




 , Xingyuan Chen 1, Mei Zheng 1, Jingzhe Zhu 1, Hui Mao 2,*
, Xingyuan Chen 1, Mei Zheng 1, Jingzhe Zhu 1, Hui Mao 2,*1 Hubei Key Laboratory of Renal Disease Occurrence and Intervention, Medical School, Hubei Polytechnic University, 435003 Huangshi, Hubei, China
2 Department of Molecular Mechanisms Research and Treatment of Skin Tumors, Huangshi Central Hospital, 435000 Huangshi, Hubei, China
Abstract
Coenzyme A (CoA) functions as a crucial carrier of acyl groups within cells, playing a fundamental role in regulating acyl transfer reactions and participating in cellular metabolic processes. As the principal substrate and cofactor engaged in diverse metabolic reactions, CoA and its derivatives exert central influence over various physiological processes, primarily modulating lipid and ketone metabolism, as well as protein modification. This paper presents a comprehensive review of the molecular mechanisms by which CoA influences the onset and progression of cancer, cardiovascular disease (CVD), neurodegenerative disorders, and other illnesses. The main focal points include the following. (1) In cancer, enzymes such as acetyl-CoA synthetase 2, ATP citrate lyase, and acetyl-CoA carboxylase regulate lipid synthesis and energy metabolism by modulating acetyl-CoA levels. (2) In CVD, the effects of enzymes such as stearoyl-CoA desaturase-1, 3-hydroxy-3-methylglutaryl-CoA (HMGC) synthase 2, and HMGC reductase on the formation and advancement of these diseases are elucidated by their regulation of CoA metabolism across multiple organs. (3) In neurodegenerative disorders, the significance of CoA in maintaining cholesterol homeostasis in the brain and its implications on the development of such disorders are thoroughly discussed. The metabolic processes involving CoA and its derivatives span all physiological aspects within cells, playing a critical role in the onset and progression of various diseases. Elucidating the role of CoA in these conditions yields important insights that can serve as valuable references and guidance for disease diagnosis, treatment, and drug development.
Keywords
- cancer
- cardiovascular disease
- coenzyme A
- lipid metabolism
- neurodegenerative disorders
Coenzyme A (CoA) plays a pivotal role in cellular processes and is synthesized from L-Cysteine, ATP, and vitamin B5 (also known as pantothenic acid) [1]. Its primary function is to facilitate the formation of acyl-CoA compounds through thioester binding with various acyl groups including acetyl, propionyl, succinyl, and 3-hydroxy-3-methylglutaryl (HMG) [2]. These acyl-CoA derivatives serve as essential participants in diverse metabolic pathways within the human body. The metabolism of CoA and its thioester derivatives is intricately linked to a spectrum of diseases [2].
Acetyl-CoA serves as a pivotal intermediary in carbon metabolism, playing a dual role by supporting the tricarboxylic acid (TCA) cycle for ATP generation and facilitating the synthesis of fatty acids (FAs). Moreover, it serves as a crucial substrate for lysine acetylation reactions within cells, which are indispensable for cell growth and survival [3]. Enzymes such as ATP citrate lyase (ACLY), pyruvate dehydrogenase complex (PDC), and acyl-CoA short chain synthetase (ACSS) are responsible for the conversion of citrate, pyruvate, and acetate into acetyl-CoA, with these processes occurring in the cytoplasm and mitochondria [4].
ACLY, which is localized in both the cytoplasm and nucleus, is responsible for converting citrate into acetyl-CoA in the cytoplasm [5]. Another significant contributor to acetyl-CoA in both the nucleus and cytoplasm is ACSS2, which synthesizes acetyl-CoA from acetate [6]. In mammalian cells, ACSS2 is implicated in cytoplasmic lipid synthesis, whereas ACSS1 facilitates ATP production in the mitochondria [7]. ACLY plays a critical role in glucose-dependent fatty acids (FA) synthesis and histone acetylation. The upregulation of ACSS2 and conversion of acetate are the primary compensatory mechanisms in response to ACLY defects. Interestingly, despite the elevated acetyl-CoA levels and increased ACSS2 expression, defects in ACLY lead to reduced acetylation levels of total histone proteins, impaired proliferation, and altered gene expression patterns [8].
The rate-limiting enzyme governing long-chain FA synthesis and mitochondrial FA
oxidation (FAO) is acetyl-CoA carboxylase (ACC) [9]. ACC1, predominantly
localized in the cytoplasm, generates malonyl-CoA, a key substrate for FA
biosynthesis, and acts as an allosteric inhibitor of carnitine
palmitoyl-transferase, a key mediator in transporting FAs into the mitochondria
for
Serving as a carrier molecule, CoA actively engages in transferring acyl groups across various metabolic pathways, playing pivotal roles in processes such as the TCA cycle, cholesterol synthesis, and protein modification [12, 13, 14]. The crucial involvement of CoA in cellular metabolism tightly correlates with the onset and progression of cancer [15]. Understanding the intricate interplay between CoA and cancer is paramount, potentially paving the way for novel research avenues, personalized diagnostic tools, and therapeutic approaches in cancer treatment [16, 17, 18]. Accumulating evidence indicates that disturbances in CoA metabolism contribute to the genesis and exacerbation of cardiovascular diseases (CVDs), impacting vital pathways such as the TCA cycle, lipid metabolism, and ketone body utilization [19, 20, 21]. Moreover, neuronal genetic defects in CoA metabolism detrimentally affect cellular energy generation and oxidative stress, leading to neuronal dysfunction and neurodegeneration [22, 23]. Diseases such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease, which frequently show disruptions in cholesterol metabolism, energy pathways, and mitochondrial function, are intricately interconnected with CoA-dependent processes [24, 25, 26, 27].
CoA and its derivatives play pivotal roles in multiple diseases. Further exploration of their functions could pave the way for innovative therapeutic interventions and strategies.
Dysregulation of metabolic processes involving diverse substances contributes to the initiation and progression of cancer [28]. While normal cells predominantly rely on mitochondrial oxidative phosphorylation for energy production, cancer cells exhibit a preference for aerobic glycolysis, a phenomenon crucial in cancer cell proliferation, commonly known as the “Warburg effect” [29, 30].
The metabolism of acetyl-CoA metabolism is governed by three enzymes, ACSS2, ACLY, and ACC, all of which exert an impact on lipid metabolism [20, 31]. These enzymes are significantly overexpressed in various cancers. ACSS2 and ACLY enhance acetyl-CoA production, while ACC improves the utilization rate of acetyl-CoA, thereby promoting cell lipid metabolism, the TCA cycle, and histone acetylation. These processes collectively facilitate tumor growth (Fig. 1) [14].
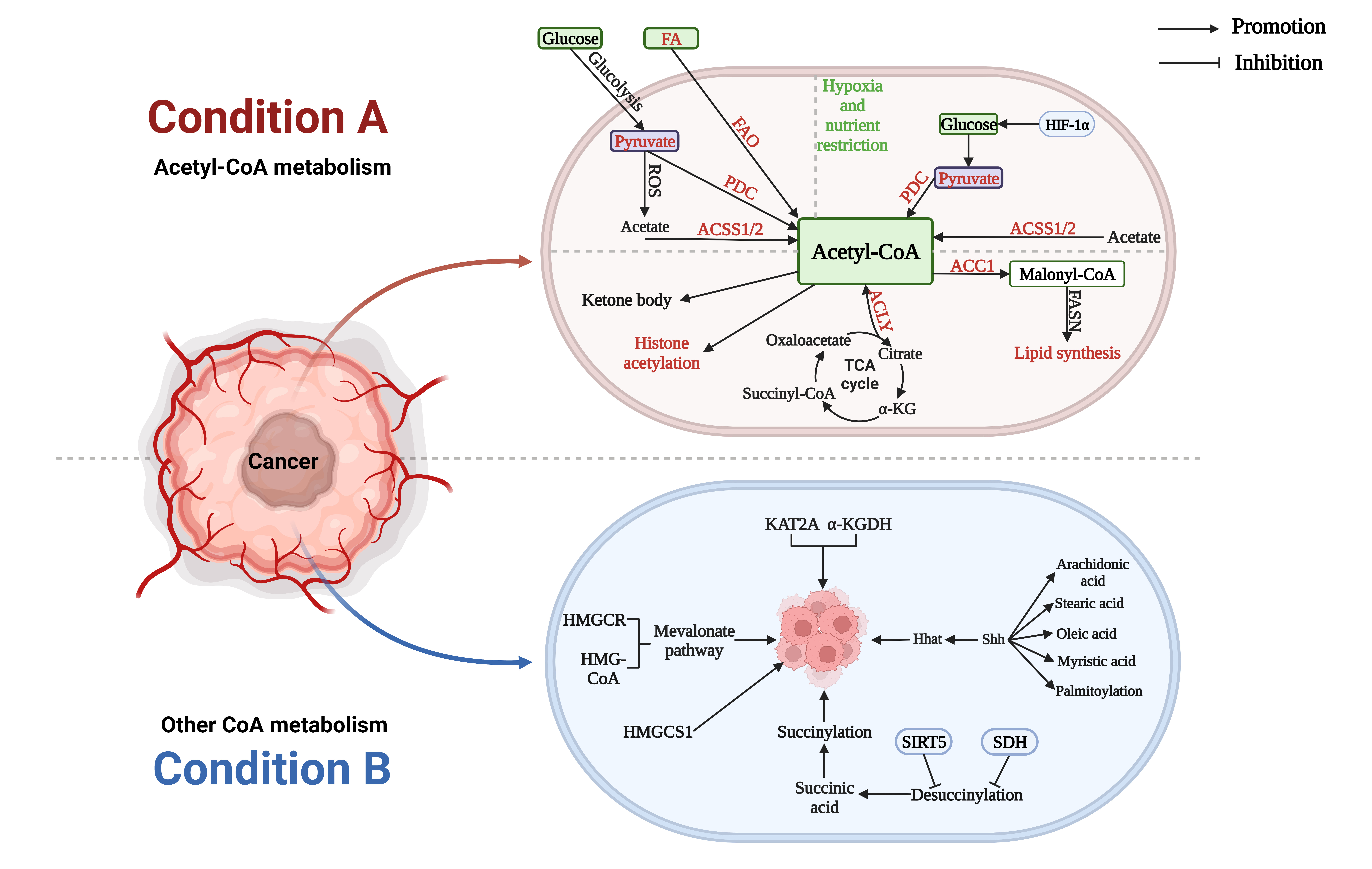 Fig. 1.
Fig. 1.Coenzyme A (CoA) and cancer. Condition A delineates acetyl-CoA metabolism in cancer, while condition B describes other CoA metabolism types in cancer. The solid arrow denotes the promotion process, whereas the blunt arrow signifies inhibition. The metabolism of acetyl-CoA in cancer is intricately regulated by acyl-CoA short chain synthetase (ACSS), ATP citrate lyase (ACLY), and acetyl-CoA carboxylase (ACC). These enzymes exert a profound influence on the metabolism of fatty acids (FAs), pyruvate, and other lipids, as well as the tricarboxylic acid (TCA) cycle and histone acetylation—critical processes for cancer cell proliferation and survival. Under conditions of hypoxia and nutrient restriction, cancer cells release hypoxia inducible factor (HIF), a pivotal regulator of acetyl-CoA metabolism. Hypoxia inducible factor acts to foster the growth of cancer cells by modulating acetyl-CoA utilization. Additionally, Hedgehog acyltransferase (Hhat), succinyl-CoA, and HMG-CoA play crucial roles in regulating other CoA metabolism pathways in cancer cells. Notably, HMG-CoA serves as a precursor for mevalonate synthesis through 3-hydroxy-3-methylglutaryl (HMG) reductase. The mevalonate pathway catalytically influences multiple cancer pathways, underscoring the significance of CoA metabolism in cancer development and progression. ROS, Reactive Oxygen Species.
ACSS2 is integral to a diverse array of cellular physiological processes. Initially, it catalyzes the synthesis of acetyl-CoA. Then it exerts regulatory control over signal transduction pathways. Lastly, within the nucleus, ACSS2 participates in the recycling of acetate generated by lysine deacetylase (Kdac) activity, thereby contributing to the energy supply essential for cellular metabolism [32, 33]. Comparative metabolomics and lipidomics studies have revealed that cancer cells utilize acetate as a nutritional source in an ACSS2-dependent manner. Under conditions of low oxygen and lipid depletion, ACSS2 exhibits the capacity to catalyze the reverse reaction from acetyl-CoA to acetate in tumor cells. This suggests that ACSS2 functions as a bidirectional enzyme in cancer cells, potentially playing a buffering role in the metabolism of acetyl-CoA/acetate in cancer [34, 35, 36]. Numerous studies have identified increased expression levels of ACSS2 across various cancer types, contributing to the maintenance of histone acetylation modifications [37, 38]. Conversely, research has demonstrated that diminishing ACSS2-mediated autophagy may promote breast cancer growth [39]. In colorectal cancer, thymine–guanine-interacting factor inhibits ACSS2, resulting in reduced acetyl-CoA production. However, cancer cells exhibit continued growth through the reprogramming of glucose transporter 1 (GLUT1) [40, 41].
In obesity-associated myeloma, elevated ACSS2 expression shows a positive correlation with increased body mass index. ACSS2 promotes the development of obesity-induced myeloma by stabilizing interferon regulatory factor 4. Given its substantial role in cell metabolism and regulation of gene expression, targeting the inhibition of ACSS2 expression may represent a promising strategy for cancer treatment [42].
ACLY, a multifunctional enzyme, not only participates in the synthesis of acetyl-CoA but also plays a pivotal role in cell signal transduction by modulating histone acetylation [43]. Functioning as a target for serine/threonine-protein kinase (AKT), ACLY contributes to the conversion of citric acid to acetyl-CoA, establishing a crucial link between glycolysis and lipid metabolism processes [44]. The AKT/ACLY signaling pathway governs histone acetylation in cancer cells, and inhibiting ACLY results in citric acid accumulation, suppression of phosphofructokinase 1 (PFK1) and PFK2 activities, thereby reducing glycolysis and inducing apoptosis in cancer cells [45, 46]. In the context of cancer-related gene regulation, nutritional restriction, and AKT metabolic recombination, the upregulation of AKT1 leads to increased phosphorylation and activation of ACLY, consequently augmenting or sustaining acetyl-CoA production [47]. Sirtuin 6 (SIRT6), on the other hand, diminishes nuclear ACLY activity and acetyl-CoA levels, impeding the histone acetylation process and inhibiting tumor cell invasion [48]. However, within the apoptosis-inducing family, caspase-10 cleaves ACLY, diminishing its activity and reducing nuclear acetyl-CoA and lipid levels, and histone acetylation, thereby preventing cancer onset [49]. The overexpression of ACLY corresponds to increased and progressive lipid synthesis in various cancers [50, 51, 52].
In colorectal cancer, cancer-derived exosomal HSPC111 phosphorylates ACLY, promoting cancer-associated fibroblast-mediated lipid metabolism and elevating acetyl-CoA levels, thereby enhancing liver cancer cell metastasis [53]. In pancreatic cancer, the acetyl-CoA generated by ACLY is utilized for histone acetylation and mevalonate metabolism, fostering the development of pancreatic ductal adenocarcinoma (PDAC) [16]. Studies have demonstrated that the upregulation of ACLY is associated with unfavorable outcomes in gastric cancer, highlighting the feasibility of targeting ACLY for treatment. ACSS2 is believed to function cooperatively with ACLY, suggesting that simultaneous inhibition of ACLY and ACSS2 could potentially hinder the progression and treatment of gastric cancer [54]. Given its multifaceted roles in cell metabolism, signaling, and gene regulation, downregulating ACLY expression offers a promising strategy to mitigate cancer risk across various cell types [55].
ACC1 enzymatically converts acetyl-CoA to malonyl-CoA, contributing to FA synthesis and regulating FAO, thereby orchestrating glucose and fat metabolism [56, 57]. ACC1 demonstrates elevated expression across numerous cancer types [58]. In breast cancer, phosphorylation of ACC1 leads to decreased lipid synthesis and elevated acetyl-CoA levels. This triggers increased histone H3 acetylation, activating SMAD2-mediated acetylation, and promoting the epithelial-mesenchymal transition (EMT), ultimately promoting cancer cell metastasis [59]. In prostate cancer, valproic acid inhibits ACC1 expression through the CCAAT enhancer-binding protein alpha and sterol regulatory element-binding protein 1 (SREBP-1) pathways, effectively curbing lipid accumulation and inducing apoptosis [60, 61]. In liver cancer, inhibition of ACC1 or the use of ND-654 inhibitors effectively suppresses de novo lipogenesis (DNL), thereby impeding cancer cell growth [62]. Additionally, in pancreatic cancer, eicosapentaenoic acid reduces ACC1 expression and ACC1-mediated DNL, leading to the inhibition of cell proliferation and survival [63]. These significant findings strongly underscore the crucial role of ACC1 in maintaining cellular energy balance and overall metabolic regulation. Targeting ACC1 expression levels can effectively reduce the proliferation of various cancer cell types.
Histone acetylation constitutes an intricate biochemical process with a profound influence on cellular function and gene expression regulation. First, it initiates structural alterations in chromatin, facilitating a more open configuration. This modification enhances DNA’s affinity for transcription factors, thereby elevating gene transcriptional activity and increasing gene expression [64]. Second, histone acetylation plays a pivotal role in DNA repair and replication processes, contributing to the preservation of genomic integrity. This is achieved by recruiting specific proteins that collaborate with others, ensuring their effective functioning across various stages of the cell cycle [65].
A positive correlation has been established between the levels of acetyl-CoA and histone modification [66]. Acetyl-CoA synthesis is enhanced in the nucleus of cancer cells, where ACSS2 and ACLY facilitate the conversion of acetate and citric acid into acetyl-CoA, thereby promoting the process of histone acetylation [67]. The PDC elevates the levels of acetyl-CoA in the nucleus, resulting in increased acetylation of histones H2B, H3, and H4 [68]. In prostate cancer, the mitochondrial PDC increases citric acid levels for fat synthesis, while the nucleolar PDC enhances acetyl-CoA levels, thus promoting the acetylation of histone H3K9. This leads to increased expression of ACLY and lipid synthesis, maintaining the progression of prostate cancer [69].
Histone hyperacetylation emerges as a contributing factor to DNA damage,
demonstrating its potential as a catalyst for liver cancer initiation. Within the
liver cancer milieu, steatosis induces chromatin relaxation, enhances the DNA
damage marker
Moreover, in PDAC, escalated acetyl-CoA levels drive increased histone acetylation, thereby promoting cancer development [74]. In acute myeloid leukemia, AMP-activated protein kinase (AMPK) plays a pivotal role in stabilizing acetyl-CoA levels, ensuring the maintenance of histone acetylation, facilitating cell proliferation, and recruiting bromodomain containing 4 onto chromatin [75]. In a rat model of colon cancer, the intricate interplay between dietary fat and cellulose elevates the acetylation level of histone H3, induces the expression of genes related to FAO, and promotes tumor development [76]. These multifaceted functions underscore the wide-reaching and pivotal role of histone acetylation in the realms of cell biology and molecular biology.
FAs serve diverse physiological functions. First, they act as crucial energy storage molecules within living organisms, adopting the form of triacylglycerol in adipose tissue. This reservoir is subsequently catabolized to provide energy as needed [77]. Second, FAs actively participate in intracellular signal transduction and the synthesis of second messengers, significantly contributing to the regulation of cellular metabolism [78].
FAs play a pivotal role in tumor cells, contributing to cell membrane
biosynthesis and serving as a source of metabolic energy under stress conditions
[79]. FAs are exclusively utilized through their combination with CoA to form
acyl-CoA through an activation step [54]. Subsequently, FAs undergo
In breast cancer, the upregulation of FAO leads to elevated acetyl-CoA levels, consequently promoting the acetylation of histone H3 and H3K27ac. This facilitates the expression of genes associated with the EMT, a critical process in cancer progression [82]. In glioblastoma (GBM), the downregulation of diacylglycerol O-acyltransferase 1 expression results in reduced lipid storage, enhanced FAO of excess FAs, and subsequent accumulation of acetyl-CoA. This cascade of events influences the level of reactive oxygen species and induces cell apoptosis, ultimately inhibiting GBM growth [83].
Pyruvate intricately participates in various physiological processes in living organisms. First, it functions as an energy storage molecule by participating in TCA cycle within the mitochondria, thereby supplying energy to the cell. Second, pyruvate serves as an intermediate product in the TCA cycle, which is capable of further metabolism to generate essential organic compounds including FAs and cholesterol. These dual roles underscore the pivotal contribution of pyruvate in sustaining cellular energy metabolism and facilitating organic synthesis [84].
The PDC is located in the mitochondria and nuclei, facilitating the connection between glucose metabolism and the TCA cycle for the synthesis of acetyl-CoA using pyruvate as a substrate [85]. Acetyl-CoA holds significant importance in cancer metabolism [86, 87]. The PDC activity is regulated by pyruvate dehydrogenase kinase (PDK) and acetyl-CoA acetyltransferase 1 (ACAT1). Phosphorylation of PDK or acetylation of ACAT1 leads to PDC inhibition, thereby enhancing the Warburg effect and promoting cancer growth [29]. Consequently, only a limited amount of pyruvate enters the TCA cycle, resulting in reduced production of acetyl-CoA within the mitochondria. Moreover, the upregulation of PDK is closely associated with a poor prognosis in cancer [88, 89, 90]. Therefore, pyruvate assumes a distinctive and crucial role in linking aerobic and anaerobic metabolism, serving as a central determinant for sustaining cancer growth [91].
Hypoxia plays a crucial role in influencing acetyl-CoA metabolism in cancer,
contributing to the survival and proliferation of specific cancer cells while
activating hypoxia inducible factor (HIF) [92]. The upregulation of
HIF-1
Acyl-CoA constitutes a category of metabolic intermediates characterized by acyl groups forming thioester bonds with CoA. These molecules play a pivotal role in facilitating acyl transfer within various metabolic processes [104].
Hedgehog acetyltransferase (Hhat) has the capability to attach non-palmitate FAs to a recombinant protein known as sonic hedgehog (Shh), a crucial gene within the Hhat signaling pathway. It has been suggested that heterogenous fatty acylation may impact Shh signaling during development and in cancer cells [105]. Aberrant re-expression and signaling of Shh have been linked to cancer. In addition to palmitic acid, the N-terminal segment of Shh features binding sites for myristate acid, palmitoleic acid, stearic acid, oleic acid, and arachidonic acid [106, 107, 108].
Succinylation plays a significant role in cancer and inflammation. In instances
where SIRT5 is absent or succinic dehydrogenase malfunctions, the succinylation
process within the mitochondria is disrupted, resulting in succinic acid
accumulation. This, in turn, triggers an overabundance of histone succinylation,
disrupting the metabolic equilibrium and elevating the cancer risk [109]. The
enzyme alpha-ketoglutarate dehydrogenase (
Mevalonic acid (MVA) is synthesized from HMG-CoA through the action of HMG-CoA
reductase (HMGCR). The MVA pathway demonstrates increased expression in leukemia,
lymphoma, multiple myeloma, breast cancer, liver cancer, pancreatic cancer,
esophageal cancer, and prostate cancer [114]. Consequently, inhibiting MVA levels
holds promise for potentially impeding cancer growth. Statins, acting as
inhibitors of HMGCR, intervene in the MVA pathway, inhibiting the synthesis of
cholesterol, geranyl pyrophosphate, and farnesyl pyrophosphate. Early clinical
studies have underscored the therapeutic potential of statins in treating acute
myeloid leukemia [115]. In PDAC cells, there is activation of ketone body
metabolism, with
CoA and its thioester derivatives play a significant role in CVDs, exerting regulatory control over processes such as FAO, ketogenic reactions, and cholesterol synthesis [117]. Globally, CVD stands out as the foremost and deadliest health concern [118].
The key enzyme, HMG-CoA holds paramount significance in the regulation of CVDs. HMGCS2 and HMGCR, in conjunction with acetyl-CoA, stearoyl-CoA desaturase (SCD1), and fatty acyl-CoA synthetase, establish critical connections to CVDs across various dimensions. The expression levels of these enzymes can differ across different tissues of the human body [119]. The relationship between CVD and CoA metabolism can be delineated across three principal aspects: the liver, kidney, and the heart and cardiovascular system.
The liver and kidneys are vital organs involved in human metabolism and detoxification, exerting an influence on cardiovascular health through their metabolic and regulatory functions [120]. Impairment of liver function can lead to cholesterol accumulation within the liver, contributing to cardiovascular ailments like hypercholesterolemia and atherosclerosis. Anomalous renal function or kidney disorders, including chronic kidney disease and kidney failure, exhibit a close association with CVD [121]. Moreover, kidney ailments have the potential to instigate or exacerbate hypertension, thereby heightening the susceptibility to atherosclerosis and CVDs [122].
The synthesis of ketone bodies intricately intersects with the regulatory
mechanisms of CVDs, mediated by CoA [123]. HMGCS2, a rate-limiting enzyme located
in the mitochondria, orchestrates the synthesis of ketone bodies by facilitating
the production of HMG-CoA [124]. This enzyme is predominantly expressed in the
mitochondrial matrix of the liver, actively participating in ketogenesis, and
supplying lipid-derived energy during fasting [125, 126]. In mammals,
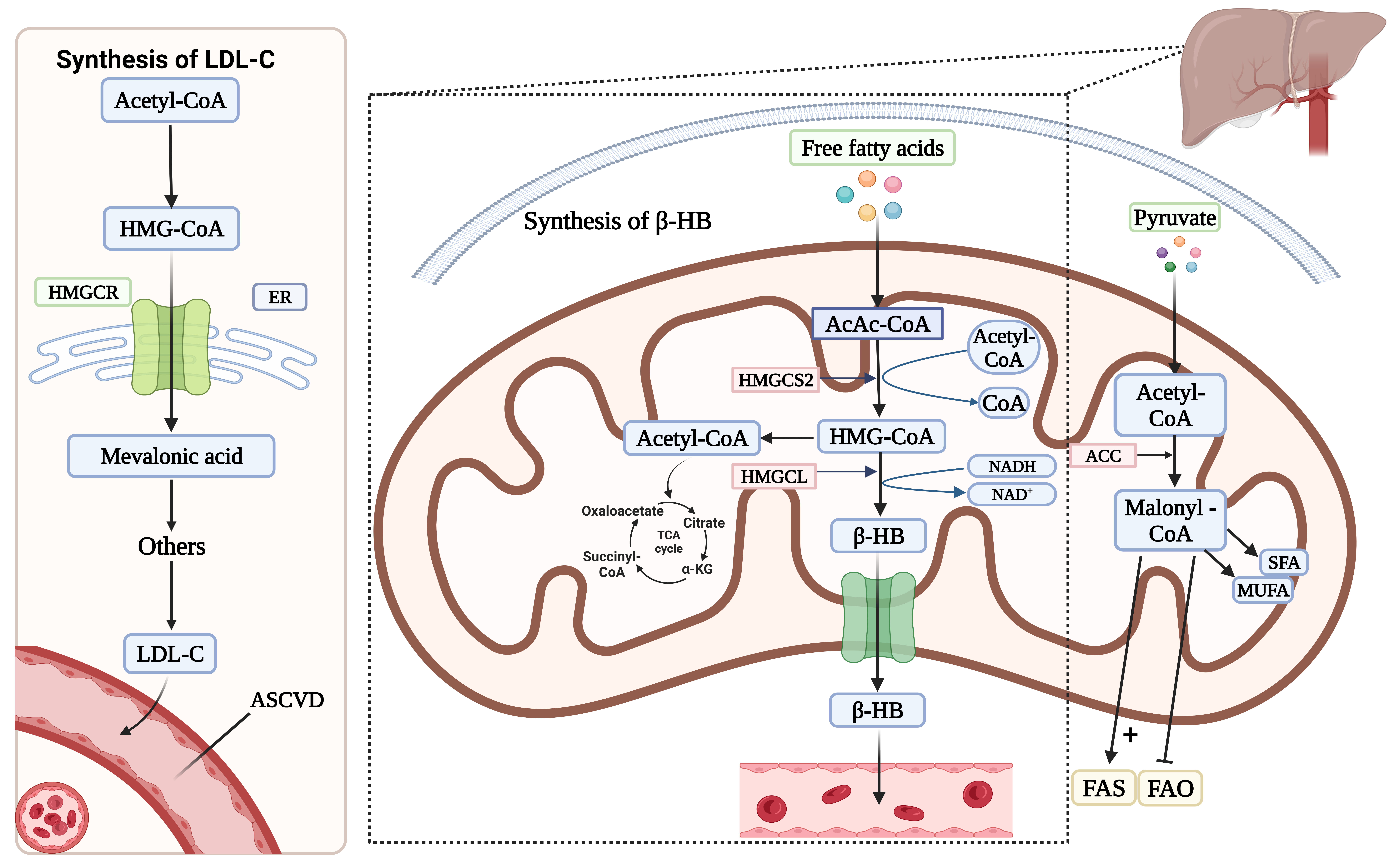 Fig. 2.
Fig. 2.Synthesis of Low-density lipoprotein cholesterol (LDL-C),
Chronic kidney disease precipitates elevated levels of triglycerides (TGs) in the body, thereby fostering dyslipidemia and CVDs [131]. Within pyruvate metabolism, ACC acts as a catalyst for converting acetyl-CoA, producing malonyl-CoA, inhibiting FAO, and supplying substrates for FA biosynthesis [132]. Both synthetic saturated FAs and monounsaturated FAs (MUFAs) become integrated into diverse lipid species, including TGs and phospholipids [133]. By diminishing the levels of synthetic substrates—acetyl-CoA and malonyl-CoA—ACC inhibitors delay the accumulation of lipids through DNL. Inhibiting ACC holds promise for reducing liver malonyl-CoA levels, mitigating lipid toxicity and bolstering FAO [134].
Stearoyl-CoA desaturase (SCD1) catalyzes the production of MUFAs, exerting influence as an energy metabolism regulator [135]. Dyslipidemia commonly observed in chronic kidney disease patients manifests as irregular levels of high-density lipoprotein cholesterol (HDL-C) and TGs [136]. SCD1 localizes alongside diacylglycerol acyltransferase 2 in the endoplasmic reticulum, tightly regulating triacylglycerol (TAG) synthesis in the liver [137]. Overexpression of SCD1 correlates with hypertriglyceridemia and heightened susceptibility to cardiovascular complications [138]. Focused exploration into the modulation of FA metabolism governed by ACC and SCD1 could reveal a deeper comprehension of the onset and progression of dyslipidemia in chronic kidney disease and cardiovascular disorders.
Cardiac dysfunction is intricately linked to modifications in cardiac substrate and energy metabolism [139]. The heart, being an organ characterized by high energy demands, heavily depends on CoA and its derivatives for the metabolic processing of FAs [140]. Concurrently, serving as the central hub of the cardiovascular system, the heart is vulnerable to various CVDs, encompassing coronary heart disease, cardiomyopathy, and heart failure [141].
Within the context of FAO, CoA plays a crucial role in converting FAs into energy. Genetic defects in CoA biosynthesis disrupt proper FA metabolism, resulting in the accumulation of related metabolites at the site of the enzyme defect. Furthermore, this genetic aberration diminishes the substrate pool available for energy production. The accumulation of metabolites, coupled with ensuing energy deficiency, can adversely impact the functionality of heart cells, culminating in the development of CVDs including cardiomyopathy and arrhythmia [142]. Deficiencies in mitochondrial trifunctional protein (MTP/TFP), stemming from mutations in the hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit alpha (HADHA) or HADHB genes responsible for critical proteins in CoA biosynthesis, can impair FAO. This impairment, in turn, affects the energy metabolism and function of heart cells. However, the specific mechanistic intricacies remain elusive, and to date, no established treatments exist for MTP/TFP deficiency [143].
Medium chain acyl-CoA dehydrogenase (MCAD) serves as a critical regulator of FAO in the heart. Genetic defects in CoA biosynthesis may lead to abnormal expression of MCAD, impacting the energy metabolism and function of the heart. Patients with heart failure exhibit disruptions in cardiac energy metabolism, prompting consideration of addressing metabolic defects as a potential treatment strategy. Consequently, utilizing the gene delivery method targeting MCAD may help restore heart energy metabolism, thereby safeguarding the heart from damage associated with pathological remodeling [144]. However, further research is needed to determine how genetic defects affect CoA biosynthesis and contribute to the development of CVD.
In the heart, elevated levels of non-histone acetylation enhance FAO in
conditions such as heart failure [145]. FA
SIRT3 plays a crucial role as a mitochondrial deacetylase, and its activation
holds the potential to restore mitochondrial metabolic homeostasis [152].
Acetyl-CoA acetylates mitochondrial proteins involved in FA beta oxidation, such
as LCAD and
Serum cholesterol emerges as a crucial focal point for CoA in the regulation of atherosclerotic CVD (ASCVD) [155, 156]. Acyl-CoA constitutes a significant element of CoA derivatives. Elevated plasma levels of acyl-CoA binding protein (ACBP) are linked to increased susceptibility to CVDs, displaying correlations with cardiovascular risk factors such as total free cholesterol and TGs. Conversely, ACBP levels exhibit a negative correlation with the protective HDL levels [157]. HDL is widely recognized as a protective factor against CVD and serves as an indicator for anti-atherosclerosis effects [158]. Globally, ASCVD stands out as the most prevalent and leading cause of mortality among CVDs [159]. Abnormal blood lipid levels, particularly elevated levels of low-density lipoprotein cholesterol (LDL-C), represent a significant risk factor for CVD and are considered a central aspect of ASCVD pathogenesis (Fig. 2). CoA itself does not directly reduce cholesterol levels. The majority of LDL-C is derived from the enzymatic activity of HMGCR, which catalyzes the conversion of HMG-CoA into mevalonate, a crucial precursor in cholesterol synthesis [160]. HMGCR serves as a typical target for statin medications, which competitively bind to the active sites of HMGCR. This inhibition results in reduced serum cholesterol levels, slowing down disease progression [161].
In addition to HMGCR inhibitors, flavonoids have demonstrated the ability to lower LDL-C levels [162]. They promote the oxidative metabolism of FAs and cholesterol by activating CoA and increasing the expression of fatty acyl-CoA oxidase. In the presence of CoA or mercaptan donors, hemoglobin converts ingested flavonoids into conjugates [163].
At lower doses, statins promote angiogenesis, whereas higher doses inhibit protein isopentenylation as well as cell and blood vessel development [161]. Xu et al. [164] discovered that alpha-ketoisovaleric acid and propionyl-CoA, metabolites of valine, strongly activate platelets, leading to propionylation of propionin-3-propionylation and subsequent thrombosis.
Neurodegenerative diseases encompass a spectrum of disorders leading to dysfunction within the nervous system, including well-known conditions such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis. An overarching characteristic shared by these diseases is the abnormal aggregation of proteins in the cytoplasm or nucleus. The genetic foundations of neurodegenerative diseases are diverse, involving both inherited disorders resulting from rare genetic mutations and the influences of common risk genes [165].
Among these conditions are pantothenate kinase-related neurodegeneration (PKAN), CoA synthase protein-associated neurodegeneration (CoPAN), protein lipoylation, and pyruvate dehydrogenase E2, four hereditary neurodegenerative diseases. Each of these disorders is linked to deficiencies in distinct metabolic processes within CoA biosynthesis [166]. Two autosomal recessive neurodegenerative diseases stem from mutations in genes encoding enzymes in the CoA pathway. PKAN and CoPAN, representing the initial and final enzymes of the CoA biosynthesis pathway related to neurodegenerative diseases, are part of the heterogeneous group of neurodegenerative diseases (neurodegeneration with brain iron accumulation) characterized by iron deposition in the brain [167].
Genetic variations lead to dysfunction of the pantothenate kinase 2 (PANK2)
gene, resulting in reduced activity of PANK2. This alteration indirectly impacts
the initial step of CoA biosynthetic pathways, where pantothenic acid is
converted to 4
Studies indicate that disruptions in cholesterol metabolism may be linked to the onset and progression of neurodegenerative diseases [170]. Intracellular cholesterol levels are tightly regulated through four processes: de novo synthesis, uptake, export, and storage [171]. Cholesterol metabolism in the brain operates relatively independently and primarily relies on the synthesis carried out by glial cells and neurons themselves [172]. Cholesterol synthesis takes place primarily in the endoplasmic reticulum and involves the conversion of acetyl-CoA into HMG-CoA through the activity of the irreversible rate-limiting enzyme HMG-CoA synthetase. This step is followed by the action of HMGCR, which converts HMG-CoA into mevalonate, squalene, and lanosterol, constituting the mevalonate pathway. In brain cholesterol synthesis, lanosterol is involved through two pathways: the Bloch pathway in astrocytes and the Kandutsch-Russel pathway in neurons (Fig. 3). The synthesized cholesterol is stored as lipid droplets and can be transported to plasma lipoproteins for the production of other substances [173, 174, 175]. Notably, adult neurons are less efficient at cholesterol synthesis compared to astrocytes [176, 177]. An imbalance in brain cholesterol levels can contribute to the development of neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease [178, 179].
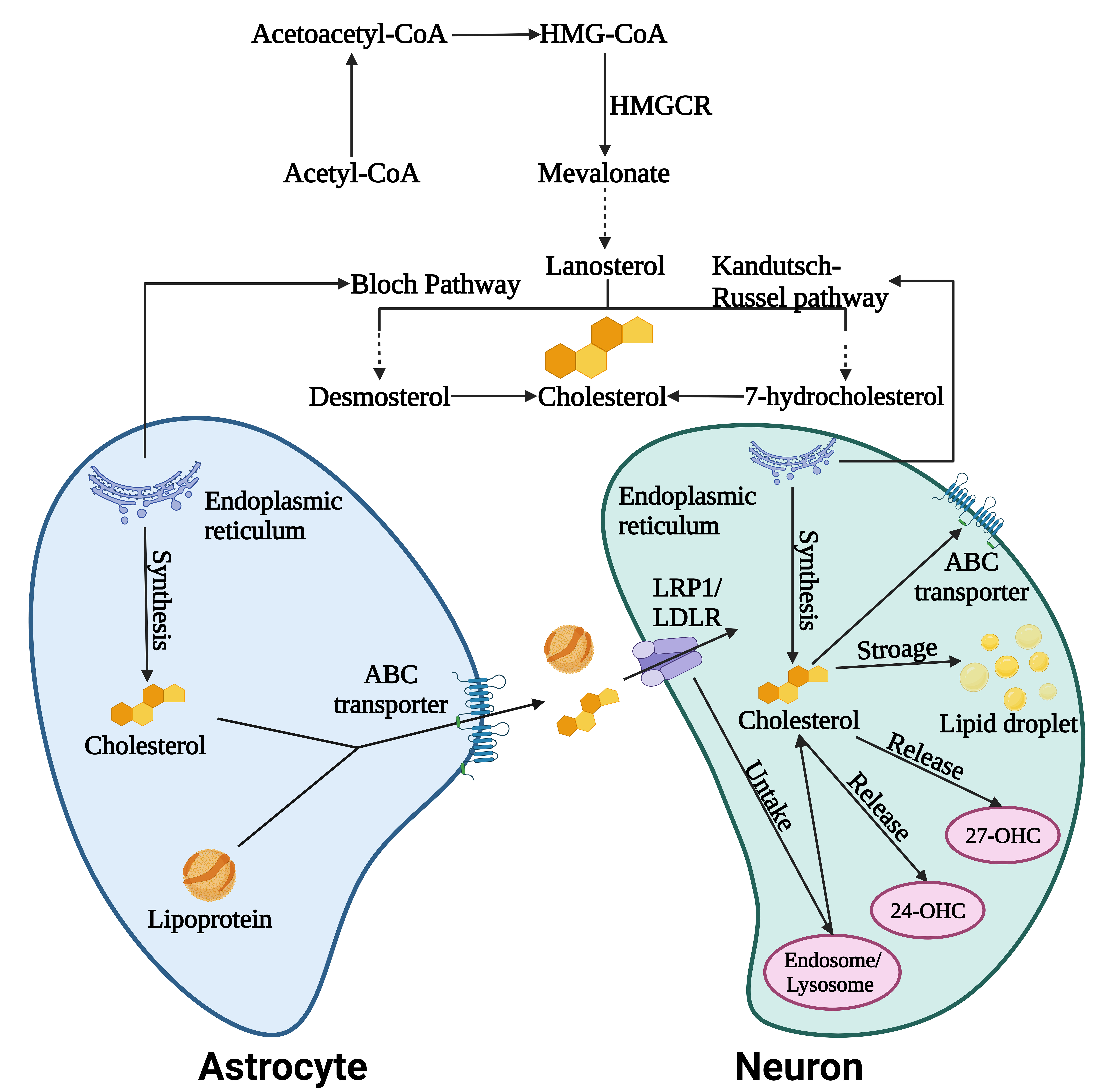 Fig. 3.
Fig. 3.Schematic illustration of CoA metabolism in neurodegenerative diseases. Cholesterol synthesis in neurons predominantly occurs through the Kandutsch-Russell pathway, while in astrocytes, it primarily takes place via the Bloch pathway. Previously, acetyl-CoA was sequentially converted to HMG-CoA, which was then transformed into mevalonate by the action of HMG-CoA reductase (HMGCR), subsequently supplying lanosterol for cholesterol synthesis. In adult neurons, the main source of cholesterol is provided by astrocytes, which supply it through the LDLR-related protein 1/LDLR in the form of apolipoproteins. Upon internalization, these apolipoproteins undergo conversion to free cholesterol within the endosome/lysosome compartment. In cases of cholesterol excess, intracellular esterification takes place, leading to the storage of cholesterol in lipid droplets. The stored cholesterol is then subject to conversion into oxysterols, particularly 24(S)-hydroxycholesterol (24-OHC).
CoA plays a crucial role in regulating lipid metabolism, influencing cellular
energy status and signal transmission. Numerous reports have underscored the
correlation between CoA-related lipid disorders and a spectrum of diseases,
including cancer, neurodegenerative diseases, and CVDs [15, 174, 180]. The
primary aspect of lipid metabolism involves the synthesis and degradation of FAs.
During FA synthesis, cellular reactions involving acetyl-CoA and malonyl-CoA lead
to the formation of long-chain FAs, such as palmitic acid [181]. This process
primarily takes place in the liver and adipose tissue, contributing to the
reduction of
AMPK plays a multifaceted role in cell metabolism, including lipid metabolism,
which is implicated in cancer, CVD, and neurodegenerative diseases (Fig. 4). AMPK
orchestrates the regulation of FFA concentration by activating FAO while
concomitantly inhibiting lipolysis and fat synthesis [186, 187]. Within the FA
metabolism, AMPK facilitates FA uptake and activates FAO by suppressing the
de novo synthesis of FAS, cholesterol, and TGs [188]. The inhibition of
FA synthesis by AMPK involves the modulation of the phosphorylation of ACC and
SREBP1c [189]. Notably, ACC1 is highly expressed in adipose tissue, regulating
the intracellular availability of acetyl-CoA involved in FA synthesis to meet the
material and energy requirements necessary for disease development [190]. The
synthesis of FAs is under the control of SREBPs, with SREBP1 primarily regulating
FA synthesis and the expression of the low density lipoprotein receptor (LDLR), while SREBP2 primarily regulates the
expression of cholesterol biosynthesis genes [191, 192]. Additionally, AMPK
inhibits cholesterol synthesis by suppressing the phosphorylation of the key
enzyme HMGCR [193]. The benefits of statins stem from their ability to inhibit
HMGCR, subsequently upregulating the cholesterol uptake gene, increasing
expression of the LDLR, and consequently reducing blood cholesterol levels [194].
Additionally, the cytoplasmic enzyme ACLY, highly expressed in
adipose-synthesizing tissues, is positioned upstream of HMGCR [195]. ACLY is
located upstream of ACC, and the acetyl-CoA produced by ACLY can be utilized for
the synthesis of cholesterol and FAs [196, 197, 198]. Likewise, the utilization of the
substrate N-acetyl-aspartate by ACSS2 is strongly involved in acetyl-CoA
production in adipocytes. Importantly, this process is not limited to adipocytes
and also occurs within the nervous system [199, 200, 201, 202, 203]. However, inhibition of ACLY
can suppress cholesterol and FA synthesis while increasing the expression of LDLR
in cells [204]. AMPK impacts TG synthesis by inhibiting glycerol-3-phosphate
acyltransferase, the enzyme responsible for catalyzing the initial step in TG
synthesis [205, 206]. Acetyl-CoA is transported intracellularly into mitochondria
for
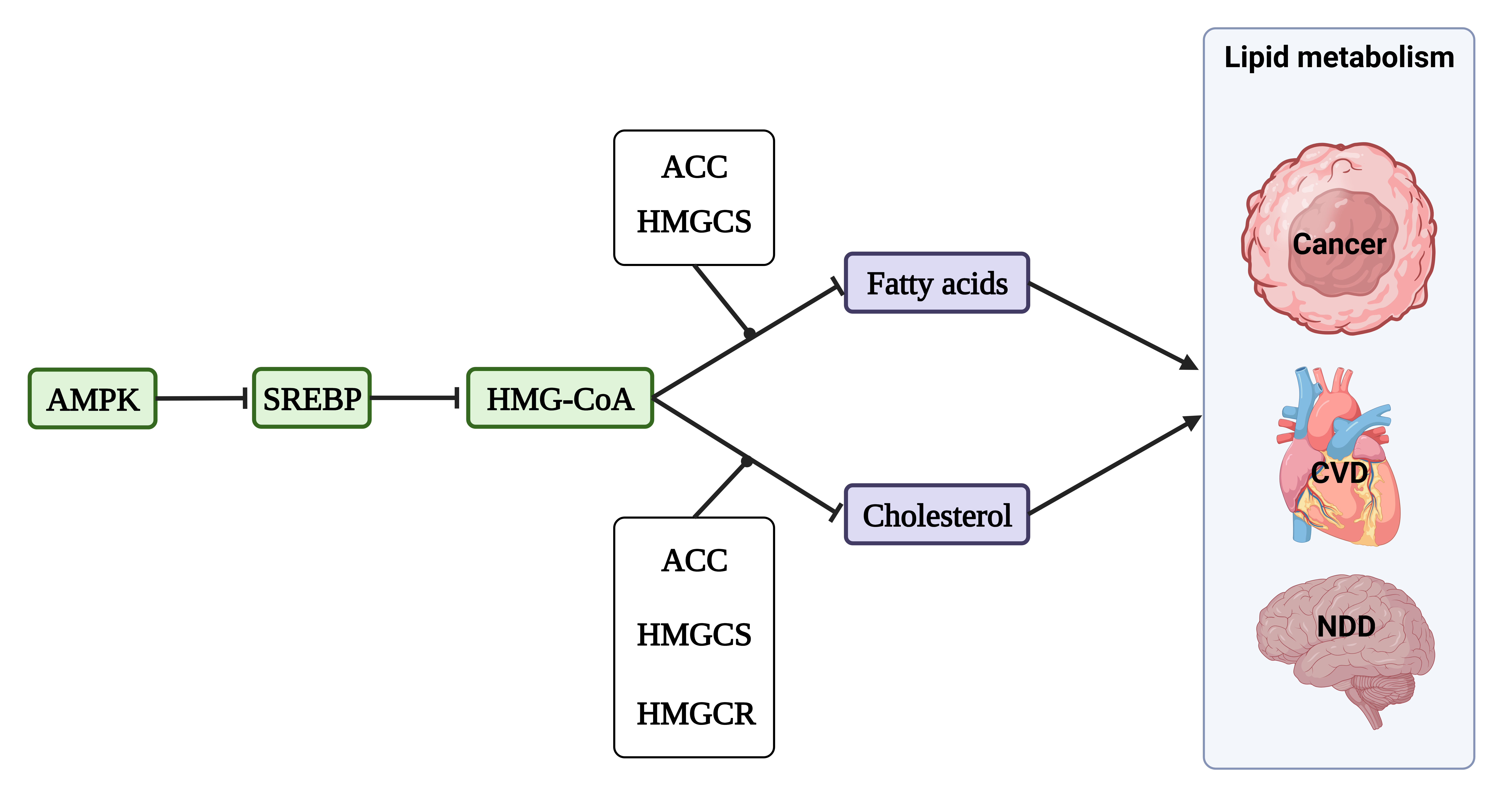 Fig. 4.
Fig. 4.Lipid metabolism in cancer, CVDs, and neurodegenerative diseases (NDD). The activation of AMP-activated protein kinase (AMPK) inhibits the activity of sterol regulatory element-binding protein (SREBP), resulting in a subsequent decrease in the expression of two key enzymes involved in HMG-CoA biosynthesis, namely HMG-CoA reductase (HMGCR) and HMG-CoA synthase (HMGCS). This negative regulation of FA and cholesterol synthesis ultimately impact the overall process of lipid metabolism in various diseases.
In conclusion, this study underscores the significant role of CoA and its derivatives in the pathogenesis and progression of various ailments such as cancer, CVDs, and neurodegenerative diseases. Moreover, this study highlights the immense potential of targeting CoA-associated processes for disease prevention and treatment.
ACAT1, Acetyl-CoA acetyltransferase 1; ACBP, Acyl-CoA binding protein; ACC,
Acetyl-CoA carboxylase; ACLY, ATP-citrate lyase; ACOT, Acyl-CoA thioesterase;
ACOT12, Acyl-CoA thioesterase 12; ACSS2, Acyl-CoA short chain synthetase family
member 2; AKT, Serine/threonine-protein kinases; AML, Acute myeloid leukemia;
AMPK, AMP-activated protein kinase; ASCVD, Atherosclerotic cardiovascular
disease; BMI, Body mass index; BRD4, Bromodomain containing 4; CAF,
Cancer-associated fibroblasts; CoA, Coenzyme A; COASY, CoA synthetase; CoPAN, CoA
synthase protein associated neurodegeneration; CPT1, Carnitine palmitoyl
transferase 1; CVD, Cardiovascular diseases; DGAT1, Diacylglycerol
O-acyltransferase 1; DGAT2, Diacylglycerol acyltransferase 2; DNL, De
novo lipogenesis; EMT, Epithelial-mesenchymal transition; FAO, Fatty acid
oxidation; FAS, Fatty acid synthetase; GBM, Glioblastoma; GLUT1, Glucose
transporter 1; GPAT, Glycerol-3-phosphate acyltransferase; HDL-C, High-density
lipoprotein cholesterol; Hhat, Hedgehog acetyltransferase; HIF, Hypoxia inducible
factor; HMGCL, 3-hydroxy-3-methylglutaryl CoA lyase; HMG-CoA,
3-hydroxy-3-methylglutaryl CoA; HMGCS1, 3-hydroxy-3-methylglutaryl CoA synthase
1; HSIL, High-grade squamous intraepithelial lesions; IRF4, Interferon regulatory
factor 4; KAT2A, Lysine Acetyltransferase 2A; LCAD, long chain acyl CoA
dehydrogenase; LDL-C, Low-density lipoprotein cholesterol; LDLR, Low-density
lipoprotein receptor; MCAD, Medium chain acyl-CoA dehydrogenase; MePAN, Protein
lipoylation; MTP, Mitochondrial trifunctional protein; MUFA, Monounsaturated
fatty acids; MVA, Mevalonic acid; NBIA, Neurodegeneration with brain iron
accumulation; PANK2, Pantothenate kinase 2; PDA, Pancreatic ductal
adenocarcinoma; PDC, Pyruvate dehydrogenase complex; PDH-E2, Pyruvate
dehydrogenase-E2; PDK, Pyruvate dehydrogenase kinase; PFK1, Phosphofructokinase
1; PFK2, Phosphofructokinase 2; PKAN, Pantothenate kinase-related
neurodegeneration; SCD1, Stearoyl-CoA desaturase; SFA, Saturated fatty acids;
Shh, Sonic hedgehog; SIRT3, Sirtuin3; SIRT5, Sirtuin 5; SREBP1c, Sterol
regulatory element-binding protein 1c; SREBPs, Sterol regulatory element-binding
proteins; SUCLA2, Succinate-CoA ligase ADP-forming subunit beta; TAG,
Triglyceride; TCA, Tricarboxylic acid; TGIF, TG-interacting factor; TWIST2, Twist
family BHLH transcription factor 2;
JX, XC, JZ, MZ, and HM conceived the review. JX and HM supervised the review. All authors wrote the draft. JX revised the manuscript. All authors read and agreed to publish the paper. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This study was supported by Key scientific research projects of Hubei polytechnic University (No. 23xjz08A).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.