1. Introduction
Alzheimer’s disease (AD) is an age-related progressive neurodegenerative
disorder characterized by cognitive dysfunction and behavioral impairment. In
addition, AD is the primary cause of dementia [1]. The two typical
neuropathological changes in AD include neuritic plaques formed by the deposition
of beta-amyloid (A) in the brain parenchyma and intracellular Tau that makes up neurofibrillary tangles (NFTs)
formed by the accumulation of hyperphosphorylated tau proteins [2, 3].
The plaques of A also deposit on vessel walls causing cerebral amyloid
angiogenesis (CAA) [4, 5]. However, the A plaques in AD and CAA are
distinct. A1-40 and A1-42 are the most common subtypes of
A peptide. A1-42 is more prone to form insoluble aggregates in
the parenchyma [6], constituting the major component of neuritic plaques in AD.
In contrast, A1-40 aggregates more slowly and ultimately deposits in the
walls of vessels through perivascular drainage [4, 7, 8]. Abnormal perivascular
drainage is the main pathogenesis of CAA [4, 9, 10], which also explains why the
A deposited on the vascular wall in CAA is mainly A1-40. Neuron
loss and synapse dysfunction caused by the toxicity of A will ultimately
contribute to dementia and degeneration of the central nervous system (CNS) [11, 12]. Moreover, a substantial body of research has demonstrated that the
occurrence of CAA and AD largely overlap [13, 14, 15]. Cerebrovascular dysfunction
caused by CAA is related to severe cognitive impairment in AD patients [15, 16, 17, 18].
The shared role of A in AD and CAA is likely the most apparent
interaction between neurodegenerative diseases and cerebrovascular diseases.
Moreover, there is evidence indicating that vascular dysfunction plays a
significant role in the onset of AD. Recent studies have demonstrated that a
reduction in the cerebral blood flow (CBF) is the earliest detectable clinical
change in mild cognitive impairment and AD patients [19, 20, 21] and that capillaries
exhibit focal constriction. The greatest vascular resistance occurs in the
capillary bed rather than in the penetrating arterioles [22]. At the capillary
level, the neurovascular unit (NVU) is composed of endothelial cells (ECs),
pericytes, glial cells and neurons. Pericytes are the only contractile cells
responsible for regulating blood flow in capillaries. Pericytes likely plays an
important role in the pathogenesis of AD. Pericytes, which are indispensable
components of the NVU, play an essential role in the formation and maintenance of
the blood-brain barrier (BBB), the regulation of the CBF, angiogenesis and the
phagocytosis of toxic substances including A from the brain. A
significant loss of pericytes in AD patients has been observed, and the
accumulation of A may be the potential cause. Conversely, the loss of
pericytes could lead to impaired clearance of A, exacerbating the
deposition of A and leading to a vicious cycle.
Currently, there are no effective drugs that can effectively reverse cognitive
decline and there are no therapeutic strategies targeting pericytes [23]. Further
understanding of the pathological changes in pericytes in AD and the interactions
between pericytes and A may provide new therapeutic directions for the
prevention and treatment of AD. In this review, we summarize the characterization
of pericytes, the signaling pathways linking pericytes and other cells in the
NVU, the physiological effects of pericytes, the functional changes in pericytes
in AD, the pathways through which pericytes clear A, the effects of
A on pericytes and the current strategies for preventing or treating AD
targeting pericytes.
2. The Characterization of Pericytes
Pericytes were originally characterized by Eberth and Rouget in the 1870s
(Eberth, 1871; Rouget, 1873) and firstly named by Zimmermann in 1923 based on their
location within the vascular basement membrane (BM) and the extension of
cytoplasmic processes to wrap ECs. Both pericytes and vascular smooth muscle
cells (VSMCs) are called mural cells [24]. In addition to ring-shaped VSMCs with
circumferential processes on arteries and arterioles [25], pericytes are
classified into three subtypes based on their morphology and location:
ensheathing pericytes which have more circumferential processes on precapillary
arterioles; thin-strand or helical pericytes, which have protruding nuclei and
longitudinal processes on the middle capillary, which is the most widely accepted
morphology of pericytes; and stellated pericytes on the postcapillary space [26, 27].
Many cell surface proteins such as platelet-derived growth factor
receptor- (PDGFR-), neural/glial antigen 2 (NG2) and CD13
[28, 29, 30, 31, 32, 33] are expressed on both pericytes and VSMCs, and these two cell types can
be distinguished by morphology. Additionally, vitronectin (VTN) and
interferon-induced transmembrane protein 1 (Ifitm-1) label pericytes specifically
[31]. However, there is currently a lack of specific markers for distinguishing
subpopulations of pericytes. Pericytes and VSMCs exhibit contractile alpha-smooth
muscle actin (-SMA) and desmin expression [26, 30, 34]. Notably, there
is a difference in the expression level of -SMA between the subtypes of
pericytes, which may be related to their distinct functions [27, 30]. These
markers, especially PDGFR-, NG2 and -SMA, are widely applied
in studies. PDGFR- can outline the contours of pericytes [35]. Because
they are labeled by PDGFR-, pericytes are easily recognized by their
protruding soma. Therefore, relying solely on morphology is sufficient to
reliably identify pericytes [26, 27]. However, it is worth noting that adequate
experience is needed for observers [36]. NG2 is the first discovered marker of
pericytes that can be used to identify pericytes through combination with
morphology, but not all pericyte subsets express NG2 [37]. -SMA is not
sensitive enough to identify pericytes in capillary beds, because pericytes on
precapillary tubes express more -SMA while pericytes on capillary beds
may be negative [27, 30].
3. The Interactions between Pericytes and other Cells in the
Neurovascular Unit
The NVU is composed of endothelial cells, mural cells (vascular smooth muscle
cells, pericytes), glial cells (astrocytes, microglia, oligodendrocytes) and
neurons [38, 39, 40]. The cellular components vary with the branching of the cerebral
vascular tree. At the capillary level, pericytes are located centrally between
endothelial cells, the endfeet of pericytes and neurons, and the BM is shared
with pericytes [26, 41]. They communicate with their neighboring cells and
generate corresponding responses which are crucial for normal functions of the
CNS [26]. We reviewed the interactions between pericytes and ECs, astrocytes and
neurons in Table 1 (Ref. [26, 35, 38, 42, 43, 44, 45, 46, 47, 48, 49, 50]).
Table 1.The interactions between pericytes and other cells in the NVU.
| Cell type |
Signaling pathway with pericytes |
Functions |
Ref |
| ECs |
PDGF-BB-PDGFR pathway |
PDGF-BB secreted by ECs combines with PDGFR on pericytes in high affinity. PDGF-BB-PDGFR signaling promotes pericytes survival, proliferation and migration. |
[26, 35, 42] |
|
TGF-–TGFR2 pathway |
TGF- is activated though interactions between ECs and pericytes to promote proliferation and differentiation of pericytes, and stabilization of vessels. |
[43, 44] |
|
Ang-Tie2 pathway |
Ang1 secreted by pericytes combines with Tie2 on ECs. Ang-Tie2 signaling regulate angiogenesis and vascular permeability. |
[26, 35, 45] |
|
VEGF-A-VEGFR2 pathway |
VEGF-A secreted by pericytes and ECs to promote the survival and proliferation of pericytes as well as angiogenesis. |
[26, 38, 46] |
| Astrocytes |
CypA–NFB–MMP-9 pathway |
ApoE secreted by astrocytes interacts with LRP1 on pericytes triggering the degradation of the extracellular matrix and tight junction. |
[26, 47, 48] |
|
|
Pericytes regulate the AQP4 distribution of astrocytes to regulate the polarization of astrocytic endfeet. |
[49, 50] |
| Neurons |
|
Pericytes secrete neurotrophic factors to promote the survival of neurons whereas neurons secrete neurotransmitters to regulate pericytes contractility. |
[26, 50] |
Abbreviations: NVU, neurovascular unit; EC, endothelial cells; PDGF-BB,
platelet-derived growth factor-BB; PDGFR, platelet-derived growth factor
receptor-; TGF-, transforming growth factor-;
TGFR2, transforming growth factor- receptor 2; Ang1,
angiopoietin-1; Tie2, tyrosine protein kinase receptor; VEGF-A, vascular endothelial growth factor-A; VEGFR2, vascular
endothelial growth factor receptor 2; CypA, cyclophilin A; NFB, nuclear
factor kappa-B; MMP-9, matrix metalloproteinase-9; ApoE, apolipoprotein E; LRP1,
LDL receptor-related protein-1; AQP4, aquaporin 4.
4. The Functions of Pericytes and Dysfunctions in AD
As pericytes are indispensable components of the BBB and NVU, we review the
roles of pericytes in the CNS and their dysfunctions in AD.
4.1 Regulation of the Cerebral Blood Flow (CBF)
Mural cells are cellular components with contractile properties in the NVU, that
enable them to regulate vascular tone and the CBF [22]. As pial arteries branch
into arterioles and capillaries after penetrating into parenchyma, the mural cell
population composed of the NVU changes [51]. Penetrating arteries consist of one
to three layers of VSMCs while arterioles contain only one layer [41, 51]. After
descending to capillary level, pericytes replace VSMCs and embed within the
endothelial BM [51]. Previously, the CBF was shown to be regulated solely by
VSMCs [52]. However, with the study of pericytes, this viewpoint has been
challenged.
In a series of studies, pericyte were confirmed to constrict or dilate in
response to neurotransmitters [53], for example, glutamate evokes pericyte
dilation, and pericytes constrict in response to a gamma-aminobutyric acid (GABA)
receptor blocker suggesting that pericytes participate in the regulation of the
CBF [52, 54, 55, 56, 57]. Moreover, an in vivo experiment in which mice expressed
DsRed in pericytes, revealed that capillary dilation precedes penetrating
arterioles, demonstrating that capillary dilation is a result of active
relaxation of pericytes rather than a passive response to elevated blood pressure
caused by arteriole dilation [52]. In ischemic stroke, capillaries constrict
segmentally at regions near pericytes, after which pericytes contract and
subsequently die rigidly [58]. Damages to pericytes contributes to long-lasting
microcirculatory reflow impairment even after reperfusion [59].
Pericytes degeneration and neurovascular dysfunction have been observed in AD
[51, 52]. Moreover, the oxidative stress caused by A leads to capillary
constriction. A reduction in the CBF reduces the oxygen supply and glucose
availability to the brain, resulting in the impairment of neurons and
neurodegenerative changes [60]. A recent study showed that white matter lesions
(WMLs) induced by persistent cerebral hypoperfusion is a driving factor for
dementia [61]. Moreover, the two-hit vascular hypothesis suggests that prior to
neurodegeneration and cognitive impairment, genetic, vascular and environmental
factors cause vascular damage (hit1), and neurovascular dysfunction contributes
to the accumulation of A (hit 2) [40, 41]. A previous study using
APP Pdgfr mice found that the loss of pericytes results
in a series of AD-like neurodegeneration pathological changes including
accelerated A deposition, tau pathology and neuronal dysfunction [62].
Notably, vascular damage (hit1) has been observed in pericyte-deficient
APP mice, indicating that vessel damage and pericyte degeneration may be
mutually causal [62]. Together, these findings suggest that pericyte degeneration
is an early and key event in AD neurodegeneration.
However, due to differences in stimulation methods, transgenic mice, and other
factors, the function of pericytes in regulating the CBF has not been fully
elucidated.
4.2 Formation and Maintenance of the Blood-Brain Barrier (BBB)
The blood-brain barrier (BBB) is a special protective barrier that exists
between capillaries and the brain, and is composed of ECs, endothelial tight
junctions (TJs), the BM, pericytes and astrocyte endfeet [63, 64]. The BBB
protects the brain from invasion by blood-derived harmful factors, thus
maintaining the homeostasis of the CNS [65]. Using quail-chick transplantation
chimeras, BBB was shown to develop in response to the neural tissue environment
[66]. During embryogenesis in rodents, astrocytes and pericytes are required to
wrap immature vessels. It is widely accepted that immature vessels are covered
preferentially by astrocytes postnatally. However, Daneman et al. [67]
reported that pericytes were recruited during embryogenesis, more than one week
before the generation of astrocytes, thus revealing the role of pericytes in the
formation of the BBB. The permeability of the BBB is increased in
pericyte-deficient mice, indicating the essential role of pericytes in
maintaining BBB integrity [49]. Pericytes maintain the integrity of the BBB
through two pathways, forming and preserving the TJs of ECs, and transcytosing in
the CNS ECs.
The breakdown of the BBB has been observed in AD [68], which is attributed to
the loss and detachment of pericytes destroying the integral structure of the BBB
and increasing BBB permeability [69]. Interestingly, sirtuin-1 (SIRT1),
an anti-aging gene, is markedly suppressed exposed to A [70]. The
decreased expression of SIRT1 also increased the permeability of the BBB
and accelerated the process of senescence [71].
4.3 Angiogenesis
Angiogenesis is the formation of new blood vessels from existing vessels.
Angiogenesis involves three steps: initiation, sprouting/migration, and
maturation. In angiogenesis, the complex signaling pathways between ECs and
pericytes which include the platelet-derived growth factor (PDGF)-BB/PDGF
receptor beta (PDGFR) pathway [72, 73], the angiopoitin1/tyrosine
protein kinase receptor (Tie2) signaling pathway [74], the vascular endothelial
growth factor (VEGF)/VEGF receptor (VEGFR) pathway [46], the
sphingosine-1-phosphate (S1P) signaling pathway [75], transforming growth factor
beta (TGF-) [76] are the foundation for the formation and stabilization
of new blood vessels. Pericytes regulate the expression of VEGF, resulting in the
instability of blood vessels and initiating the angiogenesis [77]. Pericytes
detach from blood vessels to pave the way for endothelial sprouting [78]. The
migrating ECs secrete VEGF to stabilize nascent vessels and signal to pericytes
to recruit VEGF. Moreover, the recruited pericytes communicate with ECs to
promote the stabilization and maturation of new blood vessels [78]. The coverage
of pericytes is a marker of vascular maturation and a lack of pericytes results
in vascular hyperplasia [79].
5. Mechanisms through which Pericytes Regulate A
A is continuously generated by neurons and other cells in the healthy
brain, and is subsequently cleared through various pathways [80, 81, 82] including
receptor-dependent transport [80, 83, 84, 85, 86], cytosolic protease-mediated
intracellular degradation [87] and glymphatic clearance [88]. In AD, the
clearance of A is impaired, and an imbalance between A
production and clearance leads to the aberrant accumulation of A [7].
Moreover, pericytes play a considerable role in the clearance of A,
while the loss of pericytes in AD exacerbates the deposition of A in the
parenchyma.
5.1 Pericytes Clear A by LDL Receptor-Related Protein-1
(LRP-1)
LDL receptor-related protein-1 (LRP-1) is an apoE receptor that mediates the
clearance of A. Using freshly isolated cortical slices incubated with
A, Ma et al. [80] showed that pericytes rapidly remove
Cy3-A42. In addition, in AD and APP mice, the abundant
accumulation of A in pericytes indicates the important role of these
cells in clearing A at the BBB. Additionally, in an LRP-1 conditional
knockout model, Cy3-A42 uptake by pericytes was reduced by 80% compared
with that in the control group. The process of clearing A by pericytes
can be inhibited by antibodies against LRP-1 [80, 84], further confirming
that the clearance of A is mediated by LRP-1 (Fig. 1A). Compared with
that in adult wild-type mice, clearance in apoE knockout mice is substantially
reduced, indicating that LRP-1-mediated transport can be influenced by apoE, a
definite risk factor for AD [85]. ApoE is required for A clearance and
is isoform- specific. A study revealed that apoE3, but not apoE4, normalizes
A clearance in mouse pericytes with silenced mouse apoE [80], while
another study revealed that the binding of A to apoE3 reduces its
clearance rate at the BBB. It has also been reported that A binding to
apoJ significantly accelerates the BBB clearance rate [86, 89].
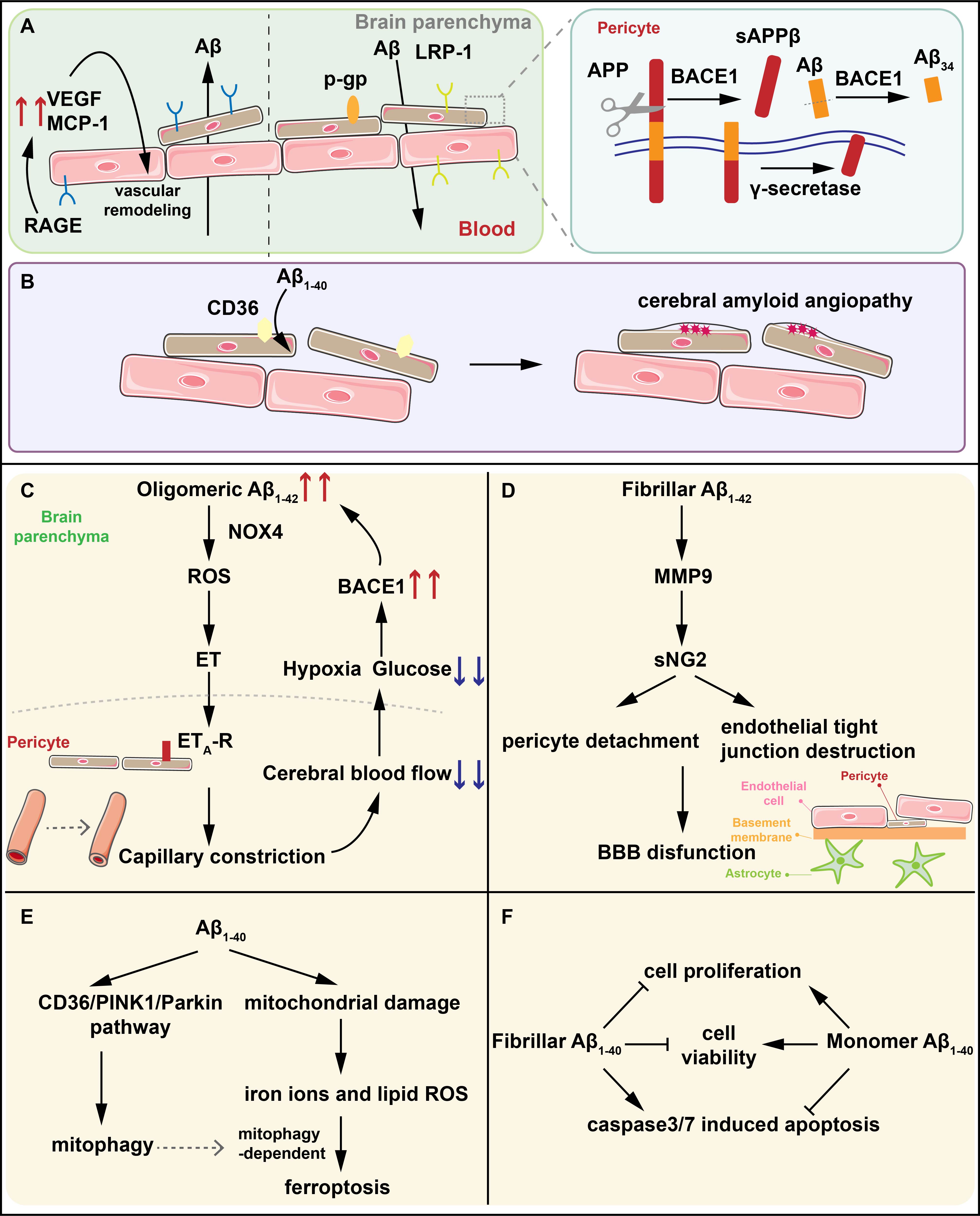 Fig. 1.
Fig. 1.
The interactions between A and pericytes. (A) LRP-1
and P-gp mediate the efflux of A while RAGE regulate the influx of
A into brain parenchyma. BACE1 on pericytes degrades A1-40 into
A34 intermediates and the A (1-42)-RAGE interaction induces the
generation of VEGF and MCP-1 contributing to the vascular remodeling. (B) CD36
mediates the clearance of A1-40 by pericytes. The reduced expression of
CD36 promotes the deposition of A1-40 resulting in CAA. (C) Oligomeric
A1-42 activates NOX4 in pericytes to produce ROS and ET in sequence, and
ET binds to ET-R on pericytes, triggering capillary constriction. Capillary
constriction results in the reduction of CBF and the glucose and oxygen it
contains. Hypoxia in turn upregulates the expression of BACE1, further increasing
the generation of A and forming an amplified positive loop, ultimately
leading to synapse dysfunction and neuron loss. (D) Fibrillar A1-42
activates MMP-9 to induce NG2 sheds from pericytes leading to the detachment of
pericytes and the destruction of endothelial TJs which is the significant part of
BBB. (E) A1-40 induces pericytes mitophagy through the CD36/PINK1/Parkin
pathway and increases oxidative stress in pericytes. The increased lipid ROS and
iron ions caused by oxidative stress in pericytes inducing pericytes ferroptosis
dependent on mitochondrial autophagy. (F) Fibrillar A1-40 reduces the
viability and proliferation of pericytes, and increases the activity of the key
apoptotic proteins caspase3/7 while the effects of monomer A1-40 are
completely opposite. LRP-1, LDL receptor-related protein-1; RAGE, receptors for
advanced glycation end products; A, beta-amyloid; BACE1, -site
amyloid precursor protein (APP) cleaving enzyme 1; VEGF, vascular endothelial
growth factor; CAA, cerebral amyloid angiogenesis; NOX4, nicotinamide adenine
dinucleotide phosphate oxidase 4; ROS, reactive oxygen species; ET, endothelin;
CBF, cerebral blood flow; MMP-9, matrix metalloproteinase-9; NG2, neural/glial
antigen 2; TJs, endothelial tight junctions; BBB, blood–brain barrier; PINK1,
PTEN-induced putative kinase 1; sAPP, soluble amyloid precursor protein
beta; MCP-1, monocyte chemoattractant protein-1; ET-R, endothelin receptor
type A.
The expression of LRP-1 is downregulated in AD patients exacerbating A
pathology [90]. By injecting pericytes into APP/PS1 mice, Tachibana et
al. [91] showed that microcirculation improved in the pericyte-injected
hemisphere and that the deposition of A decreased in a manner dependent
on the expression of LRP-1 on pericytes. However, in a recent phase I clinical
trial in which mesenchymal stem cells (MSCs) were stereotactically injected to
the brains of AD patients, no significant effects on cognitive function were
observed. The use of transplanted pericytes or MSCs in the brain to prevent or
treat AD has not been validated [92].
5.2 A40 is Degraded into A34 in Pericytes through
-Site Amyloid Precursor Protein (APP) Cleaving Enzyme 1 (BACE1)
Amyloid precursor protein (APP) is cleaved by -Site Amyloid Precursor
Protein (APP) Cleaving Enzyme 1 (BACE1) and -secretase, which
sequentially results in the formation of A peptides, including
A40 and A42 [93]. In addition to “amyloidogenic” activity,
BACE1 also possesses “amyloidolytic” activity, whereby it degrades longer
A isoforms at position 34 into A34 intermediates [94] (Fig. 1A). A previous study demonstrated that BACE1 is expressed in pericytes [95].
Treating pericytes with a BACE inhibitor resulted in a dose-dependent decrease in
A34 levels, indicating the role of BACE1 in the cleavage of A
peptides and the formation of A34 in pericytes [95]. Notably, the
substrate of BACE1 is A40, but not A42 [94, 95].
In AD, the A34/A40 ratio is decreased significantly, and the
level of A34 is correlated with disease progression [96]. The
progression of AD can be divided into six stages. AD in Braak stage I-II is
clinically silent, AD in Braak stage III–IV is incipient and AD in Braak stage
V–VI is fully developed [97, 98, 99]. Kirabali et al. [95] revealed that in
Braak stage II, A34 levels peak, while in Braak stages III and IV, the
immunoactivity of A34 significantly decreases which explains the
dysfunction and loss of brain pericytes in AD pathogenesis. Moreover, analysis of
PDGFR immunoactivity revealed that the loss of pericytes had already
occurred at Braak stage II [37].
5.3 Pericytes Regulate A Clearance via RAGE
AGEs (advanced glycation end-products) are the final products of the
nonenzymatic glycation of proteins (Maillard reaction) [100], which is
irreversible. AGEs can bind and destroy various histocytes through a process
called cross linking. A series of studies have shown that AGEs accelerate aging
and cause neurodegenerative disorders including AD [101, 102, 103]. Receptors for
advanced glycation end products (RAGE), a multiligand receptor of the
immunoglobulin family, can not only specifically bind to AGEs, but also bind to
various ligands such as high-mobility group box-1 (HMGB1), S100 and A [104, 105], and play vital roles in the occurrence and development of various
diseases. RAGE plays a critical role in regulating the influx of circulating
A into the brain via the BBB as a transporter [106]. Moreover, RAGE
promotes the generation and accumulation of A by enhancing the activity
of -secretase and -secretase [107]. In addition, RAGE induces
dysfunction of synapses and neuronal circuits, which is the structural foundation
of cognition [108, 109]. According to its structure, RAGE is classified into
three isoforms: N-truncated, and C-truncated, which are also called endogenous
secretory RAGE (esRAGE) [110]. Moreover, RAGE can be cleaved by proteolytic
enzymes to form cRAGE. The soluble form of RAGE (sRAGE) is composed of esRAGE and
cRAGE. Notably, sRAGE can interact with A to form sRAGE-A
interactions which can inhibit the neurotoxicity of RAGE and promote A
clearance from the brain [110].
It has been proven that RAGE is expressed on pericytes [111]. Using small
interfering RNA (siRNA) technology to suppress the expression of the pericyte
RAGE gene, Lue et al. [111] showed that the level of A
dramatically decreased, indicating that the A (1-42)-RAGE interaction
may function by tethering A to the cell surface of pericytes, advancing
the A-A interaction and further promoting fibrillogenesis. By
blocking RAGE with an anti-RAGE antibody, the levels of A-induced VEGF
and monocyte chemoattractant protein-1 (MCP-1) were decreased, indicating that
RAGE-A interactions in pericytes contribute to the vascular remodeling
that is observed in AD (Fig. 1A). The combination of A and pericyte RAGE
can induce oxidative stress and a subsequent inflammatory response by activating
a variety of signaling pathways, including mitogen-activated protein kinase
(MAPK), glycogen synthase kinase 3 (GSK-3) and nuclear factor kappa-B
(NF-B) [112]. Oxidative stress and inflammatory reactions can thicken
the BM and increase the deposition of A, eventually leading to vascular
amyloidosis and disruption of the BBB [111]. In addition, the A-RAGE
interaction results in cognitive impairment by accelerating the aging process and
inducing oxidative stress [113]. In AD, elevated RAGE levels may account for
neuronal death and cognitive impairment. However, the level of sRAGE is lower
[114]. In view of this, RAGE inhibitors may be potential targets for treating AD
and these agents have been proven to be effective in preclinical and clinical
studies [115], although the results have been unsatisfactory.
5.4 Pericytes Efflux A via P-Glycoprotein (P-gp)
P-gp, a subtype of the ATP binding cassette (ABC) transporter family, is an
ATP-dependent transporter responsible for the efflux of various substrates from
the brain to the blood [116, 117]. Using an immunogold technique with monoclonal
anti-P-gp antibodies, Bendayan et al. [118] showed that gold particles
are present in ECs, astrocytes and pericytes, suggesting that pericytes can
express P-gp. In human studies, the expression level of P-gp was shown to be
negatively correlated with the accumulation of A [119, 120].
Moreover, it has been proven that inhibiting P-gp leads to increased
intracellular deposition of A in brain capillaries [121, 122],
suggesting that P-gp plays a crucial role in the clearance of A.
However, the mechanism by which P-gp affects A transport remains
controversial. A variety of studies support the notion that A stimulates
the ATPase activity of P-gp [123, 124]. However, Bello et al. [125]
reported that A has no effect on the adenosine triphosphate (ATP)
hydrolysis activity of P-gp. In previous research, McCormick et al.
[123] noted that the activation of P-gp ATPase by A depends on the lipid
environment, which may account for the differences between those studies. In
addition, A can affect P-gp conversely. By treating transgenic human
amyloid precursor protein (hAPP) overexpressing mice with an irreversible
inhibitor of the ubiquitin-activating enzyme E1, Hartz et al. [126]
showed that retained P-gp results in a decreased level of A, suggesting
that A induces P-gp degeneration through the ubiquitination pathway. In
addition, the brains of AD patients exhibit marked decrease in P-gp and a
significant increase in A deposition and ubiquitinated A [126, 127, 128].
5.5 Pericytes Uptake A1-40 via CD36
CD36, a glycosylated membrane protein, is widely expressed in the nervous
system, including in pericytes [129]. CD36 is involved in a variety of
pathological processes, such as vascular oxidative stress, the inflammatory
response, mitochondrial dysfunction and neurovascular uncoupling [129, 130, 131].
Immunofluorescence staining revealed that, CD36 and A1-40 colocalize
with PDGFR, a marker of pericytes, suggesting that CD36 may be involved
in the clearance of A1-40 by pericytes [132]. In transgenic mice lacking
CD36, Li et al. [132] showed a reduction in A1-40 and cerebral
amyloid angiopathy (CAA), suggesting that CD36 promotes the deposition of
A1-40 resulting in vascular dysfunction (Fig. 1B). Moreover, the
transcription and expression levels of CD36 in pericytes treated with
A1-40 increased in a concentration-dependent manner aggravating vascular
dysfunction [132]. A1-40 increases the permeability of the BBB
in vitro, which can be reversed by inhibiting the expression of CD36 in
pericytes, suggesting that inhibiting the expression of CD36 increases BBB
tightness [133], providing a new therapeutic target for preventing BBB
destruction during AD progression.
6. The Effects of A on Pericytes
A exerts toxic effects on pericytes through various pathways, and a
significant loss of pericytes has been observed in AD patients. We have
summarized the pathological effects of interactions among targets of pericytes
and different species of A, as well as the pathological changes observed
in AD, in Table 2 (Ref. [36, 37, 128, 134, 135, 136, 137, 138, 139, 140, 141]).
Table 2.Interactions among targets in pericytes and different species
of A (f: fibrillar; o: oligomeric; m: monomeric), pathological effects
and pathological changes in AD.
| Study type |
Markers of pericytes |
Targets |
The species of A |
Pathological effects |
Pathological changes in AD |
Ref. |
| In vitro |
PDGFR |
NOX4 |
oA1-42 |
NOX4 activated by A1-42 induce oxidative stress in pericytes. ROS trigger the generation of ET, which interact with ET-R on pericytes, triggering strong capillary constriction. |
The level of ET increases. |
[36, 134, 135, 136] |
| In vitro |
NG2, PDGFR and αSMA |
MMP-9 |
fA1-42 |
f A1-42 decreases the activity of MMP-9, preventing the detachment of pericytes. |
In early stage of AD, the level of MMP-9 and sNG2 in CSF are increased. |
[137, 138, 139] |
|
|
|
oA1-42 |
oA1-42 increases the activity of MMP-9, promoting the detachment of pericytes. |
|
|
| Both in vitro and vivo |
PDGFR and NG2 |
CD36/PINK/Parkin |
A1-40 |
A1-40 induces ferroptosis of pericytes by activating mitochondrial autophagy. |
Pericytes exposed to A1-40 exhibit ferroptosis in TEM. |
[128, 140] |
| In vitro |
PDGFR and NG2 |
Caspase3/7 |
fA1-40 |
f A1-40 reduces the viability and proliferation of pericytes by increasing the activity of caspase 3/7. |
Aggregated A1-40, the major component of deposition in CAA may account for the loss of pericytes in AD. |
[37, 141] |
|
|
|
mA1-40 |
m A1-40 decreasing the mortality of pericytes by decreasing the activity of caspase 3/7. |
|
Abbreviations: AD, Alzheimer’s disease; PDGFR, platelet-derived growth
factor receptor-; NOX4, nicotinamide adenine dinucleotide phosphate
oxidase 4; ROS, reactive oxygen species; ET, endothelin; NG2, neural/glial
antigen 2; SMA, alpha-smooth muscle actin; MMP-9, matrix
metalloproteinase-9; CSF, cerebrospinal fluid; TEM, transmission electron
microscopy; CAA, cerebral amyloid angiogenesis.
6.1 A1-42 Evokes the Constriction of Pericytes
Previous studies have shown that a decrease in the CBF is the earliest change in
AD patients [19], and capillaries exhibit focal constriction [142]. Vascular
resistance in the brain mainly occurs in capillaries, and the CBF is regulated by
pericytes, indicating that pericytes dysfunction contributes to vascular
disturbances in AD [22]. One of the most characteristic changes in AD is the
aberrant deposition of A, which results in the formation of A
plaques in the brain parenchyma, suggesting that A may be the latent
culprit.
In human brain slices, A1-42 (oligomeric and monomeric) can trigger a
slowly progressive constriction of capillaries near pericytes, suggesting that
A1-42 induces the constriction of human pericytes in a
concentration-dependent manner within limits [36]. Reactive oxygen species (ROS)
are generated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and
can be removed by superoxide dismutase 1 (SOD1) [134]. Recently, Nortley
et al. [36] discovered that oligomeric A1-42 evoked capillary
constriction could be blocked by endothelin receptor type A (ET-R), SOD1 and NADPH oxidase inhibitors,
suggesting that ROS and endothelin (ET) participate in the constriction induced
by A1-42. Moreover, evidence has shown that ROS in pericytes are
produced by reduced nicotinamide adenine dinucleotide phosphate oxidase 4 (NOX4)
[135] rather than other isoforms. After ET was applied along with SOD1, the
capillaries remained constricted, demonstrating that ET functions downstream of
ROS. Moreover, ET combined with ET-Rs on pericytes triggers intense
constriction [143]. In summary, A1-42 activates NOX4 in pericytes to
generate ROS, and these ROS induce the generation of downstream ETs, which then
interact with ET-Rs on pericytes, triggering strong capillary constriction
[36, 144]. The constriction of capillaries increases vascular resistance, causing
a reduction in the CBF, which leads to a decrease in glucose and oxygen supplies,
ultimately leading to synapse dysfunction and neuron loss [145]. In addition,
hypoxia in turn upregulates the expression of BACE1, further increasing the
generation of A and forming an amplified positive loop [143] (Fig. 1C).
The discovery of this mechanism overturns the previous view that the reduction in
the CBF is the result of arteriole constriction.
It has been observed that the level of ET increases in AD with the upregulation
of enzymes responsible for synthesizing ET [136]. Similarly, compared to those in
nondemented controls, Sengillo et al. [69] reported a remarkable loss of
pericytes in the cortex and hippocampus of AD patients, which may be attributed
to the fact that chronic exposure to A1-42 results in the constriction
and rigid death of pericytes. Fortunately, blocking NOX4 and ET-Rs prevent
further contraction caused by A, although the capillary diameter does
not return to baseline levels. Moreover, C-type natriuretic peptide (CNP)
successfully reverses the A-evoked constriction [136] and may be applied
in the treatment of AD in the future.
6.2 A1-42 Induces the Detachment of Pericytes by Activating
MMP-9 to Induce NG2 Shedding from Pericytes
Neural/glial antigen 2 (NG2), a transmembrane proteoglycan, is an original
marker of pericytes [30]. NG2 not only plays a significant role in the
proliferation and motility of pericytes, but also promotes the formation and
maturation of endothelial TJs [38].
A1-42 is the principal component of neuritic plaques characterized in
AD. Different aggregated forms of A1-42 have been proven to influence
the shedding of NG2 from pericytes differently. NG2 sheds pericytes to form
soluble NG2 (sNG2). Fibrillar A1-42 decreases the level of sNG2, while
the level of sNG2 increases after exposure to oligomer-enriched preparations of
A1-42 [137]. After exerting inhibitors of matrix metalloproteinase (MMP,
an angiogenic factor secreted by pericytes) [138], the consequences of fibrillar
A1-42 remain, while increase in sNG2 resulting from oligomeric
A1-42 is eliminated, suggesting that the shedding of NG2 induced by
A is mediated by MMP-9. Moreover, fibrillar A1-42 decreases the
activity of MMP-9 while oligomeric A1-42 increased MMP-9 activity [137].
However, oligomeric A1-42 does not alter the concentration of MMP-9,
indicating that A1-42 affects the activity rather than the secretion of
MMP-9.
Taken together, in the early stage of AD, A1-42 exists as oligomers and
activates MMP-9, which subsequently increases the level of sNG2. The finding
coincides with the discovery of increased MMP-9 in cerebrospinal fluid (CSF)
during early AD pathogenesis [139]. sNG2 has been demonstrated to promote
angiogenesis, resulting in unstable blood vessels and dysfunction of the BBB
[146], and the level of sNG2 is increased in the CSF of AD patients [147]. In
addition, the release of NG2 from the cell surface of pericytes drives pericyte
detachment and contributes to the loss and dysfunction of TJs. Even worse, this
effect could be aggravated by the enhanced degradation of the extracellular
matrix and TJs caused by increased MMP-9 activity [148]. The detachment of
pericytes and destruction of endothelial TJs destroy the integral structure of
the BBB and increase the permeability of the BBB (Fig. 1D). Subsequently, the
hazardous substances enter the brain and neurotoxicity caused by A could
cause neuronal loss and cognitive decline. As the disease progresses, fibrillar
A1-42 accumulates, which may be the protective mechanism of the body
[149].
Researchers found that the SIRT1 activator, resveratrol, reduced serum
MMP-9 levels in AD patients, thus reducing neuro-inflammation [141].
Additionally, resveratrol was able to slow down the progressive decline in daily
living scores (ADLs) in AD patients [141]. In spite of this, the relationship
between the delay in cognitive decline and the decrease in MMP-9 remains unknown.
The discovery that A1-42 influences the shedding of NG2 on pericytes via
MMP-9 may explain the pathology of AD and provide a new therapeutic strategy,
such as specifically inhibiting MMP-9, to prevent AD dysfunction.
6.3 A1-40 Induces Pericyte Mitophagy-Dependent Ferroptosis
through the CD36/PINK/Parkin Pathway
A1-42 (10%) and A1-40 (80%) are two typical soluble
monomeric subtypes of A [49]. A1-42 is more likely to form
insoluble aggregates than A1-40 [6], but A1-40 can be deposited
in the vascular system to form CAA [150]. Recently, it has been reported that
more than 90% of patients with AD have CAA and CAA usually precedes the
formation of neuritic plaques that are composed mainly of A1-42 [151].
A1-40 is presumed to have a particular toxicity to pericytes [152]
and could accelerate the disruption of the BBB.
A recent study showed that pericytes treated with A1-40 exhibit a
prominent decrease in proliferation and a marked increas in mitochondrial ROS
(Mito SOX) in a manner dependent on both time and concentration [132]. The
results also demonstrated that A1-40 activates mitochondrial autophagy
through the CD36/PINK/Parkin pathway (PINK, PTEN-induced putative kinase) [132]. Mitochondrial damage and autophagy
induced by A1-40 often lead to apoptosis [140]. Notably, A1-40
rather than apoptosis increases oxidative stress in pericytes. An increase in
lipid ROS is accompanied by an increase in the concentration of iron ions,
indicating that A1-40 may induce pericyte ferroptosis [153]. The
morphological features of pericytes exposed to A1-40 according to
transmission electron microscopy (TEM) also correspond with ferroptosis.
Moreover, inhibiting mitochondrial autophagy prevents pericyte ferroptosis and
ferroptosis inhibitors could prevent mitochondrial autophagy evoked by
A1-40, suggesting that ferroptosis is dependent on mitochondrial-related
autophagy [132] (Fig. 1E).
6.4 A1-40 Affects Pericytes in an Aggregation-Dependent
Manner
The expression of pericyte markers is dynamic, and depends on the functional
state of pericytes; for example, PDGFR labels relatively immature
pericytes [33], NG2 stimulates the proliferation and migration of pericytes [34],
and laminin is expressed in active or mature subsets of pericytes [154]. In the
hippocampus of AD patients, a significant reduction in the number of pericytes
expressing NG2 [37], CD13 and PDGFR [70] is observed, while the number
of pericytes expressing other markers is not affected. It can be concluded that
the subsets of pericytes that participate in activation, migration, and
proliferation are affected by the pathology of AD. A previous study demonstrated
that the CAA formed by A1-40 is associated with pericyte degenerative
changes [150]. Interestingly, A1-40 levels are associated with the
number of pericytes [37]. Since aggregated A1-40 is toxic [150], it is
speculated that monomeric A1-40 may be beneficial.
Schultz et al. [37] showed that fibrillar A1-40 reduces the
viability and proliferation of pericytes in vitro, and increases the
activity of the key apoptotic proteins caspase3/7. These findings are consistent
with the discovery of pericyte degeneration near fibrillar A1-40 [150].
Moreover, the monomer A1-40 decreases the mortality of pericytes by
decreasing the activity of caspase3/7, and promoting pericyte proliferation,
which could explain the positive correlation between the levels of monomeric
A1-40 and the number of pericytes (Fig. 1F).
The occurrence of CAA and AD largely overlap [13, 14, 15] and the occurrence of
advanced CAA is related to more severe cognitive impairment in patients with AD
[15, 16]. Aggregated A1-40, the major component of deposition in the CAA
may account for the loss of pericytes in AD patients [155].
7. Strategies to Prevent or Treat AD via Pericytes
Further understanding of the interactions between pericytes and A could
lead to new insights for the treatment of AD. Superoxide dismutase-1 (SOD1) can
eliminate ROS generated by NOX4 activated by A [36]. Using SOD1 to
eliminate ROS may be effective at preventing the constriction of pericytes.
Indeed, overexpression of SOD1 or topical application of exogenous SOD could
reverse vascular dysfunction and premature mortality in transgenic mice
overexpressing APP [156]. Pterostilbene (PTE), the natural dimethylated analog of
resveratrol, can upregulate the expression of SIRT1 and SOD to exert
neuroprotective effects [157]. Moreover, since A cannot induce the
constriction of pericytes without ET [143], reducing the generation of ET or
blocking the combination of ET and ET-R may be effective. It has been
proven that blocking NOX4 or ET-Rs could prevent further constriction of
capillaries evoked by A and CNPs could successfully reverse the
constriction of capillaries mediated by ET [36]. In addition, A induces
endothelial dysfunction characterized by attenuated endothelium-dependent
relaxation and increased endothelium-dependent constriction since A
inactivates vasodilators produced by the endothelium and increases the production
and release of ET [158]. In APP overexpressing mice, bosentan, an antagonist of
both ET and the ET receptor, was shown to preserve the endothelial
function of the aorta and carotids [159]. Autopsy evidence has shown that the
majority of Alzheimer’s disease patients suffer from vascular diseases such as
CAA [13, 15]. Several ET receptor antagonists, including bosentan have been
applied to treat pulmonary hypertension, and recent studies have demonstrated the
beneficial effects of bosentan in restoring the cerebrovascular function of
diabetic rats and preventing coronary endothelial functions in
hypercholesterolemic pigs [160, 161, 162]. The ability of bosentan to preserve
endothelial functions in A overexpressing Tg2576 mice demonstrated the
potential of ET receptor antagonists for the prevention and treatment of AD.
Another promising approach might be to implant pericytes or mesenchymal stem
cells (MSCs) into the brain. In APP/PS1 mice, the CBF was increased and
A plaques were significantly reduced in the pericyte-injected hemisphere
[91]. However, no significant effects on cognitive function were observed in a
recent phase I clinical trial in which stereotactically injected MSCs were
administered to the brains of AD patients [92]. Many studies have shown that RAGE
inhibitors may be potential targets for treating AD [107, 163, 164]. Recently,
PF-04494700, an oral inhibitor of RAGE has attracted widespread attention.
However, two clinical trials on PF-04494700 have shown that although PF-04494700
is safe and well-tolerated, it has no apparent benefit in improving cognitive
decline [165, 166]. Notably, high-dose PF-04494700 could also increase adverse
reactions and exacerbate cognitive impairment [165].
8. Conclusion
Pericytes are multifunctional cells of the vascular system and important
components of the BBB and NVU. Pericytes regulate the CBF in response to
neurotransmitters and neuronal activity and are essential for endothelial TJs and
they are necessary for the formation and maintenance of the BBB. Bidirectional
communication between ECs and pericytes is necessary for angiogenesis. Moreover,
pericytes mediate phagocytosis to maintain homeostasis in the brain.
The aberrant deposition of A is the predominant pathological change in
AD. Pericytes can clear A via LRP-1, RAGE, P-gp, and CD36, and
A is degraded in pericytes via BACE1. The impairment of these pathways
may account for the pathogenesis of AD. A1-42 evokes the constriction of
pericytes and causes death after chronic exposure. A decrease in the CBF caused
by capillary constriction leads to hypoxia and glucose deficiency, contributing
to the neuronal dysfunction and cognitive decline. A1-42 also induces
the detachment of pericytes. A1-40 induces pericyte mitophagy-dependent
ferroptosis through the CD36/PINK/Parkin pathway. The loss of pericytes
exacerbates the aggregation of A in AD. Conversely, the loss of
pericytes is a result of the action of A. Further exploration of the
interactions between pericytes and A is beneficial for understanding AD
and provides new therapeutic targets for the prevention and treatment of AD.
Author Contributions
YYL—literature collection, literature analysis and drafting the manuscript;
DDG—literature collection and literature analysis; RND—editing, preparing the
figures, reviewing and giving final approval of the version; YL—editing,
preparing the figures, reviewing and giving final approval of the version. All
authors contributed to editorial changes in the manuscript. All authors read and
approved the final manuscript. All authors have participated sufficiently in the
work and agreed to be accountable for all aspects of the work.
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
Not applicable.
Funding
This work is supported by National Natural Science Foundation of China (No.
82101487), Natural Science Foundation of Shandong Province, China (ZR2021QH161)
and Taishan Scholar Program of Shandong Province (tsqn202211318).
Conflict of Interest
The authors declare no conflict of interest.


 , Yi Li 1,*
, Yi Li 1,*