
- Academic Editors
Background: Lactiplantibacillus plantarum 12-3 holds great promise as a probiotic bacterial strain, yet its full potential remains untapped. This study aimed to better understand this potential therapeutic strain by exploring its genomic landscape, genetic diversity, CRISPR-Cas mechanism, genotype, and mechanistic perspectives for probiotic functionality and safety applications. Methods: L. plantarum 12-3 was isolated from Tibetan kefir grains and, subsequently, Illumina and Single Molecule Real-Time (SMRT) technologies were used to extract and sequence genomic DNA from this organism. After performing pan-genomic and phylogenetic analysis, Average Nucleotide Identity (ANI) was used to confirm the taxonomic identity of the strain. Antibiotic resistance gene analysis was conducted using the Comprehensive Antibiotic Resistance Database (CARD). Antimicrobial susceptibility testing, and virulence gene identification were also included in our genomic analysis to evaluate food safety. Prophage, genomic islands, insertion sequences, and CRISPR-Cas sequence analyses were also carried out to gain insight into genetic components and defensive mechanisms within the bacterial genome. Results: The 3.4 Mb genome of L. plantarum 12-3, was assembled with 99.1% completeness and low contamination. A total of 3234 genes with normal length and intergenic spacing were found using gene prediction tools. Pan-genomic studies demonstrated gene diversity and provided functional annotation, whereas phylogenetic analysis verified taxonomic identity. Our food safety study revealed a profile of antibiotic resistance that is favorable for use as a probiotic. Analysis of insertional sequences, genomic islands, and prophage within the genome provided information regarding genetic components and their possible effects on evolution. Conclusions: Pivotal genetic elements uncovered in this study play a crucial role in bacterial defense mechanisms and offer intriguing prospects for future genome engineering efforts. Moreover, our findings suggest further in vitro and in vivo studies are warranted to validate the functional attributes and probiotic potential of L. plantarum 12-3. Expanding the scope of the research to encompass a broader range of L. plantarum 12-3 strains and comparative analyses with other probiotic species would enhance our understanding of this organism’s genetic diversity and functional properties.
Identification of microorganisms with potential probiotic application has become a focal point in the field of microbiology in recent years. Such efforts are motivated, in part, by the discovery that many microorganisms play a critical role in promoting health and avoiding disease. L. plantarum 12-3, a strain of lactic acid bacteria, has met with notable interest among potential microorganisms due to its reported probiotic properties. A thorough explanation of the rationale for closely examining L. plantarum 12-3 as a probiotic would substantially benefit the strain’s potential therapeutic contribution [1].
Understanding probiotic microbe activity and safety applications is inextricably tied to their genetic diversity. In keeping with this principal, the genetic composition of L. plantarum 12-3 has potential to understanding its probiotic efficacy and safety. Indeed, Mao et al. [1] emphasized the need to investigate the genetic landscape of L. plantarum 12-3, emphasizing the need for a more detailed understanding of its genomic composition, and Parlindungan et al. [2] commented that understanding the genetic variability of L. plantarum strain 12-3 is of importance in the investigation of its probiotic efficacy and safety implications. Here we sought to uncover subtle mechanisms that govern this bacterium’s probiotic potency and safety by better understanding its genetic diversity. Furthermore, the use of advanced genotyping technologies provides a more detailed understanding of the strain’s genetic variations, and may provide insights into its hereditary traits. As the role of probiotics in health promotion becomes more apparent, this work serves as a fundamental investigation, offering critical knowledge for exploiting the probiotic potential of L. plantarum 12-3 as this may result in an understanding of its genetic variability and possible implications in the area of probiotic use and risk evaluation [3].
The L. plantarum 12-3 species is recognized for its adaptable characteristics and extensive prevalence in diverse ecological habitats. L. plantarum 12-3 strains are frequently detected in fermented food items including dairy products, pickles, and sauerkraut where they play a significant role in preserving the food and enhancing its flavor profile [4]. Furthermore, L. plantarum 12-3 has been acknowledged for its prospective health advantages and probiotic functionality. Probiotics are living microorganisms that, upon being administered in sufficient quantities, provide advantageous health effects to the host. The probiotic potential of L. plantarum 12-3 has garnered significant interest, owing to its ability to withstand the harsh conditions of the gastrointestinal tract and elicit favorable impact on gut health.
The use of modern genotyping techniques can provide valuable insight into genomic variations and hereditary traits, thereby shedding light on its potential adaptability and advantageous features. Applications of these technologies will provide a thorough understanding of L. plantarum 12-3 can be attained, by better understanding the genetic basis of its probiotic properties and ensuring its appropriateness for human consumption [5]. The principal aim of this study was to specifically investigate the genetic diversity, CRISPR-Cas system, and genotype of L. plantarum 12-3, and place emphasis on the potential probiotic properties and safety implications associated with this particular microorganism.
Investigation of L. plantarum 12-3 has yielded useful insight into its genetic diversity, probiotic potential, and genomic features. The dynamic character of this strain’s genome has been revealed through predictive analysis of transposases, mobile elements, and genomic islands, and provides a solid platform for future research [6]. Needed information should include improved genomic analysis, resolve prior analytical flaws, and conduct in-depth functional studies. Furthermore, the prospective applications of L. plantarum 12-3 in probiotics, biotechnology, and medicines require further investigation as such additional understanding will surely advance our understanding of probiotic bacteria and their potential uses in health and biotechnology [7].
L. plantarum 12-3 was isolated from Tibetan Kefir grains in 2015 and maintained as frozen (–20 °C) stocks in MRS broth (Beijing Aoboxing Co., Ltd., Beijing, China) supplemented with 20% (v/v) glycerol. These strains were identified primarily through Gram staining, catalase assays, cell morphology, and 16S rDNA sequencing analysis as previously described [8, 9, 10, 11, 12, 13].
Genomic DNA was extracted using a Wizard® Genomic DNA Purification Kit (Promega, Madison, WI, USA) and then quantified using a TBS-380 fluorometer (Turner Bio Systems Inc., Sunnyvale, CA, USA). An OD260/280 ratio between 1.8 and 2.0 was obtained and a DNA quantity greater than 20 micrograms was utilized. Using the NEXTflex Rapid DNA-Seq Kit (Revvity, Waltham, MA, USA), sheared fragments were utilized to create Illumina sequencing libraries. Briefly, DNA ends were subjected to initial end repair and phosphorylation, and termini were subsequently A-tailed and ligated with sequencing adapters. Next, polymerase chain reaction (PCR)-based amplification of the adapter-ligated products was conducted for enrichment purposes. The constructed libraries were then sequenced utilizing a paired-end approach on an Illumina HiSeq X Ten platform, with 150 bp read length at each end. The genome of L. plantarum 12-3 was sequenced utilizing Single Molecule Real-Time (SMRT) technology and Illumina sequencing platforms. The accession number given to the completed and assembled L. plantarum 12-3 genome is GCF_004028335.1. The complexity of the genome was measured by examining the information provided by Illumina [14].
To verify the taxonomic identity of the L. plantarum 12-3 strain, an analysis of the Average Nucleotide Identity (ANI) was conducted on various strains of L. plantarum 12-3 (Table 1). After plasmid removal, ANI analysis was undertaken using the publicly-available JSpecies Web Server (http://jspecies.ribohost.com/jspeciesws/) [15]. HemI version 2.0 (http://hemi.biocuckoo.org/) was utilized to create gene clustering and heatmap [16]. In addition, fully assembled genomes of L. plantarum 12-3 isolates available on PanX (https://pangenome.org/;accessedon28May2023) were used for pan-genomic analysis. All genomes were re-annotated with the Prokka tool to eliminate biases in comparisons caused by the various annotation methods used. The output general feature format (GFF) was used to perform a pan-genome analysis using a BLASTp (https://blast.ncbi.nlm.nih.gov/Blast.cgi) threshold of 95% to identify core, accessory, and unique genes. As a result, four distinct gene classes were identified: core (99% strain 100%), soft-core (95% strain 99%), shell (15% strain 95%), and cloud (0% strain 15%) [17].
| STRAINS | SIZE | CONTINGS | GC% |
| L. plantarum 12-3 | 3,403,608 | 8 | 44.4 |
| L. plantarum CMPG5300 | 3,507,828 | 7 | 44.15 |
| L. plantarum 2025 | 3,334,257 | 164 | 44.44 |
| L. plantarum Nizo2802 | 3,372,514 | 89 | 44.45 |
| L. plantarum 16 | 3,361,015 | 11 | 44.34 |
| L. plantarum E2C2 | 3,603,563 | 121 | 43.99 |
| L. plantarum IPLA88 | 3,254,055 | 208 | 44.43 |
| L. plantarum L31-1 | 3,216,471 | 45 | 44.50 |
| L. plantarum 90sk | 3,371,458 | 47 | 44.26 |
| L. plantarum subsp. plantarum CGMCC | 3,273,801 | 4 | 44.42 |
| L. plantarum WJL | 3,503,067 | 13 | 44.21 |
| L. plantarum SRCM101060 | 3,263,056 | 101 | 44.33 |
| L. plantarum subsp. plantarum P -8plantarum | 3,246,630 | 8 | 44.55 |
| L. plantarum JSA22 | 3,455,529 | 3 | 44.29 |
| L. plantarum ZJ316 | 3,299,755 | 4 | 44.49 |
| L. plantarum WCFS1 | 3,348,624 | 4 | 44.42 |
| L. plantarum 80 | 3,224,773 | 67 | 44.36 |
| L. plantarum WJL | 3,477,495 | 102 | 44.24 |
| L. plantarum DSM 13273 | 3,439,800 | 90 | 44.22 |
| L. plantarum UCMA 3037 | 3,108,278 | 68 | 44.52 |
| L. plantarum Nizo2891 | 3,469,171 | 78 | 44.13 |
| L. plantarum B21 | 3,284,260 | 1 | 44.47 |
| L. plantarum 8RA -3 | 3,330,093 | 18 | 44.37 |
| L. plantarum ST -III | 3,307,936 | 2 | 44.48 |
| L. plantarum PS128 | 3,325,806 | 11 | 44.41 |
| L. plantarum WLPL04 | 3,185,263 | 51 | 44.52 |
| L. plantarum JDM1 | 3,197,759 | 1 | 44.66 |
| L. plantarum DSM 20174 | 3,250,154 | 2 | 44.50 |
| L. plantarum 2165 | 3,179,972 | 192 | 44.53 |
ANI, average nucleotide identity; GC, Guanine and Cytosine.
The Comprehensive Antibiotic Resistance Database (CARD) (https://card.mcmaster.ca/) was used as this database has a large number of annotated antibiotic resistance genes. Antibiotic resistance genes were identified by comparing the predicted genes with the CARD database using specific algorithms and tools. Additional analysis and filtering were performed on the discovered matches based on parameters such as sequence similarity, functional annotation, and resistance mechanisms. Based on their resistance patterns, the discovered antibiotic resistance genes were categorized and characterized [18]. Antimicrobial susceptibility tests and comparison with previously identified resistance genes were also included as validation phases.
Collectively, this approach enabled the prediction, and characterization, of antibiotic resistance genes and shed light on resistance profiles of bacterial isolates and added to our knowledge of antibiotic resistance. Human host pathogenicity predictions and the identification of acquired virulence genes were conducted using VirulenceFinder 2.0 (https://cge.food.dtu.dk/services/VirulenceFinder/) to provide a safety assessment of L. plantarum 12-3 [19, 20].
Phage Search Tool Enhanced Release (PHASTER) (http://phaster.ca) was utilized for the identification of genomic regions containing prophage. This tool provided the length, localization, guanine and cytosine (GC) content, and gene annotation of the prophage [21]. A comparison was made between the predicted intact prophage sequences and the Virus-Host DB database (https://www.genome.jp/virushostdb) to validate detected prophages. Using the VIPtree application, a proteomics tree of the viral genome sequence was also generated. This tree was constructed using tBLASTx-calculated genome-wide sequence similarities. This analysis helped clarify the evolutionary relationships between viral genomes.
The presence of genomic islands (GI) was analyzed by searching multiple databases with Island Viewer 4 (https://www.pathogenomics.sfu.ca/islandviewer). This made it possible to identify and characterize genomic islands within the bacterial genome. Using these methods and databases, a thorough analysis of prophage regions and genomic islands was conducted. These efforts yielded valuable insights into genetic elements present in the L. plantarum 12-3 genome and their potential impact on bacterial evolution and virulence. ISFinder and ISsaga were subsequently utilized for the annotation of transposase and mobile elements [22].
The identification and verification of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated genes (Cas) was accomplished with the aid of specialized bioinformatic tools. The CRISPRCasFinder v.1.1.2 (https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index) tool was used to search for CRISPR and Cas genes within the genome’s coding sequences and enabled the detection of arrays and associated Cas genes in genomic sequences of interest. For validating the identified arrays, the CRISPRdb database (https://crisprdb.org) was queried. The predicted results were cross-referenced with the CRISPRdb database to assure their accuracy and alignment with previously identified CRISPR sequences. This methodology enabled the identification and confirmation of arrays and associated Cas genes within the studied genomes, and provided valuable information concerning the presence and organization of essential genetic elements involved in bacterial defense and potential approaches to future genome engineering applications [23].
Mobile elements and repetitive sequences present in the L. plantarum 12-3 genome make it challenging to assemble the entire genome using only short-read sequences obtained using the Illumina NextGen sequencing platform. The G+C content of the 3,403,608 bp L. plantarum 12-3 whole genome was 44.4% within 8 assembled contigs. The genome’s quality analysis was estimated at 99.1% genome completeness with only 1.39% contamination. Fig. 1 indicates the L. plantarum 12-3 reference sequence (RefSeq) assembly completeness. The circular map of the whole genome is given below in Fig. 2.
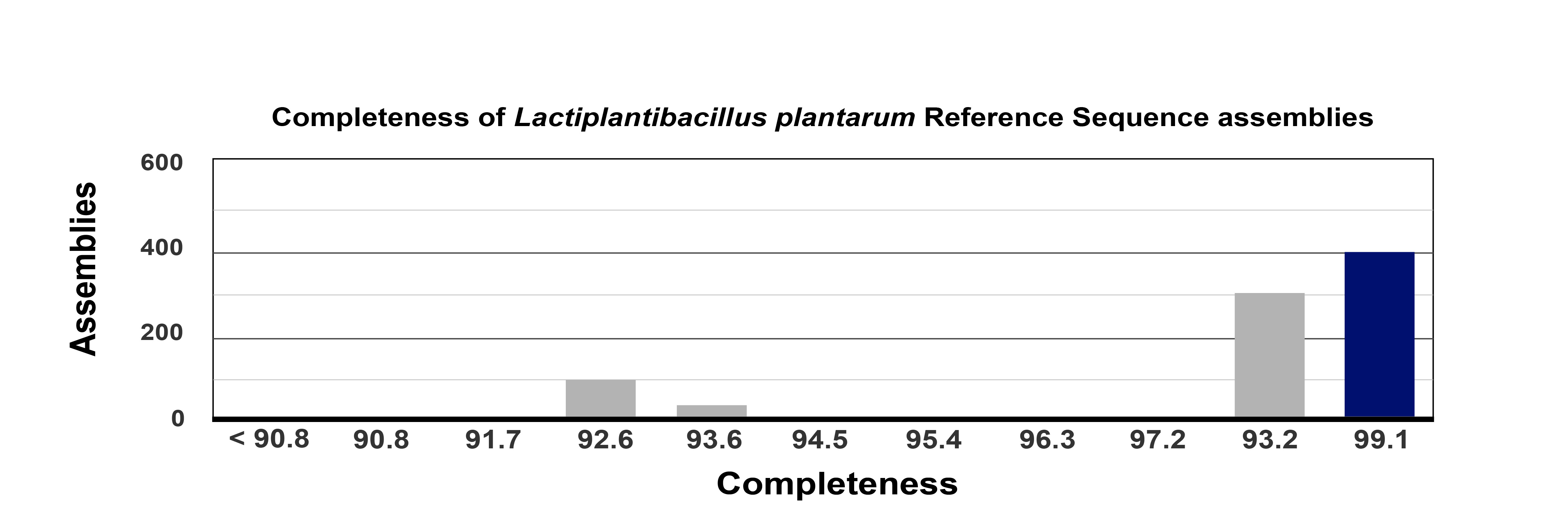 Fig. 1.
Fig. 1.The genome completeness overview of the genomic assemblies from L. plantarum 12-3. RefSeq, Reference Sequence Database.
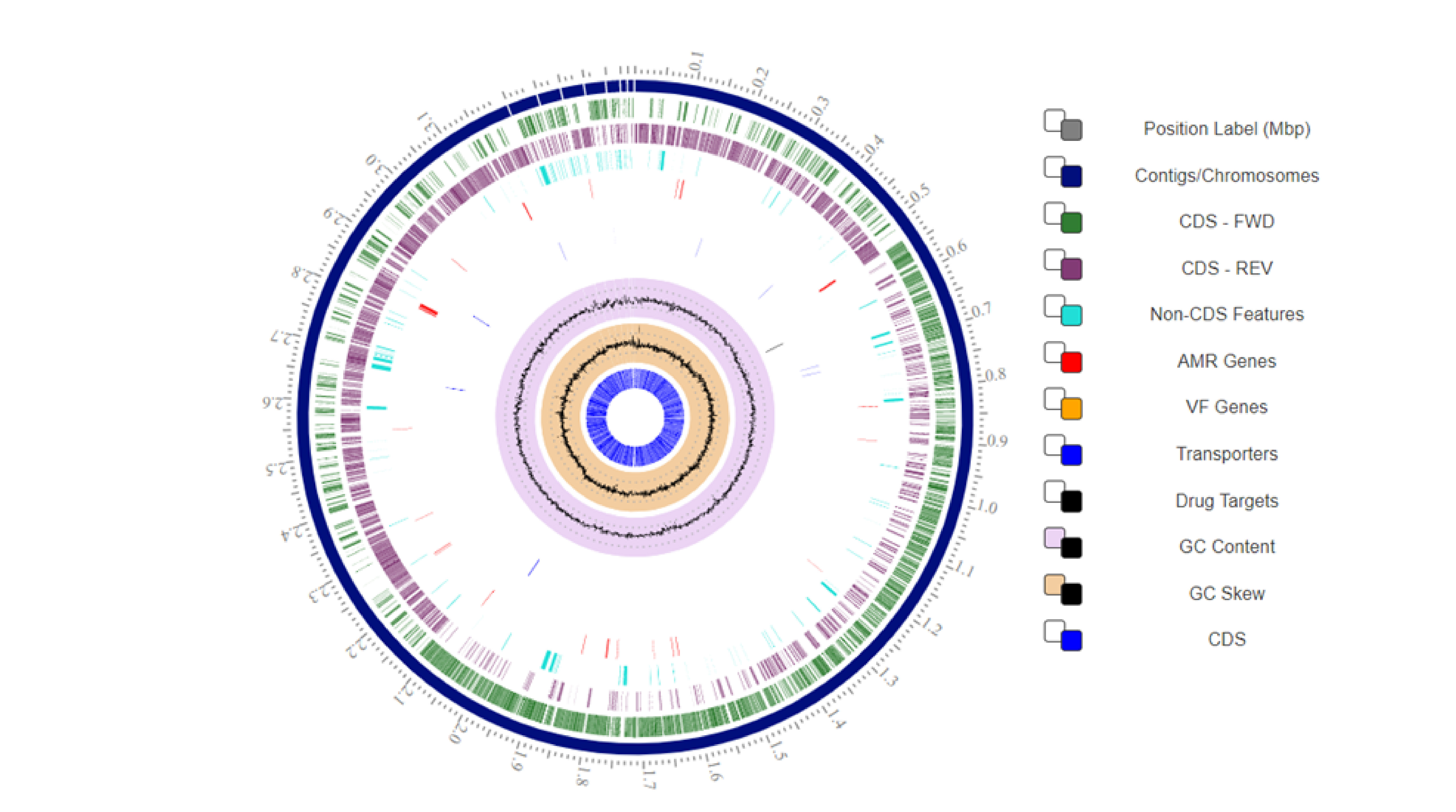 Fig. 2.
Fig. 2.The genetic characteristics and circular map of the L. plantarum 12-3 genome. CDS, Coding Sequence; AMR, Antimicrobial Resistance; VF, Virulence Factor; FWD, Forward; REV, Reverese; GC, Guanine-Cytosine; Mbp, Million Base Pairs.
MetaGeneMark (http://exon.gatech.edu/meta_gmhmmp.cgi) was used to predict the location of genes within the L. plantarum 12-3 genome. A total of 3234 genes were predicted, with typical lengths ranging from 1020 to 2500 base pairs. An average intergenic spacing of 1000 bp indicates that the genome underwent gene dispersion.
Closer examination of the L. plantarum 12-3 genome revealed a wide variety of tRNA, tmRNA, and rRNA sequences. There are several different varieties of tRNA, including tRNA-Val, tRNA-Met, and tRNA-Ala genes. Examining the predicted tRNA secondary structures revealed sequence differences in the anticodon loops and stems. In addition to transfer RNA, transfer-messenger RNA (tmRNA) genes were also expected to be present in the genome. The breakdown of abnormal proteins can occur during translation and tmRNA is essential in this process, and the mechanisms directing protein quality control can be better understood by examining tmRNA. The genome of L. plantarum 12-3 has 43 specialty genes which are specialized for various bioactivities (Supplementary Table 1).
ANI analysis was used to confirm the taxonomy and phylogenetic association of
L. plantarum 12-3, utilizing the platform’s comprehensive genome
assembly tool. In our ANI analysis, various strains of Lactobacillus
plantarum were incorporated (see Supplementary Table 1). The results of
the ANI analysis indicated that LPG1 exhibited a high degree of similarity with
other L. plantarum 12-3 strains, with an ANI value exceeding
the established threshold for species identity (
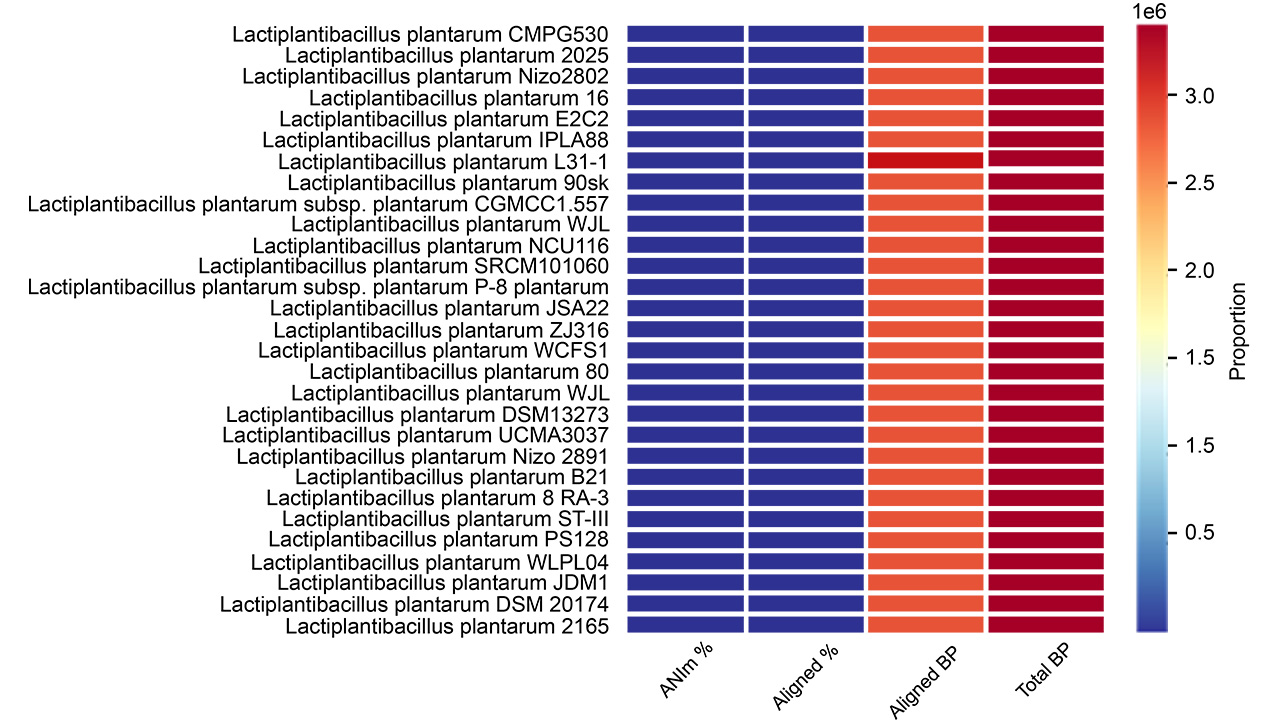 Fig. 3.
Fig. 3.Clustering and heatmaps of various strains of L. plantarum 12-3 indicating the ANIm percentage, aligned percentage, aligned basepairs, and total basepairs. BP, basepairs; ANIm, Average Nucleotide Identity based on MUMmer.
Pan-genome analysis revealed a considerable magnitude and heterogeneity in L. plantarum 12-3. We observed that genomes can be categorized into distinct types, including a collection of genes that are universally present across all living organisms and another collection of genes that are exclusively shared among a particular subset of organisms. Discrepancies observed in the analysis of prokaryotic organism genomes imply variations in their genetic makeup and evolutionary trajectories. Pan-genome functional annotation has exposed numerous pathways that are involved in diverse biological processes, including metabolism, transport, and regulation. Moreover, such variability has been observed in the distribution of genes associated with virulence and antibiotic resistance across species [24].
The utilization of the pan-genome to construct a phylogenetic strain tree can provide significant insights into the evolutionary relationships among the organisms. The results obtained from the analysis of the strain tree can uncover discernible clades, which can be interpreted as distinct manifestations of genomic diversity and evolutionary history. The results of our study suggest that, for the most part, the taxonomic categorization of the organism aligns with its evolutionary connections, with only a limited number of notable exceptions. The observed gene count rank distribution predicted a maximum value of 80 genes, accompanied by an estimated length of 6000 base pairs. Fig. 4 illustrates the distribution of gene count rank, strain count rank, and the strain tree of genomes relevant to L. plantarum 12-3 [25].
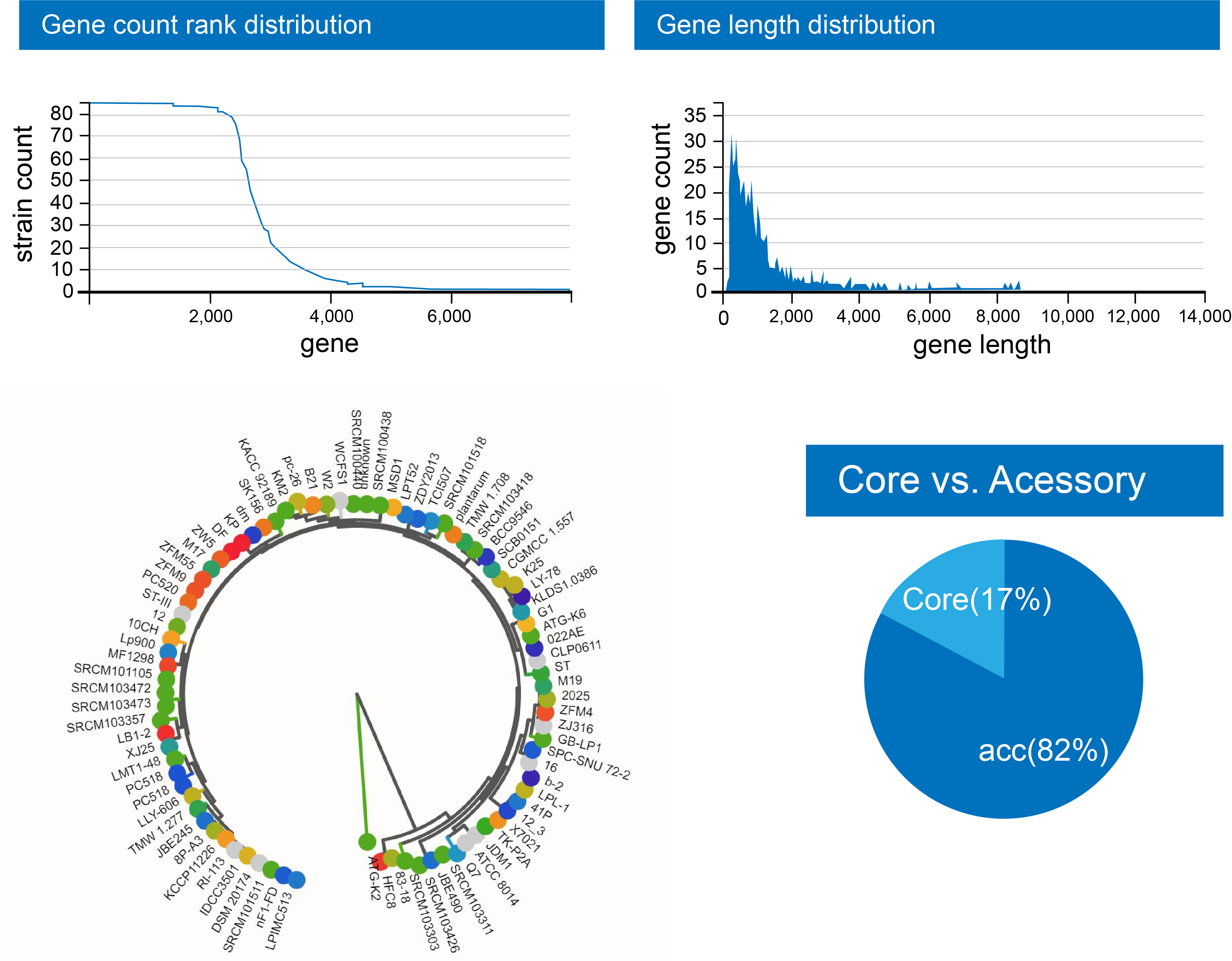 Fig. 4.
Fig. 4.The distribution of gene count rank, strain count rank, and the strain tree of genomes pertinent to L. plantarum 12-3.
Using the Comprehensive Antibiotic Resistance Database (CARD), a food safety assessment of the L. plantarum 12-3 genome was performed. Through CARD analysis, a thorough evaluation of the antibiotic resistance genes present in the L. plantarum 12-3 genome was conducted. The results indicated that the strain lacked known antibiotic resistance genes, indicating a favorable resistance profile. Only VanY and Ant-6-la genes were found, with a low criterion, in the genome that may suggest a risk of Vancomycin and STR resistance, respectively. The presence of antibiotic-resistance genes in probiotic strains can raise concerns regarding the potential transfer of resistance to pathogenic bacteria, making this finding of great importance in terms of food safety. By utilizing CARD, the study provided insightful information regarding the presence of two antibiotic resistance genes, thereby bolstering the safety of L. plantarum 12-3 as a potential probiotic candidate. These results provide, in part, an understanding of the strain’s potential safety and use in food applications such as a beneficial probiotic ingredient. Furthermore, there were no hits uncovered when VirulenceFinder 2.0 was used to categorize L. plantarum 12-3 as safe and non-pathogenic for humans [26]. Fig. 5 provides an antibiotic resistance wheel for L. plantarum 12-3 obtained from CARD analysis.
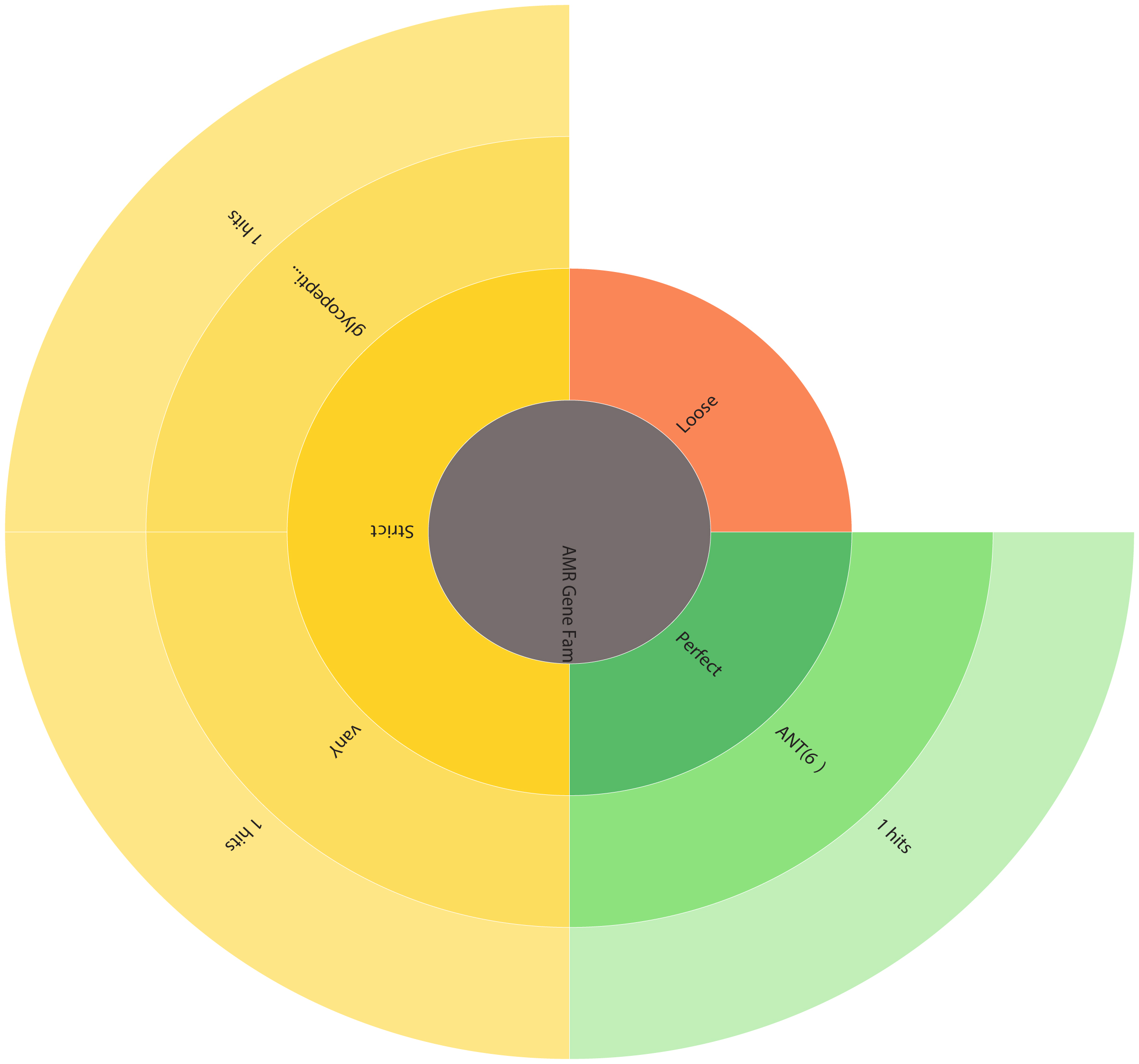 Fig. 5.
Fig. 5.Antibiotic resistance wheel depicting the antimicrobial resistance (AMR) gene families in L. plantarum 12-3. ARO, antibiotic resistance ontology.
Using the Phage Search Tool Enhanced Release (PHASTER), the identification and characterization of prophage regions within the L. plantarum 12-3 genome was performed. This potent instrument can accurately predict the length, localization, GC content, and gene annotation of prophage genomes, among other characteristics. To validate the predicted intact prophages, the identified regions were further compared to the Virus-Host database. This comparison confirmed the prophages uncovered and shed further light on their potential host range. A total 5 prophage regions were identified as outlined in Table 2. Furthermore, a circular genome map depicting the genomic regions containing prophage is provided in Fig. 6.
| Region | Region length | Completeness | Score | Proteins | Region position | Most common phage | GC% |
| 1 | 16.2 Kb | incomplete | 30 | 26 | 14366-30637 info_outline | PHAGE_Lactob_LfeSau_NC_029068 | 40.68% |
| 2 | 9.6 Kb | incomplete | 60 | 14 | 1296322-1306018 info_outline | PHAGE_Staphy_phiPV83_NC_00248 | 44.51% |
| 3 | 75.2 Kb | intact | 150 | 71 | 1529017-1604260 info_outline | PHAGE_Lactob_phig1e_NC_004305 | 41.99% |
| 4 | 40.3 Kb | intact | 150 | 55 | 1869418-1909799 info_outline | PHAGE_Lactob_Sha1_NC_019489 | 41.46% |
| 5 | 40.4 Kb | intact | 150 | 55 | 2671326-2711736 info_outline | PHAGE_Lactob_Sha1_NC_019489 | 40.67% |
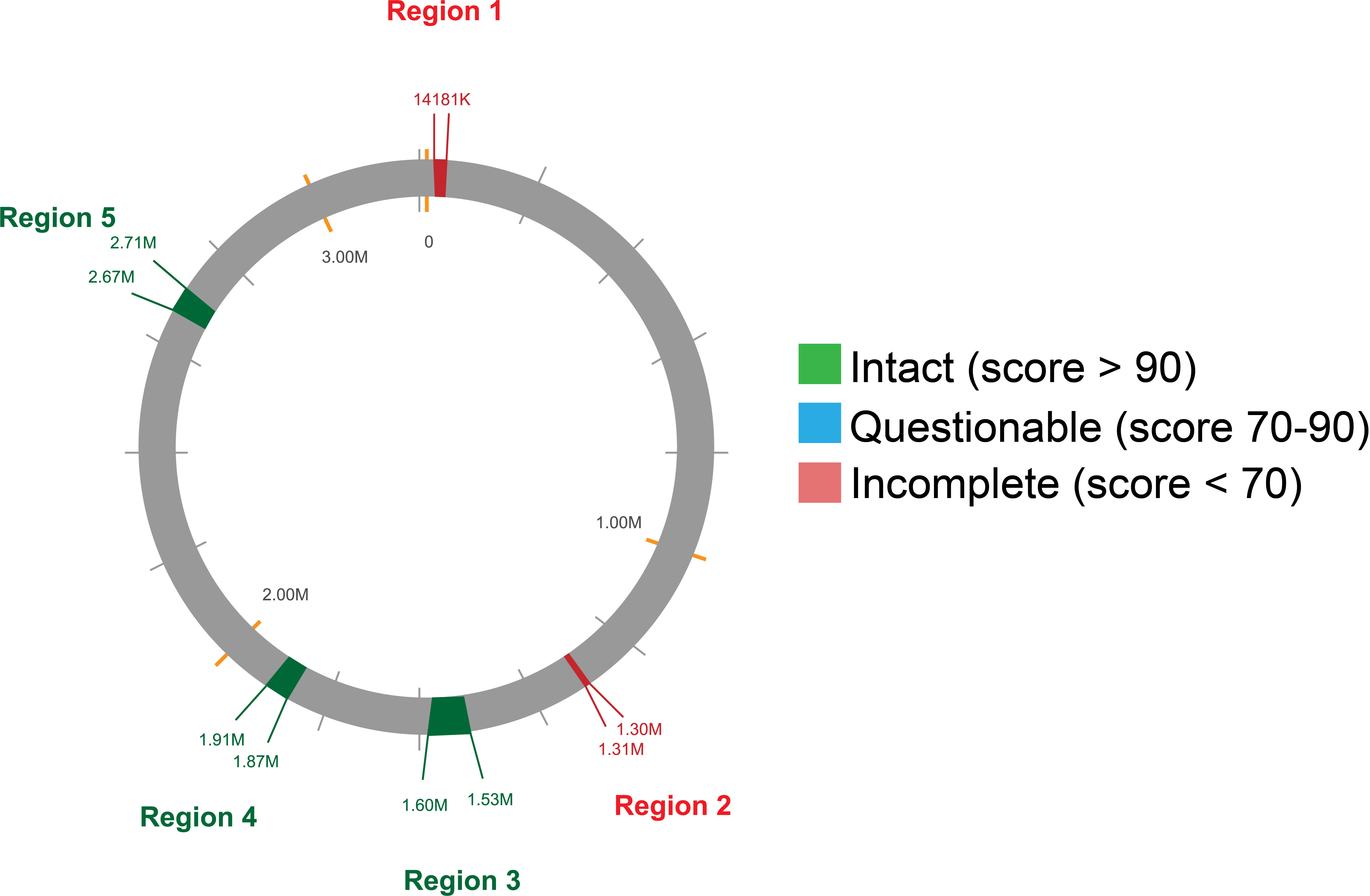 Fig. 6.
Fig. 6.Circular genome map highlighting the predicted prophage regions within the L. plantarum 12-3 genome.
Using the VIPtree tool (https://www.genome.jp/viptree), a proteomic tree of the viral genome sequence was also generated. This tree, which is based on genome-wide sequence similarities, visualized the evolutionary relationships between viral genomes. By using the PHASTER tool to perform database comparisons in conjunction with VIPtree, a comprehensive analysis of prophage regions was conducted. This analysis provided an enhanced understanding of the genetic elements present in the investigated genomes and their potential impact on bacterial evolution and host-pathogen interactions. The circular proteomic tree of the viral genome sequences is provided in Fig. 7.
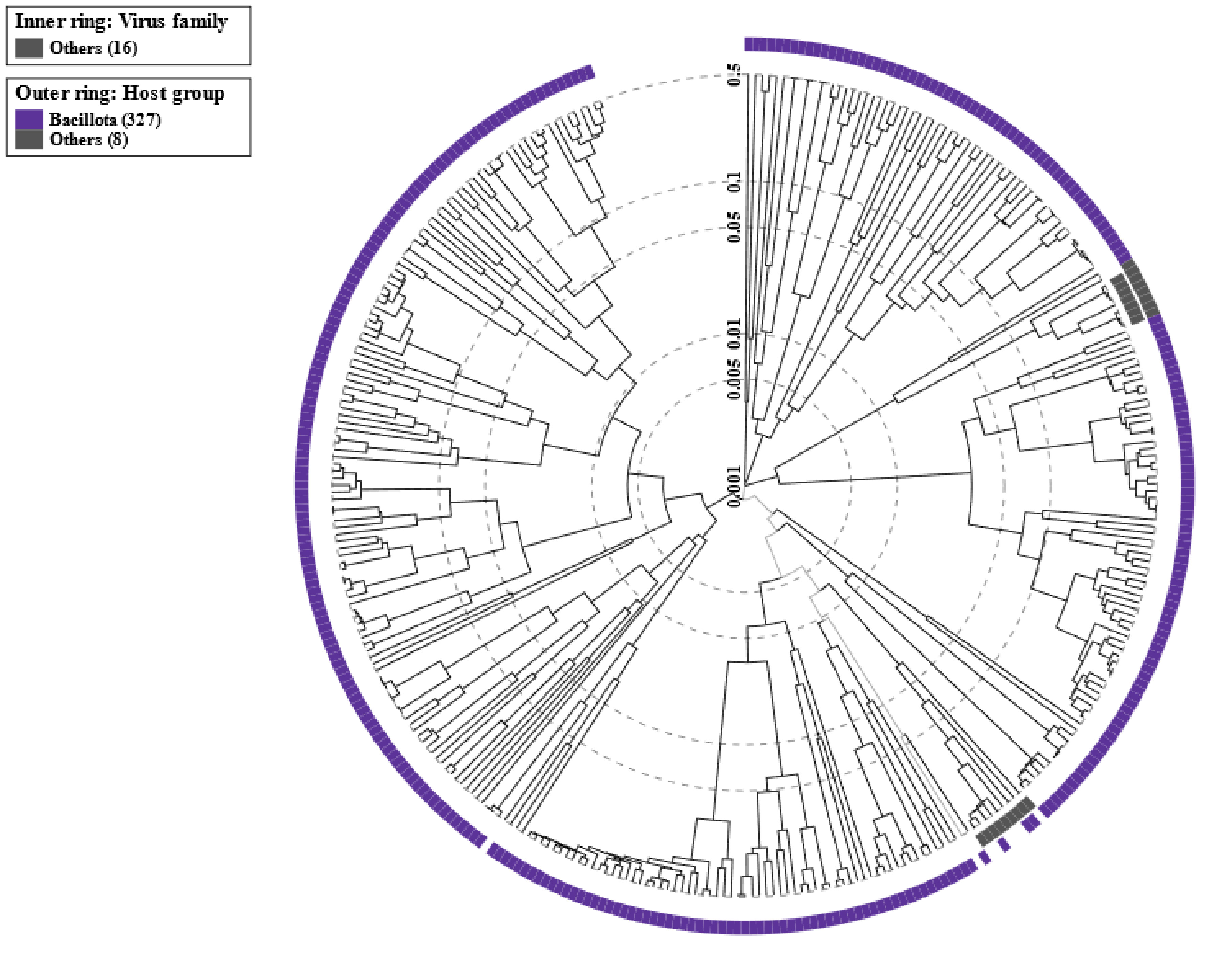 Fig. 7.
Fig. 7.Circular proteomic tree of viral genomes. The outermost ring outlines the host group of bacteriophages, while the innermost ring outlines the various phage families. The outermost ring provides a panoramic perspective of the bacteriophage host groups, highlighting the different habitats these viruses inhabit. Meanwhile, the innermost ring methodically categorizes the phages according to their viral family, providing a thorough understanding of the taxonomic relationships among these microbial predators.
Using the IslandViewer 4 tool, the presence of genomic islands (GI) within the L. plantarum 12-3 genome was investigated. To identify and characterize GI within the genome, multiple databases were queried. This analysis enabled the comprehensive identification of GIs and provided insight into their potential structural and functional functions. The results of this study advance our understanding of the genomic landscape of L. plantarum 12-3 and cast light on the potential role of GI in bacterial evolution, adaptation, and virulence [27]. A total of 18 GI were predicted by IslandViewer 4, and the various region sizes and GI prediction methods are provided in Table 3. Furthermore, a circular map, which depicts the GI within the L. plantarum 12-3 genome is given in Fig. 8.
| Start | End | Size | Gi Prediction Method |
| 351,551 | 393,860 | 42,309 | Predicted by at least one method |
| 389,368 | 393,860 | 4492 | Predicted by at least one method |
| 695,664 | 753,081 | 57,417 | Predicted by at least one method |
| 731,210 | 736,351 | 5141 | Predicted by at least one method |
| 1,691,750 | 1,749,038 | 57,288 | Predicted by at least one method |
| 1,749,032 | 1,777,308 | 28,276 | Predicted by at least one method |
| 2,170,145 | 2,178,397 | 8252 | Predicted by at least one method |
| 2,638,102 | 2,648,174 | 10,072 | Predicted by at least one method |
| 2,920,631 | 2,929,067 | 8436 | Predicted by at least one method |
| 1,749,032 | 1,777,308 | 28,276 | IslandPick |
| 389,368 | 393,860 | 4492 | SIGI-HMM |
| 731,210 | 736,351 | 5141 | SIGI-HMM |
| 2,170,145 | 2,178,397 | 8252 | SIGI-HMM |
| 351,551 | 393,860 | 42,309 | IslandPath-DIMOB |
| 695,664 | 753,081 | 57,417 | IslandPath-DIMOB |
| 1,691,750 | 1,749,038 | 57,288 | IslandPath-DIMOB |
| 2,638,102 | 2,648,174 | 10,072 | IslandPath-DIMOB |
| 2,920,631 | 2,929,067 | 8436 | IslandPath-DIMOB |
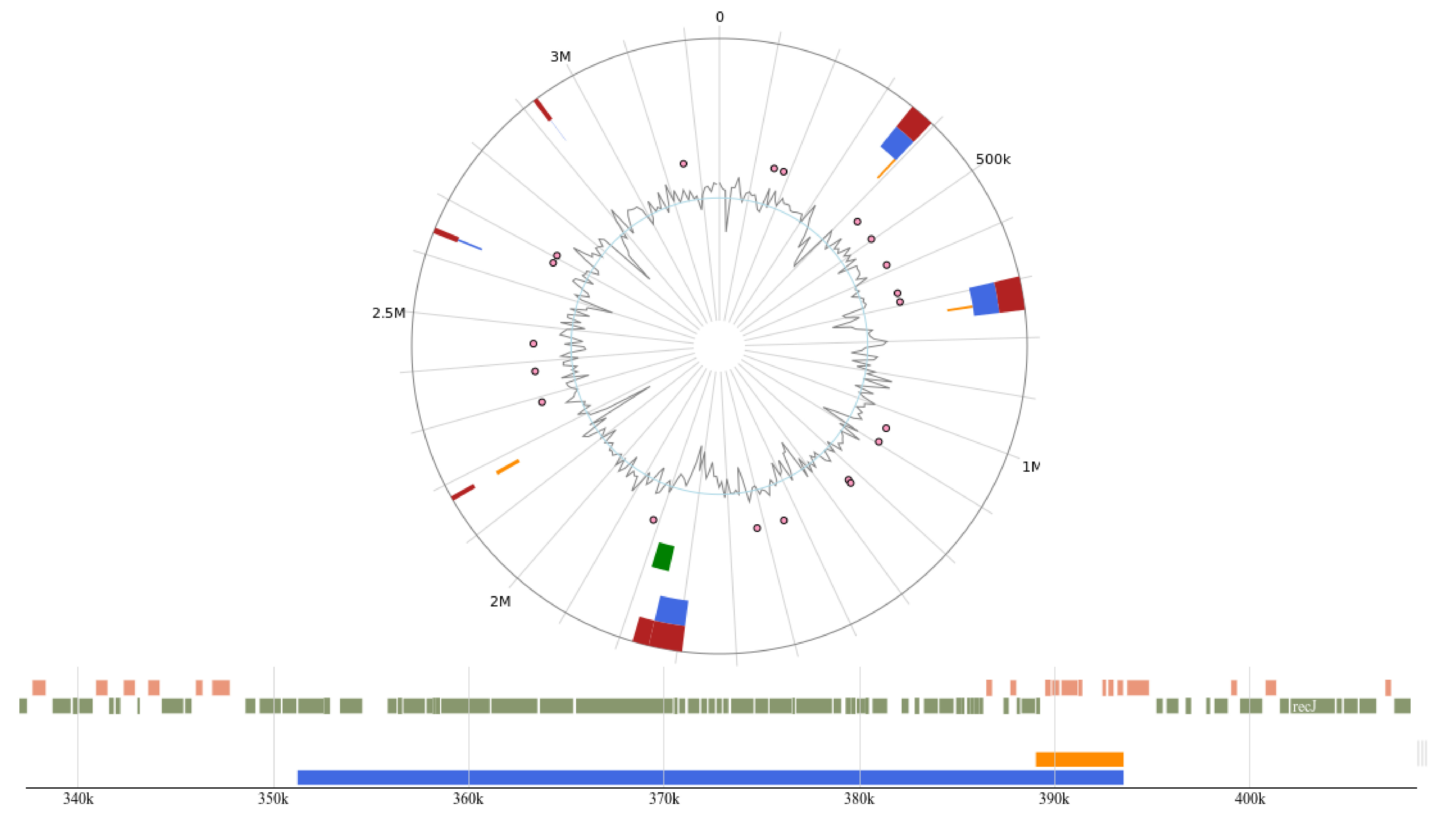 Fig. 8.
Fig. 8.Circular map of the genomic islands predicted using IslandViewer 4.
Transposases, the enzymatic catalyst of genetic transposition, and mobile elements are critical in orchestrating dynamic gene positioning within the genome. ISFinder and ISsaga are well-established bioinformatic resources designed to identify and annotate insertion sequences (IS) and other mobile genetic elements within a queried genome. Transposase genes and other mobile elements present within the L. plantarum 12-3 genome were accurately identified and annotated using these tools [28]. This annotation procedure yielded useful information regarding the diversity and distribution of transposase genes in the L. plantarum 12-3 genome. ISFinder and ISsaga facilitated a comprehensive analysis of mobile genetic elements and their potential influence on the evolution and genetic plasticity of L. plantarum 12-3. The predicted mobile elements are provided in Table 4.
| Sequences producing significant alignments | Is family | Group | Origin | Score (bits) | E. Value |
| ISP2 | IS1182 | Lactobacillus plantarum | 3418 | 0.0 | |
| ISP1 | ISL3 | Lactobacillus plantarum | 2833 | 0.0 | |
| ISLpl1 | IS30 | Lactobacillus plantarum | 1972 | 0.0 | |
| ISPp1 | IS30 | Pediococcus pentosaceus | 1853 | 0.0 | |
| ISLpl3 | IS5 | IS427 | Lactobacillus plantarum | 1651 | 0.0 |
| ISDha13 | IS200/IS605 | IS200 | Desulfitobacterium hafniense | 89.7 | 3 × 10 |
| ISSsu4 | IS200/IS605 | IS200 | Streptococcus suis | 71.9 | 7 × 10 |
| ISStin10 | IS200/IS605 | IS200 | Streptococcus iniae | 67.9 | 1 × 10 |
| ISLpl2 | IS3 | IS150 | Lactobacillus plantarum | 61.9 | 6 × 10 |
| ISLdl3 | IS30 | Lactobacillus delbrueckii | 61.9 | 6 × 10 | |
| ISPlu5 | IS200/IS605 | IS200 | Photorhabdus luminescens | 58.0 | 1 × 10 |
| ISEcl11 | IS30 | Enterobacter cloacae | 56.0 | 4 × 10 | |
| ISPlu7 | IS5 | IS427 | Photorhabdus luminescens | 52.0 | 0.006 |
| ISEnfa200 | IS200/IS605 | IS200 | Enterococcus faecium | 52.0 | 0.006 |
| ISWco1 | IS3 | IS150 | Weissella confusa | 50.1 | 0.024 |
| ISPye52 | IS3 | IS51 | Paracoccus yeei | 48.1 | 0.095 |
| ISSmi2 | IS1182 | Streptococcus mitis | 46.1 | 0.37 | |
| ISBce18 | IS3 | IS150 | Bacillus cereus | 46.1 | 0.37 |
| ISHar1 | IS3 | IS150 | Herminiimonas arsenicoxydans | 46.1 | 0.37 |
| ISSth6 | IS3 | IS150 | Streptococcus thermophilus | 46.1 | 0.37 |
| ISLpn1 | IS4 | IS10 | Legionella pneumophila | 46.1 | 0.37 |
| IS1648 | IS5 | IS427 | Streptomyces coelicolor | 46.1 | 0.37 |
| ISClta1 | IS1202 | IS1202 | Clostridium tagluense | 44.1 | 1.5 |
| ISLmo11 | IS3 | IS150 | Listeria monocytogenes | 44.1 | 1.5 |
| ISAlw31 | IS5 | IS427 | Acinetobacter lwoffii | 44.1 | 1.5 |
| ISSlu1 | IS30 | Streptococcus lutetiensis | 44.1 | 1.5 | |
| ISWsu1 | IS3 | IS407 | Wolinella succinogenes | 44.1 | 1.5 |
| IS1548 | ISAs1 | Streptococcus agalactiae | 44.1 | 1.5 | |
| ISGalb2 | IS1202 | ISTde1 | Gallionellaceae bacterium | 42.1 | 5.8 |
| ISSph16 | IS630 | Sphingopyxis sp. | 42.1 | 5.8 | |
| ISPsko1 | IS66 | Pseudomonas koreensis | 42.1 | 5.8 | |
| ISBli34 | IS3 | IS150 | Brevibacterium linens | 42.1 | 5.8 |
| ISMyr1 | IS1595 | ISSod11 | Myroides sp. | 42.1 | 5.8 |
| ISSbr1 | ISNCY | ISDol1 | Candidatus Scalindua | 42.1 | 5.8 |
| ISAsp6 | IS200/IS605 | IS605 | Anabaena sp. | 42.1 | 5.8 |
| ISHbo9 | IS200/IS605 | IS1341 | Halogeometricum borinquense | 42.1 | 5.8 |
| ISKpn26 | IS5 | IS5 | Klebsiella pneumoniae | 42.1 | 5.8 |
| ISCst1 | ISLre2 | Cylindrospermum stagnale | 42.1 | 5.8 | |
| ISRfsp3 | IS5 | ISL2 | Roseiflexus sp. | 42.1 | 5.8 |
| ISStma15 | IS3 | IS51 | Stenotrophomonas maltophilia | 42.1 | 5.8 |
| ISRel24 | IS66 | Rhizobium etli | 42.1 | 5.8 | |
| ISYen4 | IS1182 | Yersinia enterocolitica | 42.1 | 5.8 | |
| ISLsp4 | ISAzo13 | Leptospirillum sp. | 42.1 | 5.8 | |
| ISClbu1 | IS1182 | Clostridium butyricum | 42.1 | 5.8 | |
| ISCARN112 | IS1202 | ISTde1 | Metagenomic data | 42.1 | 5.8 |
| ISNha5 | IS1595 | ISNha5 | Nitrobacter hamburgensis | 42.1 | 5.8 |
| ISPto5 | ISAs1 | Psychroflexus torquis | 42.1 | 5.8 | |
| ISBthe4 | ISAs1 | Bacteroides thetaiotaomicron | 42.1 | 5.8 | |
| ISBaov2 | ISAs1 | Bacteroides ovatus | 42.1 | 5.8 | |
| ISvEsV1_1 | IS4 | IS4 | Ectocarpus siliculosus | 42.1 | 5.8 |
| ISFac2 | IS5 | IS903 | Ferroplasma acidarmanus | 42.1 | 5.8 |
| ISC1173a | IS1 | Sulfolobus solfataricus | 42.1 | 5.8 | |
| IS1419 | IS481 | Burkholderia glumae | 42.1 | 5.8 | |
| IS1070 | IS30 | Leuconostoc lactis | 42.1 | 5.8 |
Using the specialized bioinformatics tool CRISPRCasFinder v.1.1.2 (https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index), the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated genes (Cas) were identified and validated within the L. plantarum 12-3 genome. Furthermore, the presence and arrangement of CRISPR systems within the examined genome were meticulously identified and characterized [29]. Our analysis revealed 1 spacer sequence with a Conservation DR of 97.30% and a Conservation Spacer of 100%. This analysis provided insight into the molecular machinery that underlies this bacterial defense mechanism in L. plantarum 12-3. Fig. 9 outlines the spacer sequence uncovered by this analysis.
 Fig. 9.
Fig. 9.The identified CRISPR-Cas sequences in the genome of L. plantarum 12-3.
The study of the L. plantarum 12-3 genome demonstrates its potential as a probiotic strain for a variety of applications. Our genomic analysis revealed a wide variety of genes and functional elements, indicating the potential functional properties of the organism. The strain exhibited favorable probiotic properties, such as resistance to hostile environments, antimicrobial properties, epithelial cell adhesion, and immune modulation abilities [11]. The investigation of the CRISPR-Cas system disclosed the presence and organization of these essential genetic elements involved in bacterial defense mechanisms, indicating its potential for applications in genome engineering. These findings contribute to our understanding of the genetic diversity, genotype, and functional insight into L. plantarum 12-3, and amply support its potential as a safe and beneficial probiotic candidate for future use [30, 31]. These findings underscore promising attributes to L. plantarum 12-3 for probiotic function, and suggest its potential use in a variety of safety applications and, thereby, pave the way for future probiotics research. L. plantarum 12-3’s potential ranges from improving digestive health to helping produce novel functional foods and medicinal formulations.
The identification of L. plantarum 12-3 genetic diversity and favorable probiotic properties provides a foundation for further investigation into its potential therapeutic and preventative applications for human health. The strain’s resistance to severe conditions and antimicrobial properties bode well for its application in the development of probiotic formulations for specific health conditions [32]. Our findings clearly contribute to the expanding body of knowledge regarding probiotics, as well as paving the way for future research and applications in the field of probiotic functionality and safety, ultimately benefiting the health and well-being of humans [33].
It is very important to discuss the limitations of this study. First, our analysis is limited to the genomic data, and additional experimental validation is required to corroborate L. plantarum 12-3 functional characteristics and probiotic potential. Second, although bioinformatics tools were used to identify and annotate genetic elements, the accuracy and completeness of the annotations is dependent on the accuracy of databases and algorithms used [34]. Third, our investigation is restricted to a single strain of L. plantarum 12-3; thus, generalizations to other strains or species should be made with caution. Future studies using larger sample sizes, in vitro and in vivo models, and comparative analyses with other closely related strains may provide a more comprehensive understanding of the probiotic functionality, safety, and potential applications of L. plantarum 12-3 [35].
Based on the findings and limitations of this study concerning the genome of L. plantarum 12-3, several recommendations for future research can be made. As we have demonstrated in our previous studies, L. plantarum 12-3 can convert linoleic acid to various fatty acid metabolites and is also capable of producing exopolysaccharides [9]. Additional experimental evidence is required to validate the functional characteristics and probiotic potential of this L. plantarum 12-3 strain, including in vitro and in vivo evaluation with relevant model systems. In addition, broadening the scope of the study to include a variety of L. plantarum 12-3 strains and a comparative analysis with other probiotic species would improve our understanding of genetic diversity and functional properties [36, 37, 38, 39, 40, 41]. A closer investigation of the CRISPR-Cas system in L. plantarum 12-3, and its application in genome engineering, may result in the development of novel probiotic strains with improved functionalities. Additionally, comprehensive safety evaluations, including evaluation of potential allergenicity and long-term effects, are necessary to ensure the safe use of L. plantarum 12-3 and other probiotic strains. These recommendations are intended to advance our understanding of probiotics and maximize their potential health benefits for humans.
This investigation has shed light on the genetic variability, CRISPR-Cas mechanism, genotype, and functional implications of the genome of L. plantarum 12-3, thereby highlighting its potential for utilization as a probiotic agent. Our comprehensive genomic analysis unveiled a diverse array of genes and functional elements within the genome, suggesting its potential functional capabilities. This L. plantarum 12-3 strain exhibited several advantageous probiotic characteristics, including resilience to adverse surroundings, antimicrobial capability, adhesion to epithelial cells, and the ability to modulate the immune system. Additional research focused on understanding the functional characteristics and safety considerations of L. plantarum 12-3 through in vitro and in vivo investigations for its probiotic functionality and various bio-functional properties is warranted. Such studies may involve analysis of its interactions with the host microbiome, assessing its probiotic properties, and conducting a thorough safety assessment. Furthermore, a continuous exploration of the genome can provide insights into the adaptive evolution and genetic flexibility of L. plantarum 12-3 species, thereby enhancing our understanding of their potential applications as probiotic-based therapies and food safety measures.
All the data generated in this study will be available on demand from the corresponding author.
Conceptualization, TA, MN, MAS, AS, AAK, and ZY; methodology, TA, MN, MAS, AS, AH, and ZY; software, MA; validation, MA; formal analysis, TA; investigation, JMR and AZ; resources, ZY; MA and AH; data curation, TUH; writing—original draft preparation, AZ and MN; writing—review and editing, AH and MN; visualization, AH; supervision, ZY; project administration, AAS and MA; funding acquisition, ZY. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The authors extend their appreciation to the Deputyship for Research and Innovation, “Ministry of Education” in Saudi Arabia for funding this research (IFKSUOR3- 619-3).
This research work was financially supported by National Natural Science Foundation of China (Project No. 32272296) and the Deputyship for Research and Innovation, “Ministry of Education” in Saudi Arabia (IFKSUOR3- 619-3).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.