, a critical zonula adherens
protein. This process increases gut barrier permeability and activates the
The liver is the most common site of CRC metastasis, with colorectal liver metastases (CLM) developing in up to 50% of CRC patients at some point during their disease course [52]. Recent evidence suggests that a hepatic pre-metastatic niche (PMN) is formed prior to the development of overt CLM, and implicate the gut and tumoral microbiomes in its formation.
PMN are organ specific microenvironments that are primed for the implantation and outgrowth of disseminated tumor cells prior to their arrival [53]. PMNs are modulated by soluble secreted factors and extracellular vesicles from the primary tumor and are characterized by vascular permeability, extracellular matrix remodeling, angiogenesis, and an immunosuppressive microenvironment rich in regulatory T cells (Treg), myeloid derived suppressor cells (MDSCs) and fibroblasts [54].
Recent work by Bertocchi et al. [55] elucidated the role of the gut and tumoral microbiomes in hepatic PMN formation. The authors showed that gut vascular barrier impairment triggered by E. coli at the primary tumor site, as indicated by plasmalemma vesicle-associated protein-1 (PV-1) expression, correlated with bacterial levels in paired CLM samples. They further demonstrated that targeted antibiotic treatment with neomycin reduced the recruitment of innate immune cells to the liver and abrogated CLM formation [55]. Dysbiosis secondary to a high-fat diet has also been linked to liver metastasis with increased expression of multiple PMN markers, including matrix metallopeptidase 2 (MMP2), matrix metallopeptidase 9 (MMP9), fibronectin, and C-X-C chemokine ligand 12 (CXCL12), in non-tumoral hepatic tissue. This pro-tumoral cytokine signature was abrogated by antibiotic treatment, suggesting that its formation is microbiome-dependent [47]. The role of bacteria in PMN establishment was further supported in a recent study by Galeano Niño et al. [56], using spatial transcriptomics to show that tumor-associated microbial communities are not distributed randomly, but comprise organized, immunosuppressive microniches that promote tumor progression.
Epithelial-mesenchymal transition (EMT) is a highly complex process by which malignant epithelial cells lose their normal polarity and assume a mesenchymal stem cell phenotype, enabling invasion, migration, and metastasis. Studies across multiple tumor types suggest that gram negative bacteria interface with the EMT program to promote cancer dissemination.
Zhao and colleagues found that lipopolysaccharide (LPS) in the gram-negative
bacterial cell wall can induce EMT in the liver. Benign biliary epithelial cells
cultured in the presence of LPS exhibited upregulation of mesenchymal markers
including S100A and
The potential for CRC cells to migrate and metastasize is also increased in the
presence of F. nucleatum. Chen et al. [62] found that
F. nucleatum infection leads to upregulation of the non-coding antisense
RNA KRT7-AS via NF-
Fusobacterium nucleatum plays yet another role in CRC progression at
the juncture of circulating tumor cell endothelial adhesion and extravasation.
Zhang et al. [63] serendipitously found that the human CRC cell line
HCT116 exhibited markedly increased adherence to vascular endothelium in the
presence of F. nucleatum as compared to E. coli or PBS.
F. nucleatum infection also enhanced cell migration relative to
E. coli and Akkermansia muciniphilia through the upregulation
of ICAM1 via ALPK1-mediated NF-
The process by which extravasated tumor cells survive and proliferate in the target organ is exceedingly complex and involves interplay between tumor cells, the target organ microenvironment, and the host immune system. Studies in breast cancer have demonstrated that tumor cells may disseminate early in the disease course and remain in a dormant/quiescent state for years before metastatic outgrowth is triggered [64, 65]. In fact, stress from surgery to remove the primary tumor may, in some cases, precipitate metastatic outgrowth in patients with no clinical evidence of metastatic disease [66, 67].
Cytotoxic necrotizing factor 1 (CNF1), a bacterial toxin produced by E. coli, plays a role in the quiescence of disseminated CRC cells. CNF1 blocks cytokinesis, elicits endoreplication and polyploidization, and drives cells into a reversible dormant state [68]. Dormant tumor cells exhibit intrinsic resistance to chemotherapy agents that are conventionally designed to target and eradicate rapidly proliferating tumor cells [69, 70].
The multifarious triggers that cause tumor cells to exit dormancy and proliferate have not been fully elucidated, but it is known that a favorable tumor immune microenvironment (TIME) is required [71]. The gut and tumor-associated microbiomes interact with both the innate and adaptive immune systems to produce an inflammatory milieu that favors metastatic outgrowth.
Tumor-associated macrophages (TAMs) and neutrophils (TANs) play important roles in the promotion of metastatic outgrowth. TAMs exist in a state of flux between the M1 and M2 polarization states. M1 macrophages promote cytotoxicity and effectuate a tumor-suppressive microenvironment while M2 macrophages are associated with immunosuppression and the expression of cytokines that promote tumor survival and proliferation [72]. Both E. coli and F. nucleatum have been shown to shift this balance in favor of the M2 state. Li and colleagues [73] found that E. coli gavage stimulated the secretion of cathepsin K from MC38 cells implanted in the cecal mesentery of antibiotic treated mice. Binding of cathepsin K to TLR4 produced M2 polarization of TAMs and was associated with more numerous liver metastases [73]. F. nucleatum infection similarly facilitates metastasis through the downregulation of miR-1322, leading to increased CCL20 expression and M2 macrophage differentiation [74].
Activated TANs also support metastatic outgrowth through the expression of multiple cytokines including MMP-9, VEGF, CXCL4, and CCL5, and through the formation of weblike structures known as neutrophil extracellular traps (NETs). Bacteria residing in the metastatic niche activate NETosis through pathogen-associated molecular patterns (PAMPs), including LPS, fMLP, and Nigercin [75, 76, 77]. Cleavage of the extracellular matrix protein laminin by NET-associated proteases then activates dormant tumor cells through integrin signaling [75, 78]. NETs can further promote metastasis by shielding circulating tumor cells, stimulating angiogenesis, and promoting the formation of tumor thrombi [75].
Finally, tumor-associated microbes also modulate the adaptive immune response to
promote metastatic outgrowth. Sakamoto et al. [79] found that F.
nucleatum levels were associated with significantly lower cytotoxic (CD8
Systemic therapy plays an indispensable role in the modern management of metastatic CRC. In addition to mediating therapy resistance through quiescence, the microbiome can have a direct impact on response to both cytotoxic chemotherapy and immunotherapy.
Systemic chemotherapy is known to alter gut microbial composition, diversity,
and function [81]. However, the gut and tumoral microbiomes can also mediate
cytotoxic therapy metabolism and response. Multiple chemotherapeutic agents,
including cyclophosphamide, oxaliplatin, irinotecan, and 5-FU are known to
interact with the intestinal microbiome [82, 83, 84, 85]. With regard to CRC, Yu
et al. [30] found that colon cancer cell lines attained resistance to
both 5-FU and oxaliplatin when cocultured with F. nucleatum. Chloroquine
assays revealed that this process is dependent on autophagy driven by TLR4 and
MYD88 signaling [30]. F. nucleatum-induced TLR4/NF-
Immune checkpoint blockade (ICB) has revolutionized the treatment of multiple solid tumors. Several important studies have linked the gut microbiome to ICB response and toxicity in melanoma and non-small cell lung cancer. In these investigations, increased relative abundance of specific bacterial taxa, including Akkermansia muciniphila, Bacteroidaceae, Ruminococcaceae, and Bifidobacterium, were associated with improved ICB response [89, 90, 91, 92].
In CRC, the role of ICB has been largely limited to patients with microsatellite
unstable tumors [93]. These cancers, characterized by a high mutational burden
and increased expression of immune checkpoints, exhibit increased immunogenicity
and response to both CTLA-4 and PD-1 inhibition [94, 95, 96]. The impact of the
microbiome in this setting and the potential for microbe-based sensitization of
microsatellite stable tumors to ICB has not yet been established; however, early
preclinical studies provide promising results. Destefano and colleagues found
that BRAF
Peritoneal metastases (PM) occur in 5 to 10% of CRC patients and are associated with worse clinical outcomes than any other metastatic site [98]. The peritoneal microenvironment and the role of the microbiome in CRC carcinomatosis have not been well studied or defined. In ovarian cancer, another solid tumor with a strong connection to dysbiosis, local gram-negative peritoneal colonies and decreased microbiome diversity have been associated with peritoneal spread [99, 100]. In PM from appendiceal cancer, enteric bacteria, including those of the Proteobacteria, Actinobacteria, Firmicutes, and Bacteroidetes phyla, have been identified in both PM and associated mucinous ascites, implicating these taxa in the pathobiology of this disease site. Ongoing studies will reveal more about the unique TIME of CRC PM and the potential role of gut and tumoral microbes in its development.
Strong correlations between the gut microbiome and oncologic outcomes have led to the study of microbiome modulation as a therapeutic strategy. In their aforementioned study of paired CRC and CLM, Bullman and colleagues found that treatment with the antibiotic metronidazole led to reduced Fusobacterium levels and decreased tumor cell proliferation [32]. Two forthcoming trials are now evaluating the efficacy of neoadjuvant metronidazole in patients with CRC (NCT04264676, NCT05748145).
Dietary modification also presents a low-risk means to modulate the gut microbiome as well as CRC outcomes. CRC patients who consume a high-fat diet are known to have significantly shorter 5-year disease-free survival [101]. On the other hand, work by Spencer et al. [102] showed that a high-fiber diet produced taxonomic and structural changes in the gut microbiome that correlated with anti-PD-1 response in conventionally housed mice and in human patients who reported sufficient dietary fiber intake. Numerous trials are now evaluating the effect of diet-based microbiome modulation on CRC outcomes. At our institution, the Beans to Enrich the Gut Microbiome vs Obesity’s Negative Effects (BEGONE) trial is evaluating the longitudinal effect of dietary fiber on the gut microbiome and risk of CRC recurrence.
Fecal microbiota transplant (FMT) is the most direct means of altering the gut microbiome and is now widely accepted as an effective treatment for refractory Clostridium difficile colitis [103]. FMT has also been used to successfully treat inflammatory bowel disease and is under investigation in the treatment of other autoimmune disorders [104, 105]. In oncology, FMT has been used to overcome melanoma resistance to ICB in multiple preclinical studies and at least two clinical trials [106, 107]. Multiple ongoing trials are investigating the role of FMT in modulating toxicity and improving response to ICB in other solid tumors, including CRC.
While still in the preliminary stages of investigation, study of both additive (FMT) and subtractive (antibiotics and novel therapeutics) modulation of the gut and tumoral microbiome in CRC carries enormous clinical potential. As suggested by the preclinical studies covered in this review, these efforts may provide new avenues through which to improve CRC detection and increase the efficacy of existing standard treatments. As our understanding of the microbiome’s role in gut vascular permeability and modulation of the host immune response deepens, we anticipate a new class of microbiome-based interventions that may even stymie CRC dissemination and metastatic outgrowth.
RIA outlined, wrote, and edited the manuscript and created the figure. MGW was invited to contribute this review, outlined the concepts to be covered, wrote and edited the manuscript. Both authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity. Both authors read and approved the final manuscript. Both authors contributed to editorial changes in the manuscript.
Not applicable.
We thank all of the esteemed investigators whose groundbreaking research on the microbiome, cancer metastasis, and the tumor microenvironment made this review possible.
This research received no external funding.
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.