

 , Md. Zeyaullah 2,*, Abdullah M. AlShahrani 2, Adam Dawria 3, Haroon Ali 3, Ali Mohieldin 3, Abdelrhman AG Altijani 3, Ufaq Razi 4, Munzila Mehdi 5, Sabika Akram 5, Ejaz Rizvi Hussain 5
, Md. Zeyaullah 2,*, Abdullah M. AlShahrani 2, Adam Dawria 3, Haroon Ali 3, Ali Mohieldin 3, Abdelrhman AG Altijani 3, Ufaq Razi 4, Munzila Mehdi 5, Sabika Akram 5, Ejaz Rizvi Hussain 51 Hiralal Mazumdar Memorial College for Women, West Bengal State University, 700035 Kolkata, West Bengal, India
2 Department of Basic Medical Science, College of Applied Medical Sciences, Khamis Mushait Campus, King Khalid University (KKU), 62561 Abha, Saudi Arabia
3 Department of Public Health, College of Applied Medical Sciences, Khamis Mushait Campus, King Khalid University (KKU), 62561 Abha, Saudi Arabia
4 Department of Biosciences, Jamia Millia Islamia, 110025 New Delhi, India
5 Department of Botany, Aligarh Muslim University, 202001 Aligarh, India
Abstract
Innate lymphocytes, including microglial cells, astrocytes, and oligodendrocytes, play a crucial role in initiating neuroinflammatory reactions inside the central nervous system (CNS). The prime focus of this paper is on the involvement and interplay of neurons and glial cells in neurological disorders such as Alzheimer’s Disease (AD), Autism Spectrum Disorder (ASD), epilepsy, and multiple sclerosis (MS). In this review, we explore the specific contributions of microglia and astrocytes and analyzes multiple pathways implicated in neuroinflammation and disturbances in excitatory and inhibitory processes. Firstly, we elucidate the mechanisms through which toxic protein accumulation in AD results in synaptic dysfunction and deregulation of the immune system and examines the roles of microglia, astrocytes, and hereditary factors in the pathogenesis of the disease. Secondly, we focus on ASD and the involvement of glial cells in the development of the nervous system and the formation of connections between neurons and investigates the genetic connections associated with these processes. Lastly, we also address the participation of glial cells in epilepsy and MS, providing insights into their pivotal functions in both conditions. We also tried to give an overview of seven different pathways like toll-like receptor signalling pathway, MyD88-dependent and independent pathway, etc and its relevance in the context with these neurological disorders. In this review, we also explore the role of activated glial cells in AD, ASD, epilepsy, and MS which lead to neuroinflammation. Even we focus on excitatory and inhibitory imbalance in all four neurological disorders as imbalance affect the proper functioning of neuronal circuits. Finally, this review concludes that there is necessity for additional investigation on glial cells and their involvement in neurological illnesses.
Keywords
- microglia
- astrocytes
- Alzheimer's Disease
- autism spectrum disorder
- epilepsy
- multiple sclerosis
- neuroinflammation
- excitatory and inhibitory imbalance
Glial cells consist of microglia, astrocytes, oligodendrocytes and their
progenitors NG2-glia which constitute between 33 and 66% of the total mass brain
[1]. Glial cells play role in the release of gliotransmitters and cytokines, as
well as the induction of synaptic modifications and even play a crucial role in
initiating neuroinflammatory reactions [2]. Astrocytes play a crucial
role in maintaining physiological equilibrium and regulating synaptic
transmission, while microglia primarily serve an immunological function [2]. The
primary cause of the inflammatory response in the central nervous system (CNS) is
mostly attributed to factors such as infection, toxins, trauma, and ischemia.
These factors trigger the activation of microglia and astrocytes, leading to an
upregulation in the production of pro-inflammatory cytokines, chemokines,
secondary messengers, and reactive oxygen species (ROS) [1, 2, 3]. On the other
hand, the chronic inflammatory response has negative effects on neurodegenerative
diseases and is associated with functional deficits resulting from oxidative
stress, the accumulation of misfolded proteins, and inflammation in peripheral
tissues [4]. Glia has the potential to either facilitate or impede the central
nervous system’s capacity for recovery following inflammation. A1 and A2
astrocyte phenotypes, as well as M1 and M2 microglia states are simplifications
of a more complex reality. However, these classifications are not unique and
there may be more phenotypes to study [5]. The reactive astrocyte exhibits two
distinct phenotypes, namely the detrimental “A1” and the beneficial “A2”,
which are triggered by inflammatory processes and ischemic conditions. The A1
phenotype is elicited by the NF-
| Name of miRNA | Name of disorder | Target and role | References |
| miR-16 | Alzheimer’s Disease | Target APP and reduce A |
[22] |
| miR-128 | Alzheimer’s Disease | Target BAG2 and reduce tau | [23] |
| miR-9 | Alzheimer’s Disease | Target SIRT1and reduce tau | [24] |
| miR-7 | Alzheimer’s Disease | Target NLRP3 and reduce neuroinflammation | [25] |
| miR-34a | Alzheimer’s Disease | Target tau and reduce tau | [26] |
| miR-125b | Autism Spectrum Disorder | Regulates the expression of FMR1 and contributes to the alteration of synaptic plasticity as in FXS | [27] |
| miR-132 | Autism Spectrum Disorder | Involved in synaptic plasticity | [28] |
| miR-137 | Autism Spectrum Disorder | miRNA-137 is downregulated, it influences the expression of many genes implicated in neurodevelopment | [29] |
| miR-134 | Autism Spectrum Disorder | Regulates synaptic plasticity | [30] |
| miR-181c | Autism Spectrum Disorder | miRNA-181c is upregulated in the amygdala of ASD patients; its function is associated with the development of the nervous system | [31] |
| miR-146a | Epilepsy | Target IL-1 |
[32] |
| miR-155 | Epilepsy | Target TNF- |
[33] |
| miR-98 | Epilepsy | Target CCL2, CCL5, downregulated and target inflammation and blood brain barrier pathways | [34] |
| miR-218 | Epilepsy | Target GRM1, SLC1A2, ROBO1, GNAI2, downregulated and target synaptic plasticity | [35] |
| miR-21 | Epilepsy | Target NT-3, upregulated and neutrite growth pathways | [36] |
| miR-155 | Multiple Sclerosis | Upregulated and promotes microglial inflammation | [37] |
| miR-145 | Multiple Sclerosis | Downregulated and promotes microglial inflammation | [38] |
| miR-125b | Multiple Sclerosis | Upregulated and promotes microglial inflammation | [38] |
| miR-222 | Multiple Sclerosis | Upregulated and promotes microglial inflammation | [39] |
| miR-32 | Multiple Sclerosis | Upregulated and promotes microglial inflammation | [37] |
miRNA, micro-RNA; APP, Amyloid Precursor Protein; A
The terms ‘astrocytosis’, ‘astrogliosis’, ‘reactive gliosis’, ‘astrocyte activation’, ‘astrocyte reactivity’, ‘astrocyte re-activation’, and ‘astrocyte reaction’ have all been employed to characterize the responses of astrocytes to abnormal occurrences in the CNS, such as neurodegenerative and demyelinating diseases, epilepsy, trauma, ischemia, infection, and cancer [40]. Astrocyte reactivity can occur in several pathogenic situations, which can differ significantly. These contexts can be either spontaneous or genetically determined, acute or chronic, resulting from a systemic illness (such as sepsis), a specific injury or disease of the CNS, or a harmful experimental manipulation [40]. Astrocyte reactivity, as defined, is a response that occurs in response to an external signal. This response can change over time and, in many cases, can be reversed [41]. Astrocytes can also experience cell-autonomous disruptions, which occur in astrocytopathies caused by mutated versions of astrocytic genes (such as GFAP in Alexander disease), as well as from direct viral infections or exposure to toxic substances that precisely harm astrocytes (such as ammonium in hepatic encephalopathy) [41]. Reactive astrocytes experience structural, molecular, and functional alterations in reaction to abnormal conditions in the nearby tissue, such as central nervous system diseases, injuries, or harmful experimental interventions [41]. Astrocytes carrying genetic alterations that cause disease are astrocytes that originate or contribute to pathology. These astrocytes later become reactive in a manner that may differ from the typical response of astrocytes to external stimuli. Genetic variations associated with CNS disorders can also impact the functioning of astrocytes and prepare them to adopt specific reactive states [42]. There is no typical reactive astrocyte; reactive astrocytes do not divide into essential opposing characteristics, such as beneficial-harmful, toxic-protective, A1–A2, etc. Reactive astrocytes can assume several states depending on the situation, with just a tiny portion of shared alterations across distinct states [43]. Simultaneous loss of certain homeostatic functions and acquisition of certain protective or harmful functions is possible. The ultimate effect on disease, whether advantageous or harmful, will depend on the equilibrium and characteristics of impaired and acquired functions and the varying prevalence of distinct astrocyte subgroups [43]. Astrocytes affected by the disease are the primary initiators and may subsequently develop a reactive phenotype that influences the progression of the disease. Genetic mutations in genes expressed in all cells of the body, such as those seen in familial neurodegenerative disorders, or variations in genes highly expressed in astrocytes (such as APOE in AD), can result in dysfunctional astrocytes [44]. While astrocytes may not be the leading cause of the disease, their dysfunction can negatively affect the progression and outcome of the condition [41].
Glial cells, namely astrocytes and microglia, play a crucial role in the pathogenesis of neuroinflammatory and neurodegenerative disorders by means of several inflammatory mediators and pathways. In this review we have discussed seven different pathways.
The immune system can be classified into two main components, namely the
cellular and humoral systems. In general, the cellular immune system is composed
of macrophages, neutrophils, natural killer cells, and dendritic cells [45]. On
the other hand, within the CNS, the primary components of the cellular immune
system consist of glia and macrophages [46]. Glia cells have been proposed to
fulfill many functions inside the CNS, including serving as a cellular repair
mechanism, facilitating the removal of deceased cells, and contributing to
neuroinflammatory processes [47]. Microglia and astrocytes are essential
components in the preservation of neuronal integrity. Microglial cells contribute
to the neuroinflammatory response in the CNS by releasing chemokines and
cytokines with pro-inflammatory properties, including IL-1
MYD88 refers to Myeloid Differentiation Primary Response 88,
commonly known as MyD. This protein is associated with myeloid
differentiation. Simultaneously, the numerical value “88” denotes the position
of the activated gene within a list of five adaptors that interact with
downstream signaling proteins associated with IL-1 and TLRs [62, 63]. Type
I transmembrane receptors known as TLRs possess external leucine-rich repeats, a
transmembrane helix, and an internal Toll/IL-1 receptor domain
(TIR) [63]. TLRs are a subset of pattern recognition receptors
(PRRs) that are typically triggered by pathogen-associated
molecular pattern molecules (PAMPs) and damage-associated
molecular pattern molecules (DAMPs). Each TLR can identify
certain components of pathogens. For instance, TLR4 can recognize the
LPS found in the outer membrane of bacteria, whereas TLR5 can identify
flagellin, a protein present in bacterial flagella [64, 65]. Hence, TLRs
play a crucial role in the activation of innate immunity, leading to the
initiation of inflammatory reactions and the development of antigen-specific
adaptive immunity [66]. In general, TLR signaling pathways can be categorized
into two main groups: MyD88-dependent pathways and MyD88-independent
pathways, which are distinguished by the recruitment of certain adaptors. In
relation to the MyD88-dependent pathway, it is noteworthy that all Toll-like
receptors (except for TLR3) rely on MYD88 for the activation of downstream
signaling. This process, known as “Myddosome”, is responsible for activating
NF-
The TIR-domain-containing adapter-inducing interferon-
Interleukin-6 (IL-6) is a cytokine that has pleiotropic
effects and has gained significant recognition for its pivotal involvement in
maintaining the pathophysiological equilibrium of the central nervous system and
regulating glial function [95]. In the brain, under typical physiological
circumstances, there exists a diminished concentration of IL-6 [95].
Nevertheless, the protein expression and release of this particular entity
exhibited a favorable correlation with several neurological illnesses, including
brain cancer, AD, PD, MS, and brain ischemia [96]. The pleiotropic biological
effects of IL-6 on target cells are facilitated by its contact with the
non-signaling membrane-bound receptor, IL6R [97]. In comparison to the
receptor for Lipocalin-2 (LCN2), the expression of the receptor
specific to IL6R was observed to be relatively restricted inside nervous
tissue. Specifically, IL6R was mostly detected in microglia, while its presence
was not observed in oligodendrocytes or astrocytes [98]. The IL6R is also present
in a soluble form, which is produced through either alternate splicing of its
mRNA or proteolytic cleavage from the cell membrane [99]. At the molecular level,
it can be observed that IL6R does not possess the inherent ability to transmit
signals downstream. Therefore, it elicits its chemical cascades via two distinct
signaling channels, namely the canonical signaling pathway and the
trans-signaling pathway. In the conventional signaling pathway, the cytokine IL6
engages in interaction with a particular glycoprotein receptor called
IL-6 receptor-
Chemokines are a group of extracellular pro-inflammatory proteins that function
as chemotactic agents, stimulating cellular communication within the immune
response. Additionally, they have a role in the establishment of neuronal
networks, synapse development, and the regulation of cognitive function [101].
The classification of chemokine subfamilies can be determined by analyzing the
structural motifs of the N-terminal cysteine residues. These subfamilies are
categorized as CC-, CXC-, XC-, and CX3C [102]. Multiple chemokines can bind to either one receptor or multiple receptors.
These receptors can be categorized based on their ligands into chemokine
receptors CCR (1-10), CXCR (1-7),
XCR1, and CX3CR1 [103]. Therefore, chemokines have the
potential to serve as chemotactic mediators in various biological processes
[104]. The CNS possesses a distinct anatomical arrangement, which is delineated
from other bodily organs by the presence of colony-
stimulating factor (CSF). The CNS has cellular
compartments, including T cells, which exhibit a deficiency in humoral immunity
to uphold an immunosuppressive milieu. During instances of neuronal damage, glial
cells become activated and initiate inflammation in the CNS by releasing
chemokines. These chemokines serve to attract T-lymphocytes, which infiltrate the
affected area. Additionally, glial cells facilitate communication between neurons
to maintain proper brain function. However, it is worth noting that the levels of
these chemokines have been found to be linked to the development of many CNS
diseases [105]. The activation mechanism of glia cells involves the activation of
various signaling pathways through trans-membrane G protein-coupled receptors.
These pathways include the activation of mitogen-activated protein
kinases (MAPKs) and phospholipase C, which leads to the release of
intracellular Ca ions. This release of Ca ions plays a role in modulating gene
regulation, among other pathways [106, 107, 108]. Chemokines are observed within
the CNS in the form of pro-inflammatory cells, serving a protective function
against neuronal injury. Nevertheless, it has been documented that chemokines
also contribute to the development of many neurological illnesses [109].
CX3CL1, a chemokine of considerable interest in scientific research, has a
distinctive characteristic by selectively binding to a singular receptor,
CX3CR1. The central CNS concentrations of this chemokine are subject to
conflicting interpretations and opinions among the academic community. The
CX3CL1-CX3CR1 signaling pathway has the potential to stimulate microglia, leading
to the release of inflammatory substances and exacerbating the pathological
condition in various neurological illnesses, including mesial temporal lobe
epilepsy (MTLE), AD, PD, and amyotrophic lateral sclerosis (ALS). During the
occurrence of MTLE, which is distinguished by an intense inflammatory reaction,
the activation of microglia was found to be linked to the recurrent initiation of
seizures, heightened neurogenesis, and synaptic remodeling [110, 111]. Elevated
concentrations of CX3CL1 were observed in both blood and CSF of individuals
diagnosed with mesial temporal lobe epilepsy (MTLE). Experimental studies have
indicated that the absence of the CX3CR1 receptor leads to a decline in
microglial activation and a decrease in the quantity of degenerated neurons
[111]. The CX3CL1-CX3CR1 signalling route has been implicated in the pathogenesis
of neurodegenerative illnesses. It has been observed that dysfunction of this
signalling system tends to augment the activation of microglia. The chemokine
signalling system has been proposed to undergo modifications in AD, resulting in
an elevated population of activated microglia and the subsequent production of
inflammatory markers, including IL6. Thus, it was discovered that modifying the
signalling route will lead to an increasing amount of amyloid-
Lipocalin-2 (LCN2) is classified as a low molecular weight acute phase protein that is a member of the lipocalin superfamily. This superfamily encompasses a total of 20 soluble proteins [117]. The substance referred to as neutrophil gelatinase-associated lipocalin (NGAL) or 24p3 is primarily synthesized and released by astrocytes, serving as a robust indicator of reactive astrocytes. Like other proteins implicated in inflammation, LCN2 is upregulated in reaction to diverse inflammatory and harmful stimuli, including as infection and damage [118]. Furthermore, the release of LCN2 by glial cells in response to various inflammatory stimuli enhances and controls neuroinflammation by activating the transcriptional processes of inflammatory chemokines and subsequently attracting immune cells [119]. Nevertheless, the function of LCN2 is facilitated by its interaction with hydrophobic ligands known as LCN2R, which enables the regulation of several cellular processes either by autocrine or paracrine mechanisms. The cellular processes may include cellular proliferation, differentiation, migration, and survival. Furthermore, the expression profile of LCNR2 exhibits significant upregulation in several neurological tissues, including neurons, microglia, and astrocytes. Thus, LCN2 is acknowledged to be elevated in numerous clinical disorders linked with the central nervous system, such as brain injury [52, 120, 121]. At the molecular level, it has been observed that the activation of LCN2 elicits the initiation of many inflammatory signaling pathways, the majority of which are facilitated by NF-Kb, MAPK, STATs, C/EBP, and HIF-1 [122]. Furthermore, the engagement between Lcn2 and LCN2R leads to the enhancement of the molecular signaling pathways involving JAK-STAT3 and IKK/NF-kB. This, in turn, triggers the transcriptional activities of various genes associated with the maintenance of glial cell integrity, including CXCL10, GFAP (glial fibrillary acidic protein, a constituent of the cytoskeleton), and ITGB3 (integrin beta chain beta 3).
IL-6 is a cytokine with pleiotropic effects that has gained significant
recognition for its pivotal involvement in the pathophysiological regulation of
the central nervous system and glial function [123]. In the brain, there exists a
basal level of IL-6 under typical physiological circumstances [124].
Nevertheless, previous studies have established a favorable association between
the production and release of this protein and several neurological illnesses,
including brain cancer, AD, PD, MS, and brain ischemia [94]. The pleiotropic
biological effects of IL-6 on target cells are facilitated by its contact with
the non-signaling membrane-bound receptor, IL6R [95]. In comparison to
the receptor for LCN2, the expression of the receptor specific to IL-6
(IL6R) was observed to be relatively restricted inside nervous tissue.
Specifically, IL6R was predominantly detected in microglia, whereas its presence
was not identified in oligodendrocytes or astrocytes [96]. The IL6R can also be
detected in a soluble state, which is produced through two mechanisms: alternate
splicing of its mRNA or proteolytic cleavage from the cell membrane [97]. At the
molecular level, it can be observed that IL6R exhibits a deficiency in its
ability to generate intrinsic downstream signals. Therefore, it initiates its
molecular cascades by either the traditional or trans-signaling pathways. In the
conventional signaling pathway, the cytokine IL6 engages in interaction with a
particular glycoprotein receptor called IL-6 receptor-
Neuroinflammation, an often-observed symptom in inflammatory and degenerative disorders, serves as a distinguishing feature of both acute and chronic neurological conditions. The aforementioned problem primarily arises from chronically activated glial cells inside the cerebral region. Pro-inflammatory cytokines are a subset of neuroexcitatory substances that are generated by activated glial cells and have the ability to promote the release of neurotransmitters. One potential use involves the utilization of this method to inhibit glial activity as a means of effectively managing neuropathic pain.
Dementia is the term used to describe the decline in cognitive functioning that
impairs an individual’s ability to carry out typical daily activities [125].
Neurological illnesses such as brain vascular diseases, brain damage, and
dementia with Lewy’s bodies have been identified as contributing factors to its
development [126]. Around 60% to 70% of dementia instances were detected in
patients diagnosed with AD [125]. AD is a neurodegenerative disorder
characterized by progressive cognitive decline and is recognized as the primary
contributor to memory impairment [125]. In certain instances, the use of AD
medication has been associated with the development of psychological problems,
including schizophrenia. The etiology of AD remains elusive; nevertheless, a
prominent clinical characteristic is the development of senile plaques inside the
cerebral tissue [126]. The aforementioned plagues played a role in the initiation
of neuroinflammation within the brain tissue [126]. The presence of
neuroinflammation in AD was initially observed during the early 20th century
through postmortem brain examinations conducted by Alois Alzheimer, a
neuropathologist from Germany. Subsequently, there has been a comprehensive
investigation into the neuroinflammation process in patients and models of AD
[127]. This section aims to elucidate the involvement of inflammatory mediators
in the interaction between neurons and glial cells in AD. In the context of
axonal sprouting, synaptogenesis, and nervous tissue regeneration, it is crucial
to acknowledge the significant role played by chemokines, which are inflammatory
mediators produced by supporting cells and glial cells within the nervous system
[128]. During instances of brain tissue injury, the pro-inflammatory neurotoxic
cytokines, namely TNF-
ASD is distinguished by enduring impairments in both verbal and nonverbal
communication, as well as repeated behavioral patterns, hobbies, or activities
[154, 155]. According to recent research, the prevalence of ASD is approximately 1
in 100 children, with a significantly higher occurrence in boys compared to
females, with a ratio of 4–5 to 1 [156]. The etiology of ASD is multifaceted and
may involve a combination of genetic and environmental influences [157]. Several
environmental factors related to ASD have been found, including the presence of
an inflammatory reaction [158]. The interaction between the CNS and the immune
system, encompassing both innate and adaptive immunity, occurs continuously
throughout an individual’s lifespan and developmental stages. This dynamic
relationship is commonly referred to as neuroimmunity [159]. The influence of the
maternal immune system on fetal brain development is believed to have a
significant impact on the occurrence of ASD [160]. There are other variables that
contribute to the dysregulation of maternal immunity, including viral and
bacterial infections [161]. The correlation between prenatal ASD and maternal
infection is contingent upon the specific pathogen involved as well as the timing
of the infection. Research has indicated a correlation between the occurrence of
bacterial infection during the second trimester of pregnancy and the development
of ASD. Conversely, viral infection has been found to relate to the first
trimester of pregnancy in relation to ASD [162]. It is noteworthy that the virus
does not manifest in the fetal brain, indicating that the materials associated
with the maternal immune response hold greater significance compared to the
infection itself. One piece of evidence that supports the link between maternal
immune response and fetal ASD is the detection of cytokines in the blood of the
fetus. The transmission of cytokines from the mother to the fetus can occur via
the placental barrier, or alternatively, fetal cytokines may be generated because
of maternal cytokine activity [163]. The impact of cytokines on prenatal brain
development is shown regardless of whether they originate from the maternal or
fetal source [164]. An excessive production of pro-inflammatory cytokines,
including tumour necrosis factor-
Seizures refer to the occurrence of abnormal neuronal excitation or inhibition
inside the brain, resulting in the development of abnormal hypertonic or
hypotonic states [178]. The exact cause of seizures remains uncertain; however,
several risk factors have been identified as contributors to aberrant brain
activity, including but not limited to high-grade fever, head trauma, and
metabolic illnesses [179]. Epilepsy is characterized by the occurrence of
recurrent unstimulated seizures [180]. There exist various clinical
manifestations of this condition, each exhibiting distinct levels of severity and
prognosis. Examples include absence epilepsy, generalized tonic-clonic epilepsy,
and benign newborn convulsions. The etiology of epilepsy often involves brain
tissue injury, with neuroinflammation being a commonly reported pathogenic
mechanism in this disorder. The signs of inflammation, brain tissue damage and
enhanced glial cell activities have been identified in several brain locations in
epilepsy [181]. Previous research has documented a notable increase in microglial
activity, with the presence of cytokines being identified in brain tissue as soon
as 4 hours after the occurrence of status epilepticus [182]. Elevated levels of
inflammatory mediators, such as IL-1
MS is a complex neurodegenerative and inflammatory disorder that is
characterized by recurrent inflammatory episodes leading to demyelination and
damage to axons [194]. MS is characterized by three distinct patterns, namely
relapsing-remitting MS (RRMS), secondary progressive MS (SPMS), and primary
progressive MS (PPMS). RRMS is distinguished by the occurrence of recurring
episodes that are followed by periods of remission. These remission periods have
the potential to develop into SPMS. On the other hand, it is plausible that it
could manifest as PPMS. The manifestation of MS symptoms is contingent upon the
location of demyelination and might manifest as cognitive, motor, or sensory
dysfunction [195, 196, 197, 198, 199]. The CNS lesions manifest as demyelination plaques located
in both the white and grey matter [200]. Historically, MS has been widely
acknowledged as a T-cell-mediated immunological disorder [201]. The involvement
of B-cells and innate immunity in the pathophysiology of MS has been widely
recognized [202]. The detection of B-cells and macrophages in MS lesions has been
substantiated [202]. The process of pathogenesis begins with the invasion of CD4+
cells in the white matter, facilitated by the compromised integrity of the BBB.
The expansion of CD4+ cells is facilitated by the presence of
IL-1, IL-6, IL-12, and IL-23, which are
produced by dendritic cells, microglia, and macrophages derived from monocytes.
These cytokines facilitate the recruitment of extra macrophages produced from
monocytes into the CNS, resulting in the rupture of the BBB [203]. The enlarged
T-cells can be categorized into two distinct kinds, namely T helper 1
(Th1) and T helper 17 (Th17) cells.
The cells are responsible for the secretion of IL-17 and
IL-12, which in turn contribute to the amplification of the Th17 cell
through a feedback mechanism. Furthermore, Th1 and Th17 produce
granulocyte-macrophage colony-stimulating factor (GM-CSF), INF-
Excitatory and inhibitory (E/I) imbalance is regarded to be critical for the proper functioning and integrity of neural circuits and is illustrated in Fig. 1. This section covers the concept of the E/I imbalance in AD, ASD, MS, and Epilepsy.
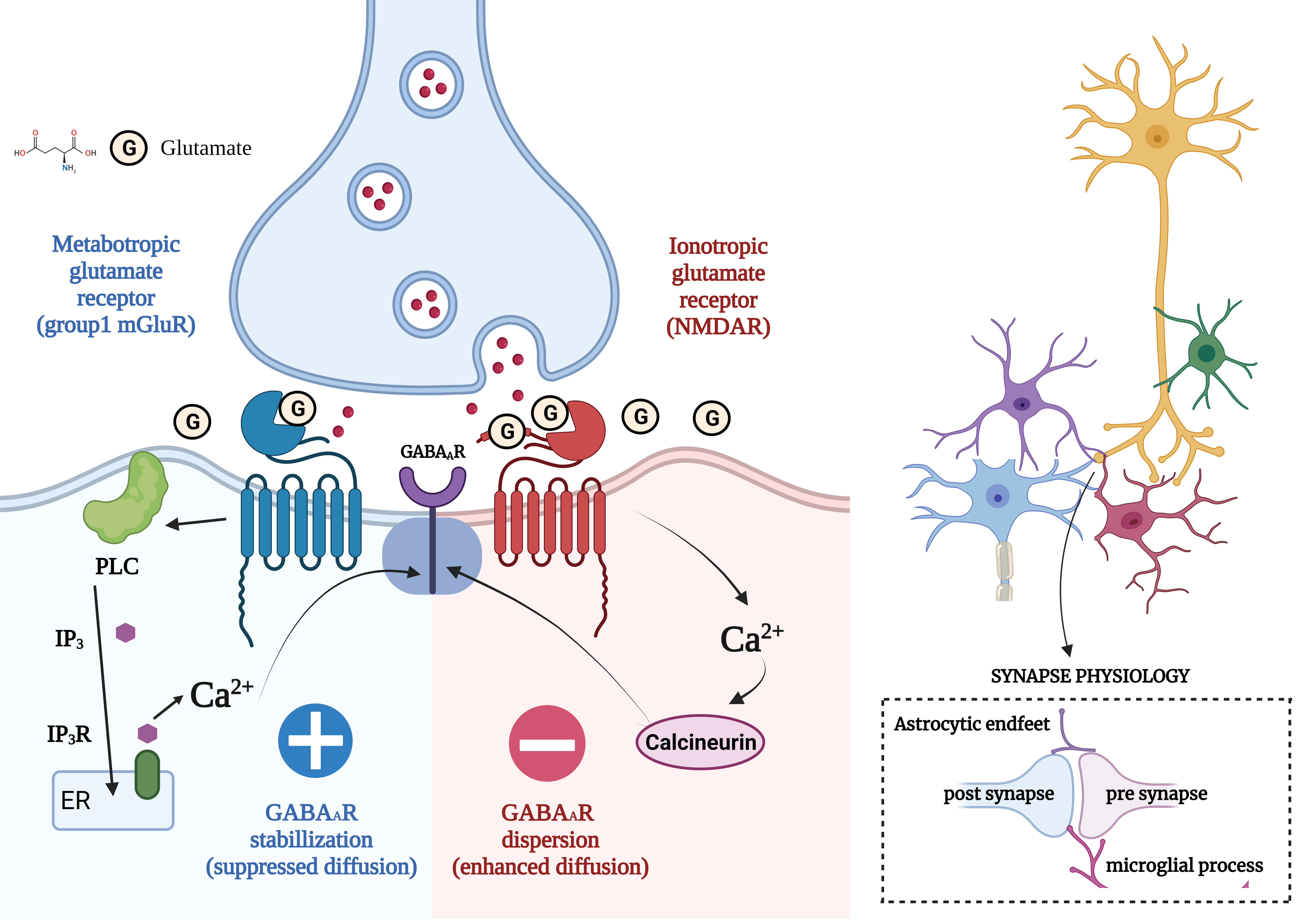
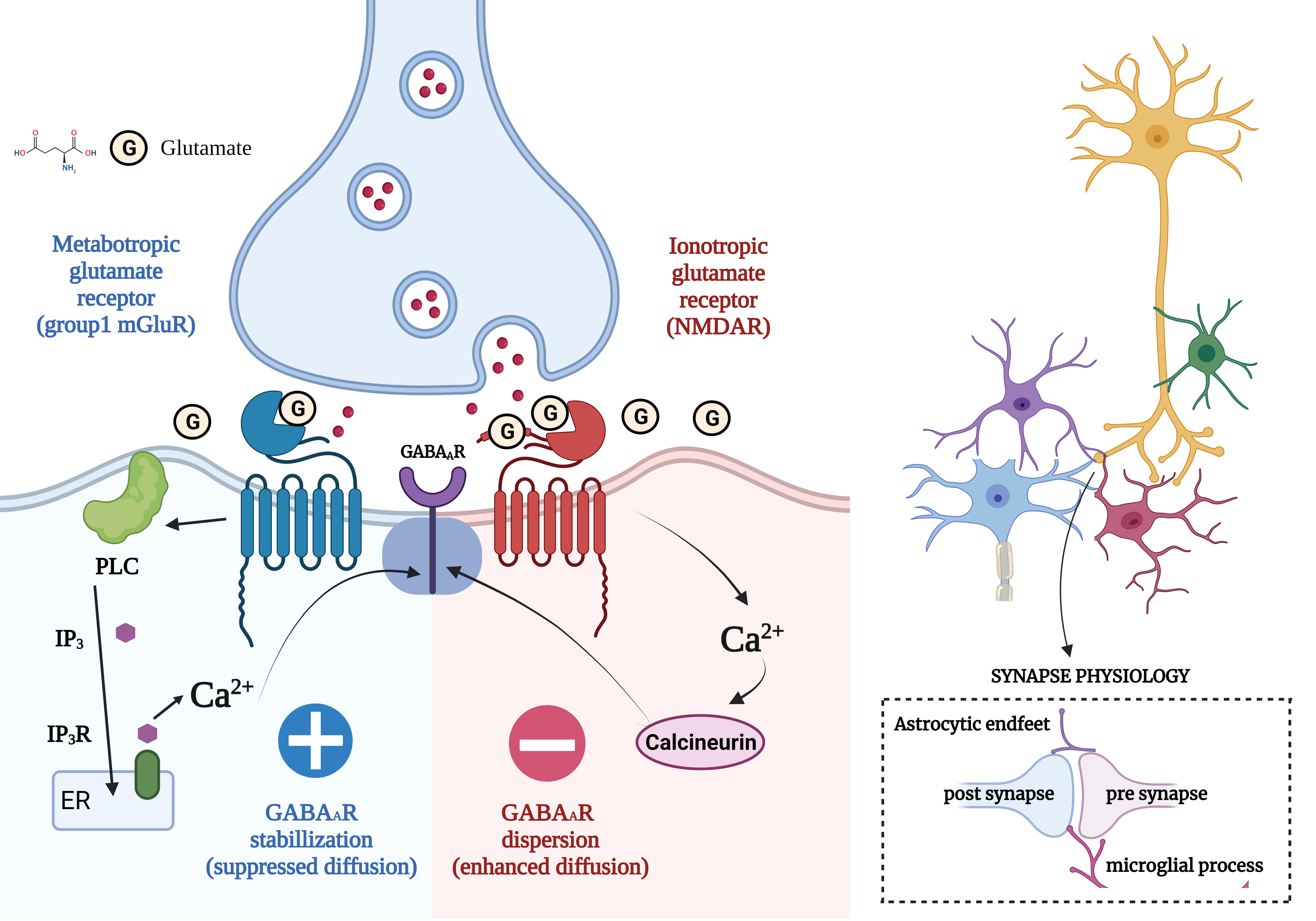 Fig. 1.
Fig. 1.Concept of Excitatory/Inhibitory Imbalance in Different
Neurological Disorders. (Figures were created with BioRender.com). Regular neuronal stimulation of N-methyl-D-aspartate
(NMDA) receptors by the neurotransmitter glutamate results above incoming
calcium, eventually promoting the sensors to be more dispersed and reducing the
amount of GABA that can suppress the neuron. GABAA synapses are generally
grouped. The receptors are continuously re-clustered to counteract this action,
maintaining the proper E/I equilibrium in the brain. In this route, glutamate
interacts with the mGluR, causing Ca to be released from internal storage and
released into the interior surroundings of the cell. This Ca interacts with
protein kinase C to encourage the aggregation of GABAAR at the post-synaptic
membrane, which has been shown using quantum dot-single particle monitoring. The
same neurotransmitter that causes GABAAR scattering from the synaptic space also
functions oppositely to stabilize GABAAR. It was also unexpected that the
procedures utilizing various Ca ions in the signalling pathways of the
cluster-forming passageway effectively prevented the distribution of GABAAR,
which is generally caused by extremely high levels of glutamatergic input, as
happens in epileptic seizures. Synapses are tri- or even quadripartite
arrangements in which glial pathways come into touch with elements of the neuron.
Additionally, the glial removes extra connections throughout maturation,
adjusting the excitatory/inhibitory balance in growing neural networks. GABA,
gamma-aminobutyric acid; GABAAR,
Research show that amyloid-beta (A
Neurons exhibit excitability and inhibition during neurodevelopment in the
forebrain are produced in distinct regions and undergo migration to atypical
locations which leads to the formation of heterotopias [137]. Heterotopias,
disorganized cortical patches and focal cortical dysplasia, are abnormalities
seen in ASD. However, E/I neocortical imbalance may be connected to pyramidal
glutamatergic neurons and the inhibitory GABAergic parvalbumin-positive
interneurons [138, 139]. ASD show modifications in gene transcription,
translation, and the degeneration of synaptic proteins known as neuroligins. This
modification has an effect on ion channels, receptors, cell adhesion, and
synaptic scaffolding proteins. Neuroligin is a cell adhesion molecule which is
specifically localized to excitatory synapses through the action of neuroligin
one, or to inhibitory synapses through the action of neuroligin 2 [140].
Neuroligin 3 is present in both excitatory and inhibitory synapses, whereas
neuroligin 4 has been observed to be specifically localized in glycinergic
synapses [141]. During in vivo study, mutation in the neuroligin 4
genes, which differs from that of humans [142, 143]. However, mice with a mutation
in neuroligin 1–3 exhibited impaired transmission of GABAergic/glycinergic
signals in the brainstem, both in response to external stimuli and spontaneously
[144, 145]. The regulation of synaptic function is influenced by neurexin, which
acts as a binding partner for neuroligin, and regulation is dependent on the
specific neurotransmitter involved [225]. The trans-synaptic adhesions formed by
neuroligin and its binding proteins, neurexins (Nrxns), have been implicated in
ASD by facilitating the recruitment of NMDARs and AMPARs to the synapse surface
[226]. A mutation in contactin-associated protein-like 2
(CNTNAP2), a protein belonging to the neurexin superfamily, is
identified as a cause of childhood epilepsy, hyperactivity, and Autism. The
absence of CNTNAP2 results in impairments in the process of neuronal migration
and the degradation of GABAergic cells [227]. Tuberous sclerosis complex (TSC)
plays a vital role as an inhibitory element within the mTORC1 pathway, which is
responsible for cellular growth and preserving the balance of synaptic activity.
The activity of the mTORC1 pathway is increased in ASD because of mutations in
the TSC gene. However, the functional examination of neurons with TSC mutations
has show conflicting results. The presence of a loss-of-function mutation in the
TSC gene has been linked to a multisystem illness is characterized by
neurological symptoms which is associated with ASD [228, 229]. Decrease in
excitability of TSC2-/- stem cell-derived neurons reported an elevation in
neuronal activity [230, 231, 232]. Significantly, the administration of rapamycin
effectively rectified aberrant excitability by suppressing mTORC1 in instances
when neuronal activity was heightened or diminished [233, 234]. The occurrence of
haploinsufficiency in the ARID1B gene, serves as a sequence-specific
DNA-binding subunit within the chromatin complexes of mammalian SWI/SNF
or Brg1-associated factors (BAF) and it contribute to
the development of ASD [235]. The occurrence of haploinsufficiency in
ARID1B is considered as a causative factor with decrease of total
GABAergic interneurons in the cerebral cortex, but cortical pyramidal neurons do
not show major alteration which is the concept of E/I [236] The cognitive
impairment observed in Arid1b-deficient mouse models resembled the behavioral
pattern associated with ASD [237]. Based on recent study, ARID1B regulatES
Wnt/
Epilepsy is distinguished by the occurrence of recurrent and unprovoked seizures
[239]. Epilepsy has a global prevalence rate of 1% among the general population
[238]. The equilibrium between E/I synapses is crucial for regulating synaptic
activity. And Epilepsy is characterized by an imbalance in electrical activity,
which arises due to a decrease in inhibitory GABA synaptic and voltage-gated
conductance, as well as an increase in excitatory glutamate synaptic and
voltage-gated conductance [239, 240]. Glutamatergic neuronal activity is referred
to as the glutamate hypothesis. And even basic anomaly in the pathogenesis of
epilepsy [240, 241]. The administration of Pilocarpine at a dosage of 40 mg/kg
intraperitoneally shows persistent seizure-like behavior in mice which is through
activation of the M1 receptor [242]. The resulting seizure-like behavior is
primarily mediated by the activation of NMDA receptors, which subsequently leads
to the development of temporal lobe epilepsy (TLE), secondarily generalized
seizures, and status epilepticus [130, 131, 132, 133, 134, 135, 136, 137, 138]. The activation of receptors has a
crucial role in the establishment of long-term synaptic plasticity and
coordinated firing, although its significance in the preservation of epileptic
convulsions is limited [139, 243]. High influx of calcium ions (Ca
Still, intense research is needed to understand the importance of glutamatergic and GABAergic systems in MS [252]. MS involves glutamate-mediated excitotoxicity, wherein neuronal death occurs due to excessive activation of glutamate receptors and subsequent increase in intraneuronal calcium levels [253, 254]. The extracellular level of glutamate is elevated due to the overproduction of glutamate by macrophages and microglial cells [255] and decrease in the breakdown of glutamate by oligodendrocytes [256]. The increase in extracellular glutamate levels initiate the glutamate signaling pathway by activating glutamate receptors present in neurons, oligodendrocytes, and astrocytes [257]. Therefore, the increase in extracellular glutamate adversely affects all of these cellular populations. Furthermore, an excessive amount of glutamate stimulates both NMDA and non-NMDA receptors, causing an aberrant buildup of intracellular calcium ions, which ultimately leads to neuronal harm [258, 259]. The utilization of magnetic resonance spectroscopy (MRS) imaging methods helps in identification of increased levels of glutamate in the active lesion located in the white matter. Conversely, individuals with MS exhibit a diminished concentration of glutamate in the grey matter when compared to individuals without the condition [260]. Based on findings, by decreasing the quantity of glutamate in the grey matter, it led to neuronal and synaptic degeneration [261, 262]. Excitotoxicity can arise not only due to an excessive concentration of glutamate, but also due to an inadequate presence of GABA to counteract excitatory effect. Even reduction in the extracellular concentration of GABA has been observed in individuals diagnosed with MS which is linked to deficits in both motor and cognitive functioning [263, 264]. The decrease in GABAergic neurotransmission is hypothesized to be facilitated by IL-1 in MS [265].
Proteinopathies, or the accumulation of toxic proteins, cause synaptic
dysfunction, which is linked to AD [266]. The amyloid hypothesis posits that
A
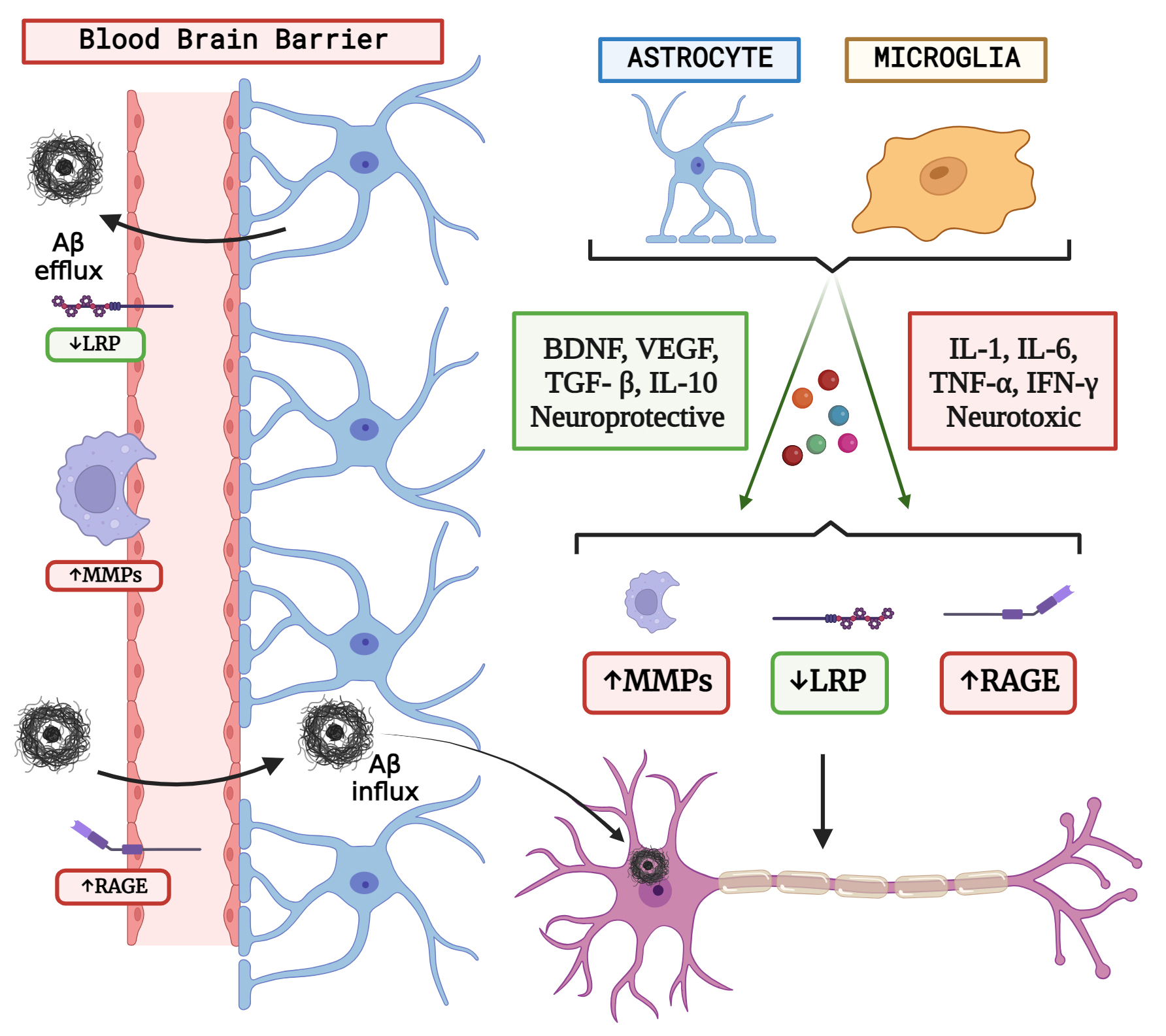 Fig. 2.
Fig. 2.The concept of neuron glial interaction in Alzheimer’s Disease. (Figures were created with BioRender.com).
A
ASD is caused by a combination of genetic and environmental factors and there is no single cause of ASD and it is thought that both genetic and environmental factor contribute to development of ASD [273, 274]. Glial cells even regulate and optimize several neurodevelopmental processes, including synaptic connectivity shaping [275]. The involvement of glial cells in the pathogenesis of ASD and the meticulous genetics in the etiology of ASD is unknown, as no solitary liable gene has been discovered thus far. Some of the genes linked to ASD risk perform a crucial part in brain development and thus are abundantly expressed in glial cells [275]. For instance, ASD has been linked to mutations in genes like PTEN, TSC1, TSC2, and NF1, linked to conditions like tuberous sclerosis and neurofibromatosis. These genes are essential for controlling the connection of neurons and the activity of glial cells. Gene changes that affect the ligand-binding molecules neuroligins (NL), for example, have been linked to ASD in humans. These NLs play critical roles in synaptic structure formation, maturation, and function. Indeed, genetic studies of rare ASD variants revealed a possible link to NL3 and NL4 mutations in humans. Transgenic mice with mutations like those found in ASD patients showed impaired social and stereotyping behaviours, indicating that NLs play a conserved role in determining ASD phenotype across species [275, 276, 277]. Increasing data shows that inflammatory NLs are disrupted in Autism, although neuroligins were also generated exclusively in astrocytic glial and oligodendrocyte progenitor cells [276, 277]. Enhanced soma, quick processes, extended filopodia, and elevated microglial cell density in the grey matter were all signs of activated microglia in ASD patients. Greater levels of pro-inflammatory cytokines/chemokines, such as IL-1 alpha, IL-6 alpha, IL-8 alpha, INF, and TNF, were identified that affect the ligand binding molecules to typically developing controls, confirming the neuroinflammatory status [277]. Glial cells are widely recognized as the primary contributor to the neuroinflammatory response mediated by IL-1 signalling. ASD is linked to mutants in the IL-1 cytokine receptor family. Individuals with Autism have both an uncommon mutant in the genome expressing for Interleukin-1 binding site accompanying associated protein, which is intensely concentrated in astrocytes, and a restriction fragment length polymorphism in the genes coding for Interleukin-1 binding site type 2, which is significantly concentrated in microglia [278]. Pre-clinical research has linked the mother’s nutrition during maternity, nursing, and child behavioural modification, as such changes appear to be linked to changes in the inflammatory response; neuroglia is most likely involved in this mechanism. Still, glial cells are highly dynamic elements of the CNS, and their implication in ASD needs further research [279]. The concept of neuron glial interaction in ASD is shown in Fig. 3.
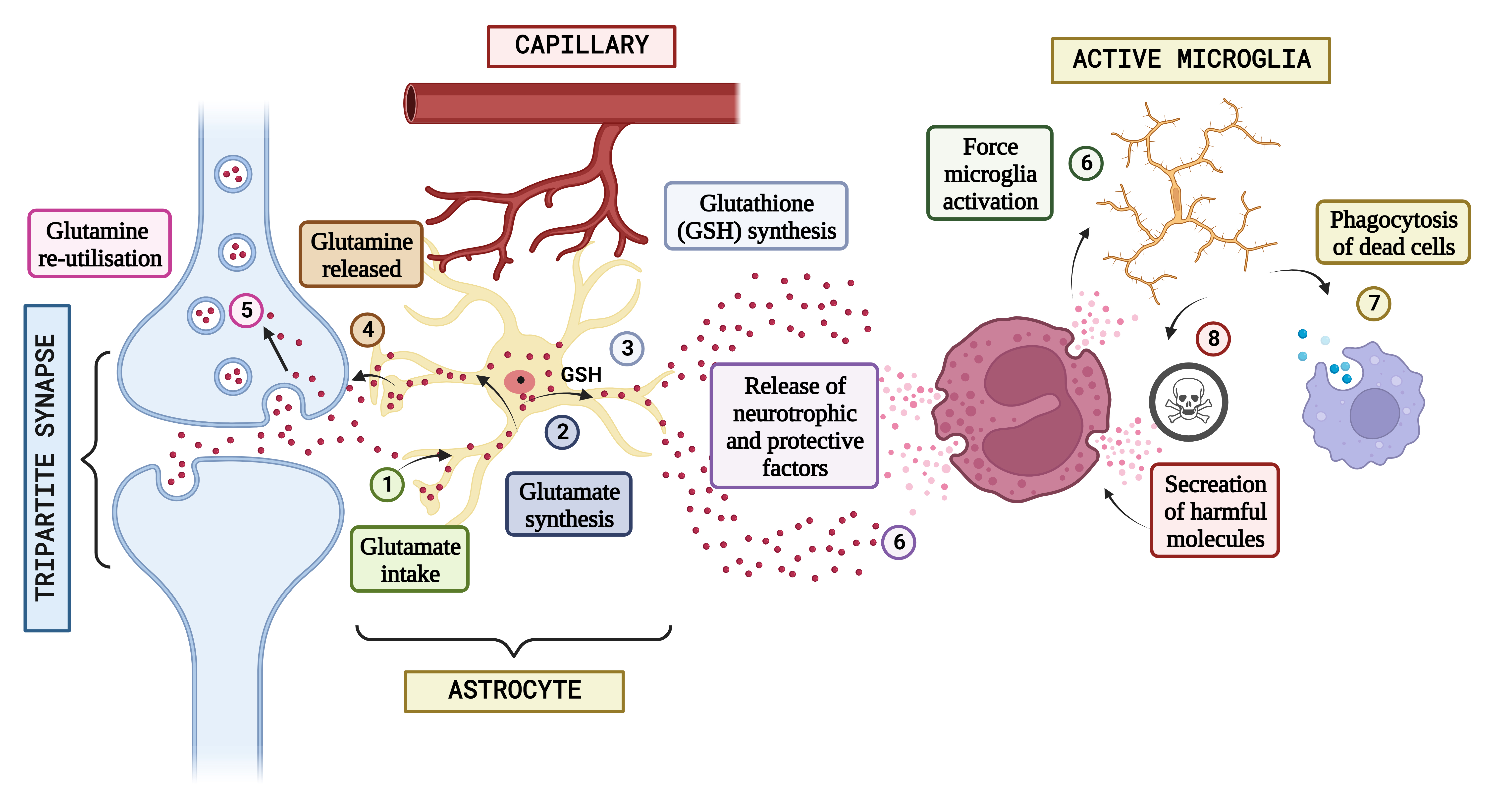
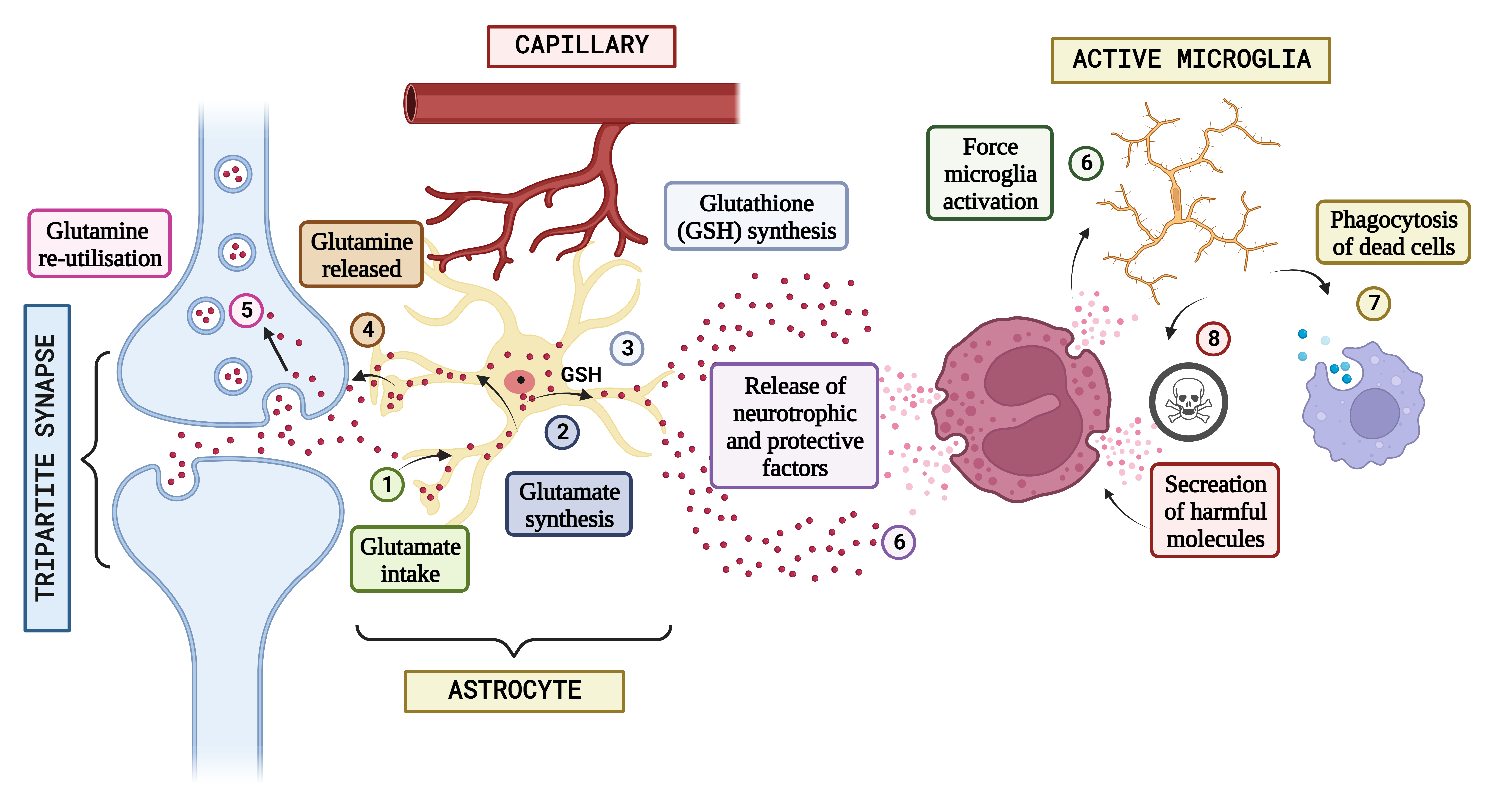 Fig. 3.
Fig. 3.Concept of neuron glial interaction in Autism Spectrum Disorder. (Figures were created with BioRender.com). Integrating neuron-glial connectivity and predicting glutamatergic transmission in the brain of patients having Autism. The pathogenesis of Autism is heavily influenced by glutamatergic communication. When glutamate is produced during neurotransmission in standard settings, astrocytes absorb it (1), which is often used to generate either glutamine (2) or glutathione (GSH) (3). After that, glutamine is liberated and used again by neural connections in the form of freshly produced glutamate (4 and 5), keeping the glutamatergic pathway. However, glial stimulation and increased astrocytic-mediated secretion of neurotrophic substances might result from epigenetic, hereditary, or environmental variables that impair neuronal propagation (6) and cause neuro-inflammatory processes that induce various amounts of stimulation in the microglia. It is hypothesized that activated microglia will scavenge cellular debris (7). In reaction to signs of neurotoxicity, the Central Nervous System (CNS) releases neuroprotective substances. Contrarily, the same microglial excitation might release potentially dangerous chemicals (8) such as NO, glutamate, pro-inflammatory mediators, free radicals, and others. The characteristic abnormal sociological phenomena seen in autistic individuals may be significantly influenced by a long-lasting interruption of neuron-glia connections and a defective release and action of neurotransmitter systems.
Based on previous studies, glial cells are linked to epilepsy pathogenesis, suggesting that targeting these cells could be used to supplement existing treatments [280]. Microglial involvement, as a major inflammatory cell in the epileptic brain and it may activate in response to the abnormal neuronal activity that occurs during epileptic seizures [281]. Neuroinflammation may result from releasing pro-inflammatory cytokines, chemokines, and other mediators by activated microglia. This neuroinflammatory response, in turn, can affect seizure activity and epileptic phenotypes in both favourable and unfavourable ways. Since microglia are central nervous system homeostasis controllers and convulsions and as seizures entwined with variations in neuronal functioning, microglial influences on epileptic phenotypes and repercussions are predicted [282]. On the one hand, epileptogenesis—the process of epileptic brain alterations and the shift from a single seizure to a chronic epileptic condition and the production of inflammatory chemicals by microglia can potentially worsen seizure activity [283, 284, 285]. On the contrary, microglia also contribute significantly to the control of synaptic connection, the promotion of tissue repair, and the resolution of inflammation. They have been demonstrated to participate in synaptic pruning, which removes extra or pointless synapses from the developing brain. Nevertheless, abnormal, or dysregulated microglial synaptic pruning may be a factor in the disruption of neuronal function in epilepsy. Microglial might well be able to manage neuronal processes in epilepsy [286]. Fractalkine receptor is expressed exclusively on microglial cells. Since chemokine (C-X3-C motif) ligand one is produced by brain cells, it is a fascinating and well-studied signalling pathway for interactions among microglial and nerve cells. Mice missing microglia ligands experience latency in the operational development of neural junctions, which disrupts neural activity [287]. The absence of postnatal excision reduced microglial colonization of the CNS, and a breakdown of the transition from Glutamate [NMDA] receptor subunit epsilon-2N to A N-methyl-D-aspartate subunits at the synaptic were all associated with chemokine signalling impairment. Impairment in anxiety, sensorimotor, and social behaviour has also been associated with chemokine receptor impairment and disturbances in neurogenesis, brain connection, and long-term stimulatory effects [288]. Microglia ablation techniques also raise concerns about local inflammation and astrogliosis. It is uncertain if additional glial purinergic channels are also implicated, even though microglial P2Y12 channels have been discovered as stimulators of epileptic movement morphologies. According to a new analysis, there was no discernible difference comparing wild-type and P2X4 defective rats undergoing abrupt convulsions, suggesting that glial P2X4 sensors do not influence rapid seizure behaviours. Finally, while P2X7 receptor function is thought to be neurotoxic during epilepsy, the evidence for the role of microglial-specific P2X7 receptors in epilepsy is currently being debated in addition to acute seizure phenotypes, ed. Microglia perform a crucial role in seizure-induced neurodegeneration [289, 290]. According to these findings, microglia are crucial in controlling both the long-term and short-term aspects of sudden seizures and their effects on neuronal maintenance and growth. After CNS is affected, activated glia is essential for the recovery of neuropathological disorders. On the contrary, if left uncontrolled, severe reactive gliosis may significantly contribute to the neuropathology of epilepsy. Finally, more study is required to thoroughly understand and comprehend the role of microglia during acute seizures [291]. The concept of neuron glial interaction in epilepsy is shown in Fig. 4.
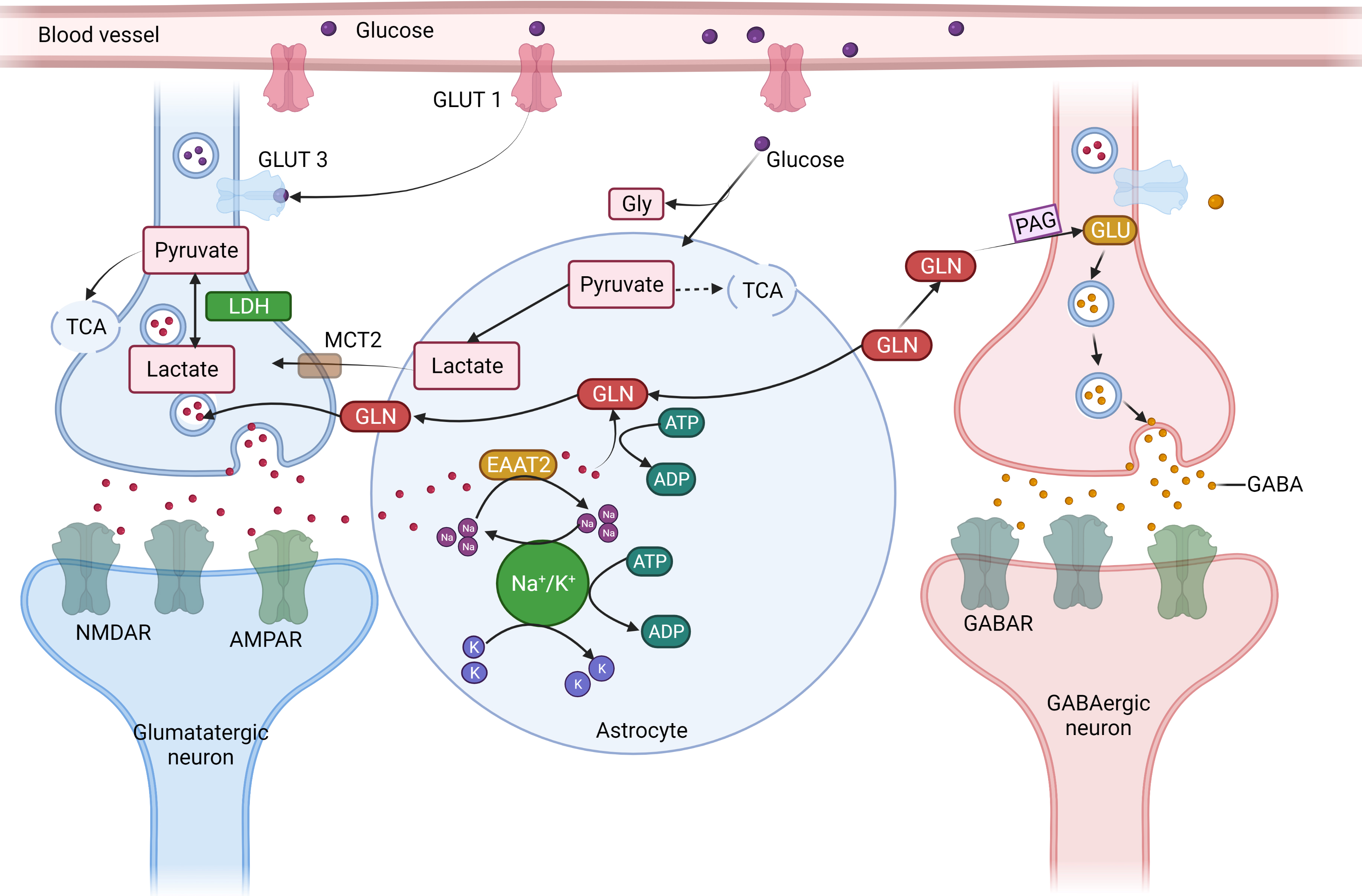
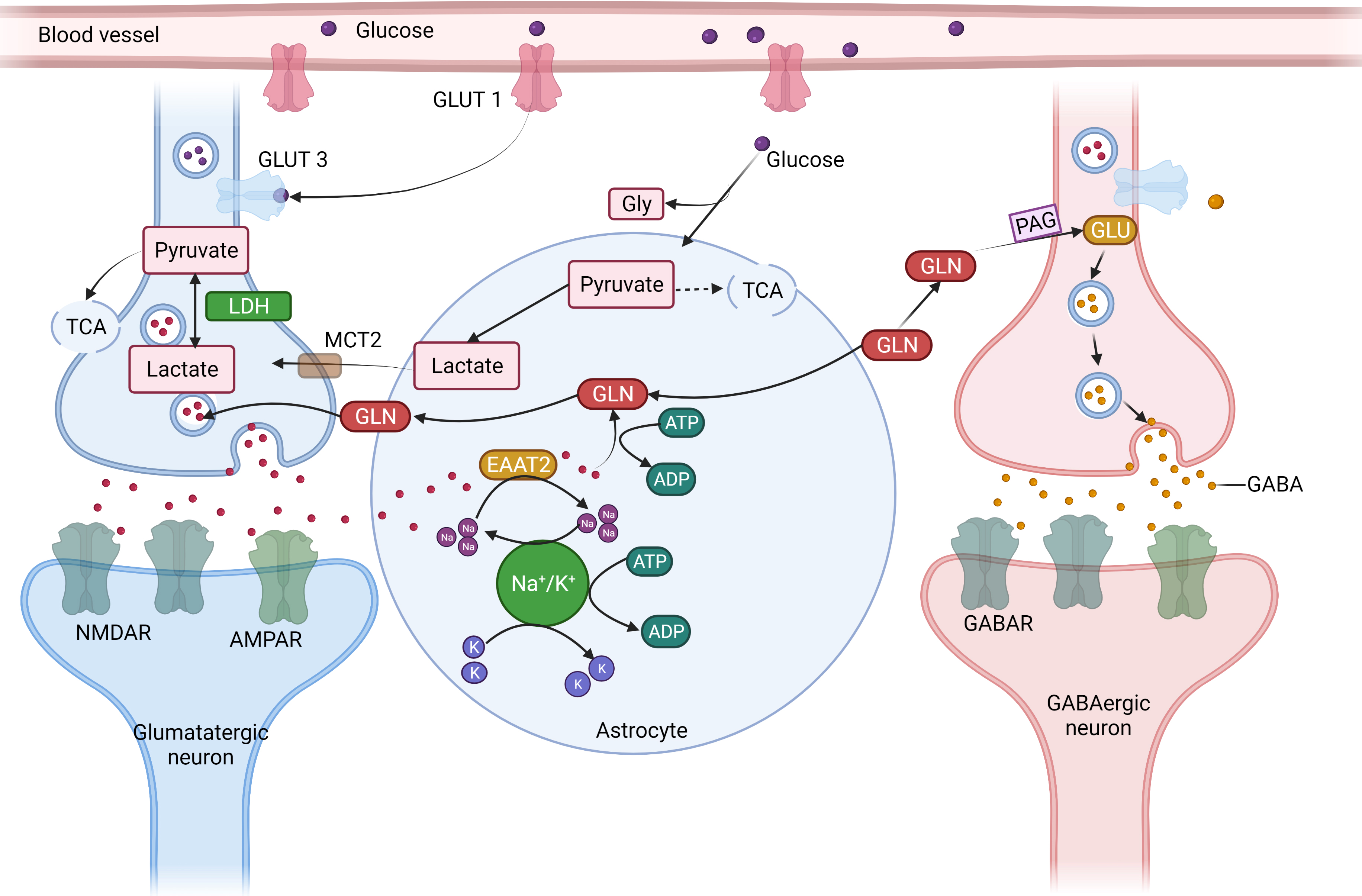 Fig. 4.
Fig. 4.The concept of neuron glial interaction in epilepsy. (Figures were created with BioRender.com). Astrocytes
control the equilibrium of synaptic transmitters and active intermediates.
Through the glutamate (GLU)-glutamine (GLN) cycle, astrocytes play a crucial role
in preserving the ideal amount of extracellular Glu. Astrocytes take up (1)
Excessive extracellular Glutamate via an excitable amino acid channel after the
signal transduction. (2) GLN synthetase (GS) produces GLN, which is then
transported to the nerve cells. (3) Phosphate-activated glutaminase (PAG)
converts GLN to GLU, which is subsequently packed in synaptic vesicles in
glutamatergic synapses. (4) GLU is further transformed by the enzyme Glu
decarboxylase (GAD) in GABAergic neurons, producing GABA, which is secreted at
the synapses during inhibiting neurotransmission due to the energy-intensive
aspect of the process and the requirement of Adenosine triphosphate for both
excitatory amino acid transporter two and GS, complete removal of extracellular
Glu following enhanced synaptic transmission places energetic stress on
astrocytes. (5) Energy mediator lactate and pyruvate are formed by the action of
the glycolytic conversion of glucose that is enzyme-derived from astrocyte
glycogen reserves and absorbed from the blood through glucose transporter 1
(GLUT1). (6) Because astrocytes have a limited ability to oxidize substances,
lactate dehydrogenase 5 (LDH5) and other enzymes primarily turn pyruvate into
lactic (7) transported by monocarboxylate transporters (MCT) into the
neighborhood neurons. (8) Lactate dehydrogenase 1 catalyzes the process in
neurons where lactate is transformed into pyruvate, which is then utilized as an
energy source by the tricarboxylic acid (TCA) cycle. Astrocyte neuronal lactate
shuttling, or ANLS, is an essential mechanism for generating power to neurons
during intense neuronal transmission, such as during epilepsy (9). Furthermore,
due to the cell’s low glycogen synthesis capitals and the absence of glycogen
reserves, neurons primarily get pyruvate from astrocytes via the ANLS route.
Glucose is also absorbed by neurons from blood arteries using GLUT3. Inflammation
which is driven by cytokines like IL-1 and TNF-
The brain has an immune system, which may encourage or block regeneration as a suitable reaction. In contrast, an inability in this response causes neuronal death, which is frequently found in MS [292, 293]. The immune system incorrectly targets and assaults the myelin sheath, which serves as the central nervous system’s safeguarding layer of nerve fibres, in MS. This immune reaction causes inflammation and demyelination, impairs nerve signal transmission and, in some cases, results in neuronal death. Microglia and invading immune cells release pro-inflammatory mediators and contribute to neuroinflammation when the neuroimmune response is dysregulated in MS. Myelin degeneration. Neuronalry potential consequences of this persistent neuroinflammation. Neurodegenerative illnesses of the CNS, such as MS, are characterized by leukocyte invasion with few T-lymphocytes and peripheral blood mononuclear cell (PBMC), as well as a significant glial activation of the CNS. Neuro-inflammatory mechanism stimulation phases cause the remyelination processes that characterize MS. There have been studies that back up the theory that the autoimmune response starts in the CNS [294, 295]. Others, on the other hand, believe the initial activation takes place outside the CNS. To develop MS, a primary neuron disorder must exist within the CNS. In an operational setup with the inoculum of neural cells, even glia produces major histocompatibility complex-II molecules following activation with IFN- when substantially isolated from electroactive neurons. Glial cells expressed MHC-II throughout the crop culture after adding tetrodotoxin (TTX) to hinder the action potential of neurons. Treatment with TTX inhibited myelination, indicating that the production of MBP by oligodendrocytes depends on neuronal electrical activity. Numerous investigations have shown that myelination is regulated by neuronal electrical activity. In these investigations, TTX was utilized to decrease electrical activity in neurons and stop neuronal action potentials. The result was a reduction in MBP synthesis and subsequent myelination. Investigations are ongoing to determine the precise methods by which neuronal electrical activity affects myelination. However, it is thought that neuronal activity can affect the release of particular signaling molecules or elements that control oligodendrocyte activity and myelin synthesis. These elements could consist of neurotransmitters, growth factors, or other substances involved in cell-cell communication. The precise developmental stage, brain location, and cell types can impact the link between neuronal activity and myelination, which is probably complex and can change. It is possible that the effects of TTX on myelination in experimental models do not completely replicate the physiological circumstances in the human brain. It has also been suggested that the first event in MS is an operational interference or damage to myelination’s ability [296, 297]. Initially, no morphological changes were associated with this, but later, myelin degeneration occurred. As a consequence, neurons, astrocytes, and endothelium may display allergenic epitopes that were initially inaccessible to the immune function in the setting of specific human leukocyte antigens molecules. Eventually, they penetrate the central nervous system, and T-lymphocytes that can recognize all of these hidden antigens start an immune condition [298, 299]. The pathogenic mechanism is sped up by cytotoxic T cells, macrophages, and cytokines, which can impede bare axon activity. The CNS has previously been viewed as a specialized system in considerations of immunological irrespective of the CNS because it tends to lessen and restrict innate immunity reactivity. This is due to the possibility of permanent neuron death if the CNS has a significant inflammatory process. Low human leukocyte antigens-I&II expression, low signal transduction effectiveness, and the emergence of solubility substances in CSF that neutralize or reverse the actions of some pro-inflammatory mediators all contribute to this result [300, 301]. Many writers propose that demyelination starts after active lymphocytes penetrate the CNS since, in this situation, the CNS would not be the ideal location to initiate an immune response that demands higher effectiveness. In this context, we found that MS individuals’ peripheral blood also began lymphocytes with phenotypically resembling encephalitogenic cells. It is suggested as a middle ground between the two possibilities that antigenic release through some (mainly olfactory) cranial nerves via lymphatics, where effective antigen-presenting would happen, happens due to an earlier engagement in the CNS. The theory proposes that lymphatic capillaries found in the meninges, the protective membranes enclosing the brain and spinal cord, may be able to trap antigenic material emitted by the olfactory nerve or other cranial nerves once it has entered the CNS. These lymphatic tubes are a component of the recently identified glymphatic system, which allows the interchange of fluid and molecules between the peripheral lymphatic system and the central nervous system, as well as the removal of waste materials. If meningeal lymphatics take in antigenic material from the CNS, it may be transferred to lymph nodes, home to antigen-presenting cells like dendritic cells. These antigen-presenting cells can process the antigens that have been collected and then deliver them to T cells to start an immune response. Excluding the target tissue, the CNS, and the existence of the BBB, the sequence of processes and components in an immune reaction are virtually the same in anybody else’s non-autoimmune responses. Many researchers support this data, and each link in the circumstances is examined to pinpoint the comparative relevance of signalling pathways or mediators that suppress innate immunity as precisely as feasible [48, 49, 50, 51]. Investigators now understand the ontogeny, diversity, and functioning of glial cell types in inflammation situations with concurrent demyelination because of recent advances in multiplexed and single-cell omics. Recent discoveries of sequences of gene activation and transcriptome tissue segmentation, which also included postmortem MS tissues, have increased our understanding of glial subgroups throughout numerous CNS locations at various states of activation, but they have also brought new challenges. Contrary to the commonly held idea that the glial reaction is solely reactive, active microglia may also serve as effector cells, regulating and prolonging the immune system response and tissue damage [298, 299, 300, 301, 302]. Reactionary glial subcategories frequently deliver antigens via major histocompatibility complex I and II proteins, demonstrating that both reacting OL and astrocyte progenitor cells play significant roles in immune-related processes. Glial cells may phagocytose antigens and display them on major histocompatibility complex-II molecules, just as expert allergen cells like dendritic cells, which cause a particular immune system response. Oligodendrocyte progenitor cells (OPCs) that exhibit MHC class I are more susceptible to immune cell cytotoxicity during inflammation than microglia and DCs, which may not be as skilled at antigen-presenting via MHC class I as Dendritic cells. Another instance of improper immunological signalling is the release of neurotoxic substances like complementing cascade constituents, which can cause synaptic disease and accelerate neurodegeneration [297, 298]. Additionally, it has been shown that a range of CNS glial subtypes exhibits several MS susceptibility gene variations in individuals that were assistive devices to peripheral immune responses, including some that control Nuclear factor-B in astrocytes. Different microglial types have different functions in the pathophysiology of MS, many of which are hostile. As indicated by abnormal immunological activation that can damage neurons through complementing mediators and self-destruction via primary histocompatibility complex class I recruitment and activation, these activities can be detrimental and, in certain circumstances, result in the development of a pro-inflammatory, tissue death, and CNS degeneration [295, 296, 297, 298, 299]. However, pro- and anti-inflammatory neuronal subgroups can be essential in removing waste and encouraging repair, as shown by pro-regenerative myeloid cells or astrocyte-mediated myelin phagocytosis. Because the single cell type is believed to play several functions, addressing these variants to slow neurodegenerative and boost healing will be challenging. Finding treatment targets unique to a specific glial subtype will need an awareness of the distinct molecular areas of glial subtypes in the pathogenesis of MS symptoms. This is where MS pathobiology varies from other predominantly aggressive or neurodegenerative illnesses, such as AD, which do not have repeated stages of inflammatory demyelinating lesions that can damage the whole CNS. The intrinsic transcriptional and operational distinctions of glial subtypes among anatomical categories and CNS regional groups must be considered to translate glial subtype variety and functioning in MS [298, 302]. Given that MS is a relapsing-remitting disease, it is also possible that glial cells cycle between different subtypes depending on the stage of the inflammatory phase. Therefore, future studies must identify these subtype-specific crucial health regulators and develop therapeutic strategies to alter glial subtype-specific networks along spatial and temporal MS development pathways. This may also enable us to comprehend and promote anti-inflammatory and pro-regenerative cell subgroups, speeding up MS recovery. The Toll-like receptors- TRIF Interferon regulatory factor 3 pathways, which regulate caspase11 transcription, produce the pro-inflammatory mediator type I-IFN and Nuclear factor, which increase caspase11 synthesis. Recent research indicates that the non-canonical transit of the pro-inflammatory mediators, which contain Caspase11, is crucial for the beginning of seizures [299, 300, 301, 302]. The concept of neuron glial interaction in Multiple Sclerosis is shown in Fig. 5.
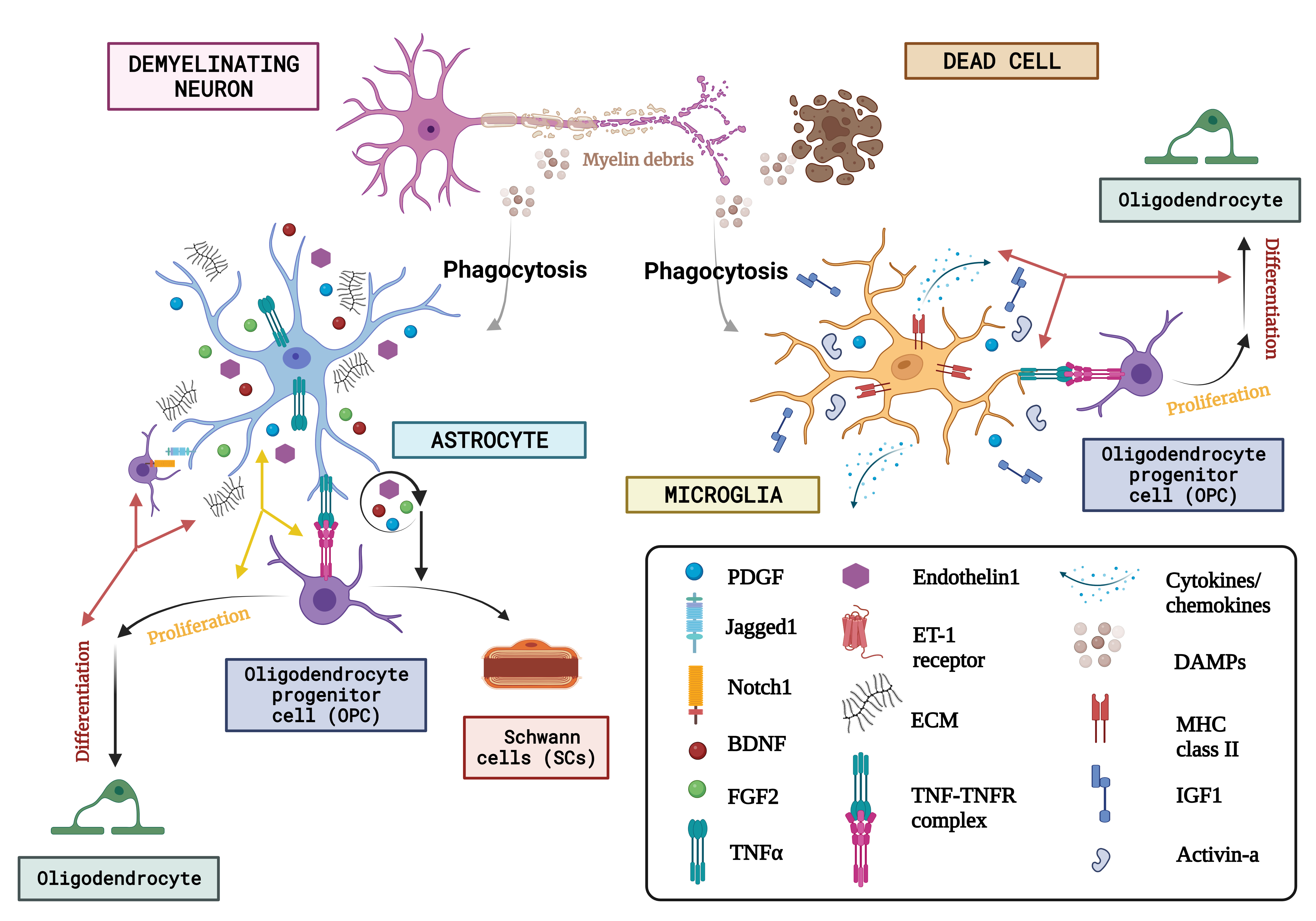 Fig. 5.
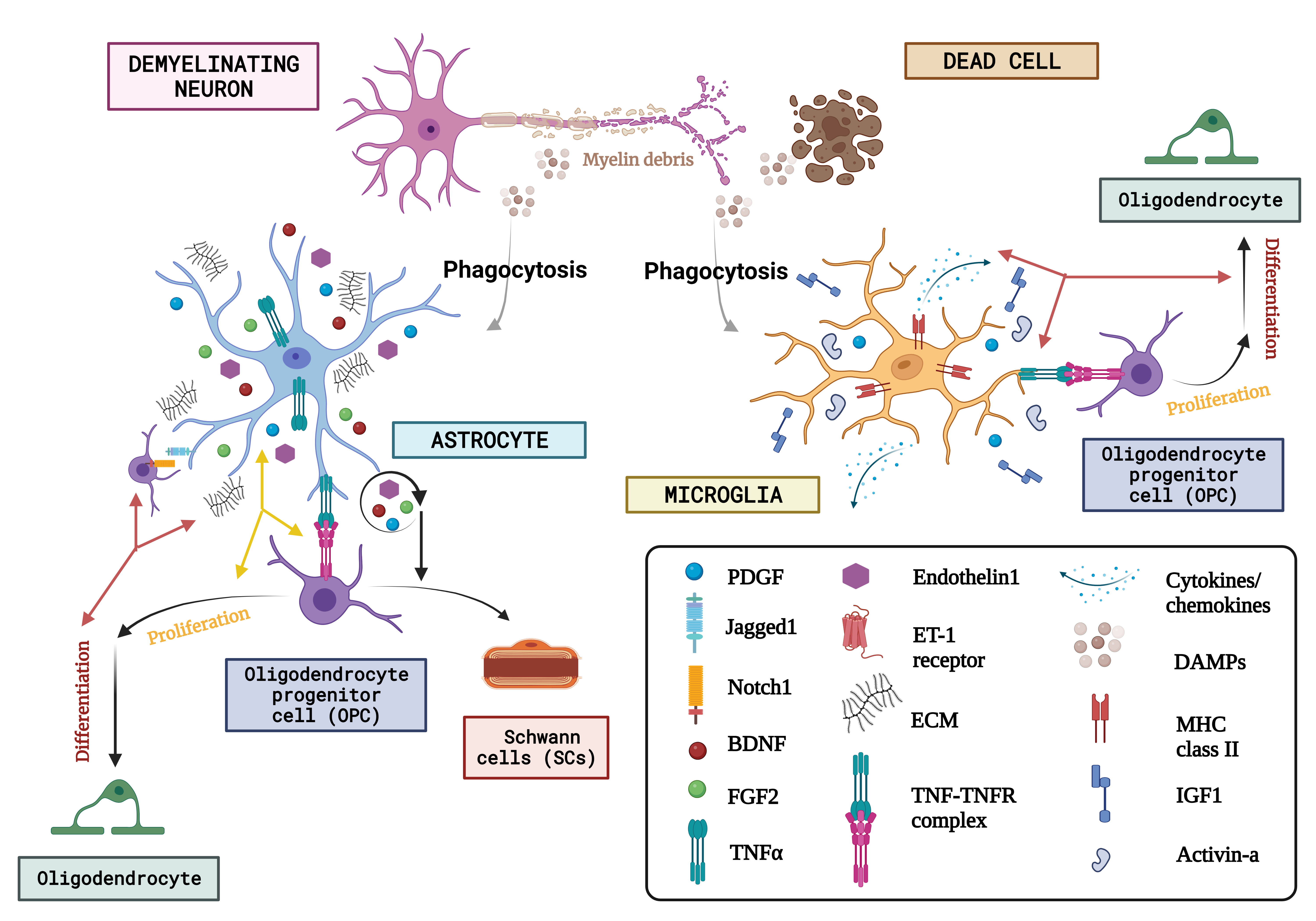
Fig. 5.The concept of neuron glial interaction in multiple sclerosis. (Figures were created with BioRender.com). Through the affirmation of extracellular matrix (ECM) compounds like chondroitin sulfate glycosaminoglycans (CSPGs), hyaluronan, fibronectin, and tenascin C, endothelin-1 and growth factor exudation including such Fibroblast growth factor-2 (FGF2), BDNF, and astrocytes, Oligodendrocyte progenitor cells behaviour is regulated after neurodegeneration. Microglia are discerned as DAMPs (damage-associated molecular patterns) via various receptor classes. Microglia recognize abnormalities in the aftermath of demyelination. Via paracrine and autocrine signalling, endothelin-1 increases the production of the notch ligand jagged. OPCs’ destiny selection after demyelination is governed by astrocytes, who oppose their development into Schwann cells (SCs). On the other hand, astrocytes have only been shown to phagocytose myelin detritus in culture. Numerous actions are induced by DAMPS, one of which is the stimulation of microglia to phagocytose myelin and dead cells. Once energized, microglia also increase the expression of the utterance of a variety of immunologic molecules like MHC-II and cytokine release, chemokines, and growth regulators like Activin-A, platelet-derived growth factor (PDGF), IL-1, and IGF-1 that control the lineage advancement of OPCs. Finally, they both produce tumour necrosis factor (TNF), which interacts with TNF receptor 2 (TNFR2) and encourages remyelination. ET-1, endothelin 1; DAMPs, danger associated molecular patterns; MHC-II, major histocompatibility complex class II; IGF1, insulin-like growth factor 1.
Overall, this review has elucidated the inherent significance of glial cells, specifically microglia and astrocytes, in the etiology of diverse neurological disorders, such as AD, ASD, epilepsy, and MS. Our comprehension of the intricate interplay between neurons and glia, as well as its involvement in neuroinflammation and neurodegeneration, has revealed potential targets for therapeutic intervention. Synaptic dysfunction may arise as a consequence of dysregulation in proteinopathies and innate immune function in AD, or as a result of neurodevelopmental processes and inflammation in ASD. An in-depth examination of epilepsy and multiple sclerosis MS has revealed the significant influence of microglia activation, leukocyte infiltration, and other inflammatory mechanisms on neurological functions. Therapeutic approaches may show potential by focusing on the targeting of neuroinflammation and inflammatory mediators. The presence of an imbalance between excitation and inhibition has been identified as a significant factor in the development of these disorders. It has been observed that changes in genes associated with neurotransmission can possibly result in either neurotoxicity or neuroprotection, depending on the specific pathway and characteristics of the disorder. The crucial involvement of GABAA receptors in the initiation of conditions such as AD and epilepsy has been firmly established. Nevertheless, further investigation is required in order to comprehensively comprehend the intricate molecular connections, imbalances, and inflammatory responses associated with various neurological disorders, with the ultimate goal of developing efficacious therapeutic interventions.
CNS, Central Nervous System; AD, Alzheimer’s Disease; ASD, Autism Spectrum
Disorder; MS, Multiple Sclerosis; ROS, Reactive Oxygen Species; NF-
SRA and MZ designed the research study; performed the research, AMA, AD, HA, AM, AAGA, UR, MM, SA, ERH analysed & interpreted the data, SRA, UR, MM, SA, ERH wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have significantly contributed to this work and agreed to be accountable for all its aspects.
Not applicable.
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University, KSA, for this support.
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University, KSA, for funding this work through large group Research Project under grant number RGP2/355/44.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.