
 , Yong Xia 2, Anqi He 2, Shuang Chen 3, Jie Zhang 4,*
, Yong Xia 2, Anqi He 2, Shuang Chen 3, Jie Zhang 4,*
1 Department of Burn and Plastic Surgery, The First Affiliated Hospital of Chongqing Medical University, 400016 Chongqing, China
2 Department of Rehabilitation Medicine, Rehabilitation Medical Center, West China Hospital, Sichuan University, 610041 Chengdu, Sichuan, China
3 Department of Dermatovenereology, The First Affiliated Hospital of Chongqing Medical University, 400016 Chongqing, China
4 Department of Oncology, The First Affiliated Hospital of Chongqing Medical University, 400016 Chongqing, China
Abstract
Background: The incidence of melanoma brain metastasis (MBM) is high and significantly compromises patient survival and quality of life. Effective treatment of MBM is made difficult by the blood-brain barrier (BBB), since it restricts the entry of drugs into the brain. Certain anti-psychotic drugs able to cross the BBB have demonstrated efficacy in suppressing brain metastasis in preclinical studies. However, the activity of zuclopenthixol against MBM is not yet clear. Methods: Cell viability assays were employed to investigate the potential of zuclopenthixol in the treatment of MBM. Subsequently, the mechanism of action was investigated by RNA-sequencing (RNAseq), flow cytometry-based cell cycle and apoptosis assays, protein expression analysis, and autophagy flux detection. Additionally, the efficacy of zuclopenthixol against tumor growth was investigated in vivo, including MBM models. Results: Zuclopenthixol inhibited the proliferation of various melanoma cell lines at minimal doses by causing cell cycle arrest in the G0/G1 phase and mitochondrial-mediated intrinsic apoptosis. Zuclopenthixol also induced cytoprotective autophagy, and inhibition of autophagy enhanced the anti-melanoma effects of zuclopenthixol. Furthermore, zuclopenthixol inhibited the growth of human melanoma tumors in nude mice, as well as the growth of intracranial metastases in a mouse model of MBM. Conclusions: These results demonstrate that zuclopenthixol has significant potential as an effective therapeutic agent for MBM.
Keywords
- zuclopenthixol
- cell cycle arrest
- brain metastasis
- apoptosis
- melanoma
Since most patients with solid cancer succumb to metastasis, inhibition of this process is crucial for improving cancer patient survival. Late-stage melanoma exhibits strong metastatic behavior, with the brain, liver, lung, bone, and peritoneum being the primary sites of metastasis [1]. Limited information on the treatment progress of melanoma brain metastases (MBM) is available, mainly due to the exclusion of patients with brain metastases from clinical trials. However, recent advances in targeted therapies and immunotherapies have significantly improved the treatment of extracranial melanoma [2]. Positive outcomes have also been observed for the treatment of MBM, with combined inhibition of serine/threonine-protein kinase B-Raf (BRAF) and mitogen-activated protein kinase kinase (MEK) leading to intracranial response rates of almost 60% [3]. The combination of nivolumab and ipilimumab also demonstrated a clinical benefit in 58.4% of patients with symptomatic MBM [4]. Despite these advances, some MBM patients do not respond favorably to treatment, while others experience relapse after an initial response. Overall, the survival of MBM patients has not been significantly prolonged, with a median overall survival ranging from 5.6 to 17.4 months [5]. Consequently, melanoma metastases, particularly fatal MBM, continue to present significant challenges for the effective treatment of advanced melanoma [6].
A major challenge in the treatment of brain metastases is the blood-brain barrier (BBB), which hinders drug entry and promotes brain efflux of anti-cancer drugs [7]. Moreover, the development of new anti-cancer drugs is limited by high costs, lengthy timelines, and elevated risks of failure [8]. Consequently, the repurposing of existing drugs has attracted the attention of academic researchers and pharmaceutical companies in recent years. Comprehensive data is already available for approved drugs, including clear targets, mechanism of action, and safety profiles [9]. Once approved drugs are identified as being suitable for standalone treatments or for synergistic effects with other approved drugs, they can be efficiently repurposed for other diseases. Successful examples of this in the cancer field include the antiemetic drug thalidomide for the treatment of multiple myeloma, and the immunosuppressive drug rapamycin used in the treatment of colorectal cancer [10].
Previous studies have suggested that patients using anti-psychotic drugs have a lower cancer incidence, suggesting these drugs may also have anti-cancer properties [11, 12]. Recent studies, including our previous research, showed that certain anti-psychotic medications effectively impede the growth and spread of cancer cells, both in vitro and in vivo. For example, penfluridol has been shown to inhibit various cancer types, including lung [13], esophageal [14], breast [15] and renal [16], while trifluoperazine has been shown to inhibit triple-negative breast cancer [17] and colorectal cancer [18]. The ability of anti-psychotic drugs to cross the BBB makes them promising candidates for treating brain metastasis. Therefore, the investigation of potential anti-cancer drugs amongst anti-psychotic medications, especially those that inhibit brain metastasis, may be quite productive.
Zuclopenthixol is a typical anti-psychotic and acts as a D1 and D2 dopamine
receptor antagonist, as well as an
Zuclopenthixol (Cat# G12762) was purchased from AstaTech BioPharmaceutical Corporation (Chengdu, China), the Annexin V-7-AAD dual-staining apoptosis detection kit (Cat# 420404) from Biolegand (San Diego, CA, USA), propidium iodide (PI, Cat# ST1569-10mg) and DCFH-DA (Cat# S0033S) from Beyotime (Shanghai, China), and Rhodamine 123 (Cat# R8004-5MG) and MTT (Cat# M5655-1G) from Sigma-Aldrich (St Louis, MO, USA). 3-methyladenine (3-MA, Cat# M129496-1g) and chloroquine diphosphate (CQ, Cat# C129284-5g) from Aladdin (Shanghai, China).
The melanoma cell lines B16, A375, SK-MEL-28, C32, W266-4, A875,
HT144, SK-MEL-2, WM115, and A2058 were bought from the American Type Culture Collection (ATCC) within 5 years. These cell lines were grown in DMEM culture medium containing
100 U/mL of penicillin and streptomycin, and 10% FBS. The cells were maintained
at 37 °C under humidified conditions and with 5% CO
The impact of zuclopenthixol on melanoma cell viability was assessed using the
MTT assay. Between 1000 to 3000 cells were placed in a 96-well plate for 24 h,
after which an increasing amount of zuclopenthixol was added to individual wells
and incubated for different times. Subsequently, 20 µL of MTT
solution (5 mg/mL) was added, and the cells incubated at 37 °C for 2–3
h. Next, the medium was removed and 150 µL of DMSO was added to each
well. The absorbance at 562 nm was then measured, thus allowing the rate of
inhibition to be calculated for the various concentrations of zuclopenthixol.
Half-maximal inhibitory concentration (IC
The colony formation assay was conducted using 6-well plates. Each well was seeded with 800 cells and these were subjected to varying concentrations of zuclopenthixol for a period of 7 to 10 days. The treatment duration for B16 cells was 7 days, while for A375 cells it was 10 days. Cells were then fixed with a 0.5% solution of crystal violet, and the number of colonies counted under a microscope to determine the rate of inhibition.
Following treatment with vehicle (0.1% DMSO) or zuclopenthixol (12.5
µM) for 24 h, A375 cells were processed using TRIzol (Invitrogen, Carlsbad, CA,
USA) in order to obtain total RNA. Three samples were harvested for each
treatment group. RNA purity and concentration were evaluated with a NanoDrop 2000
spectrophotometer (Thermo Scientific, Waltham, MA, USA), while RNA quality was assessed with
an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). PolyA
selection with oligo(dT) beads was performed to isolate mRNA. The SuperScript kit
(Invitrogen) was used to synthesize cDNA using random hexamer primers
purchased from Illumina (San Diego, CA, USA). Sequencing and analysis of the transcriptome was carried
out by Majorbio Co. Ltd. (Shanghai, China). The libraries were sequenced on the
Illumina Novaseq 6000 platform. Genes exhibiting a |log
For analysis of the cell cycle, melanoma cells were treated with zuclopenthixol for 12 h and 24 h, followed by fixation for 24 h in ice-cold 75% ethanol. Cells were then rinsed with PBS and placed in the dark for 30 minutes in a solution containing RNase, 50 µg/mL of propidium iodide (PI), and 0.1% Triton X-100. After washing in PBS, the cell cycle distribution was examined using ACEA NovoCyte and analyzed using NovoExpress software (version 1.4.1.1901, ACEA Biosciences Inc., San Diego, CA, USA).
The detection kit for analysis of apoptosis used Annexin V-7-AAD dual-staining.
The cells were placed in 6-well dishes at a concentration of 1
Increasing amounts of zuclopenthixol were administered to melanoma cells for 24
h, followed by assessment of
For the migration test, 5
After treatment with different concentrations of zuclopenthixol for 24 h,
proteins were extracted by lysing cells with a lysis buffer containing a protease
inhibitor cocktail. Proteins were then separated using sodium dodecyl-sulfate
polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred onto
nitrocellulose membranes (Millipore). Subsequently, conventional Western blot
analysis was performed to detect protein expression. The antibodies used for
Western blot analysis were as follows: anti-DRD-2 was purchased from Abclonal
(Wuhan, China). Anti-p62 (Cat#CY5546, WB: 1:6000 dilution), anti-p-p44/42 MAPK
(Thr202/Tyr204) (Cat#CY5044, WB: 1:2000 dilution), anti-CDK2 (Cat#CY5020, WB:
1:1000 dilution), and anti-GAPDH (HRP conjugated) (Cat#AB2000, WB: 1:20,000
dilution) were purchased from Abways (Shanghai, China). Anti-Bax (Cat#610982,
WB: 1:1000 dilution) and anti-Bcl2 (Cat#2610538, WB: 1:1000 dilution) were
purchased from BD Bioscience (Piscataway, NJ, USA). Anti-AKT (Cat#4685s, WB: 1:1000
dilution), anti-p-AKT (Cat#4060s, WB: 1:2000 dilution), anti-cyclin D1
(Cat#2978, WB: 1:1000 dilution), anti-p27 (Cat#3698, WB: 1:1000 dilution),
anti-cleaved caspase 3 (Cat#9664s, WB: 1:1000 dilution), anti-p44/42 MAPK
(Cat#4696, WB: 1:2000 dilution), and anti-p-
Transfection of the GFP-RFP-LC3 plasmid into A375 cells was performed using Lipofectamine 3000 (Invitrogen). Cells were placed on Millicell EZ slides (Merck Millipore, Darmstadt, Germany) for 24 h and then exposed to zuclopenthixol (12.5 µM) or CQ (20 µM) for 24 h. They were subsequently immobilized with 4% paraformaldehyde at room temperature for 30 minutes and stained with 4’,6-diamidino-2-phenylindole (DAPI) solution (5 µg/mL) for 5 minutes. Confocal microscopy (LSM 880, Zeiss Microsystems, Jena, Germany) was utilized to observe the presence of LC3 in cells.
Approval for the ethical use of animal models was obtained from the Animal Care
and Use Committee, Sichuan University. In order to create the xenograft model for
subcutaneous melanoma, 5
Data is shown as the average
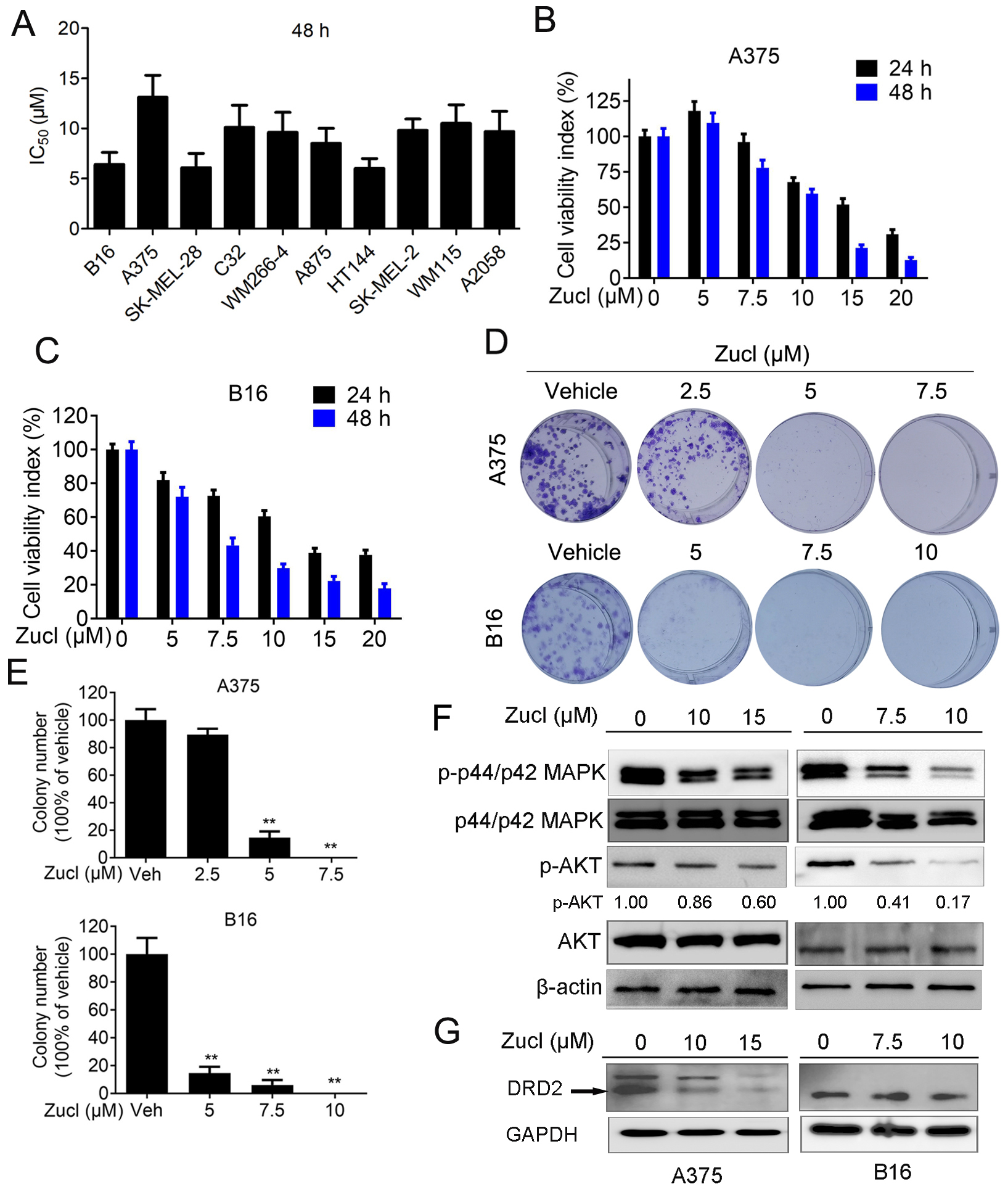
In our previous study [21], zuclopenthixol showed strong inhibitory activity
against B16 cells, prompting us to expand testing to 9 other melanoma cell lines.
Interestingly, zuclopenthixol inhibited all melanoma cell lines examined, with
IC
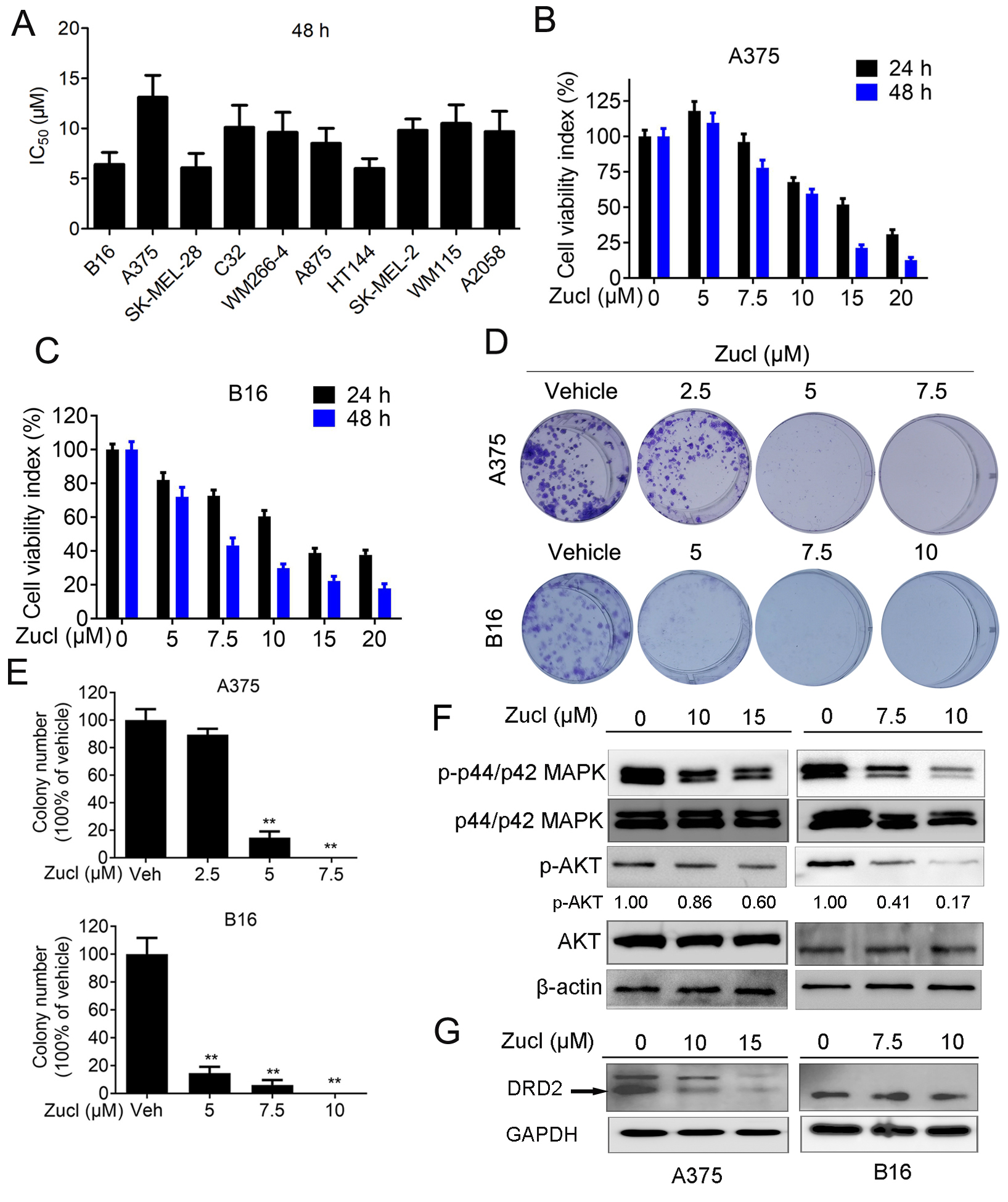 Fig. 1.
Fig. 1.Zuclopenthixol suppresses melanoma growth and viability
in vitro. (A) The Half-maximal inhibitory concentration (IC
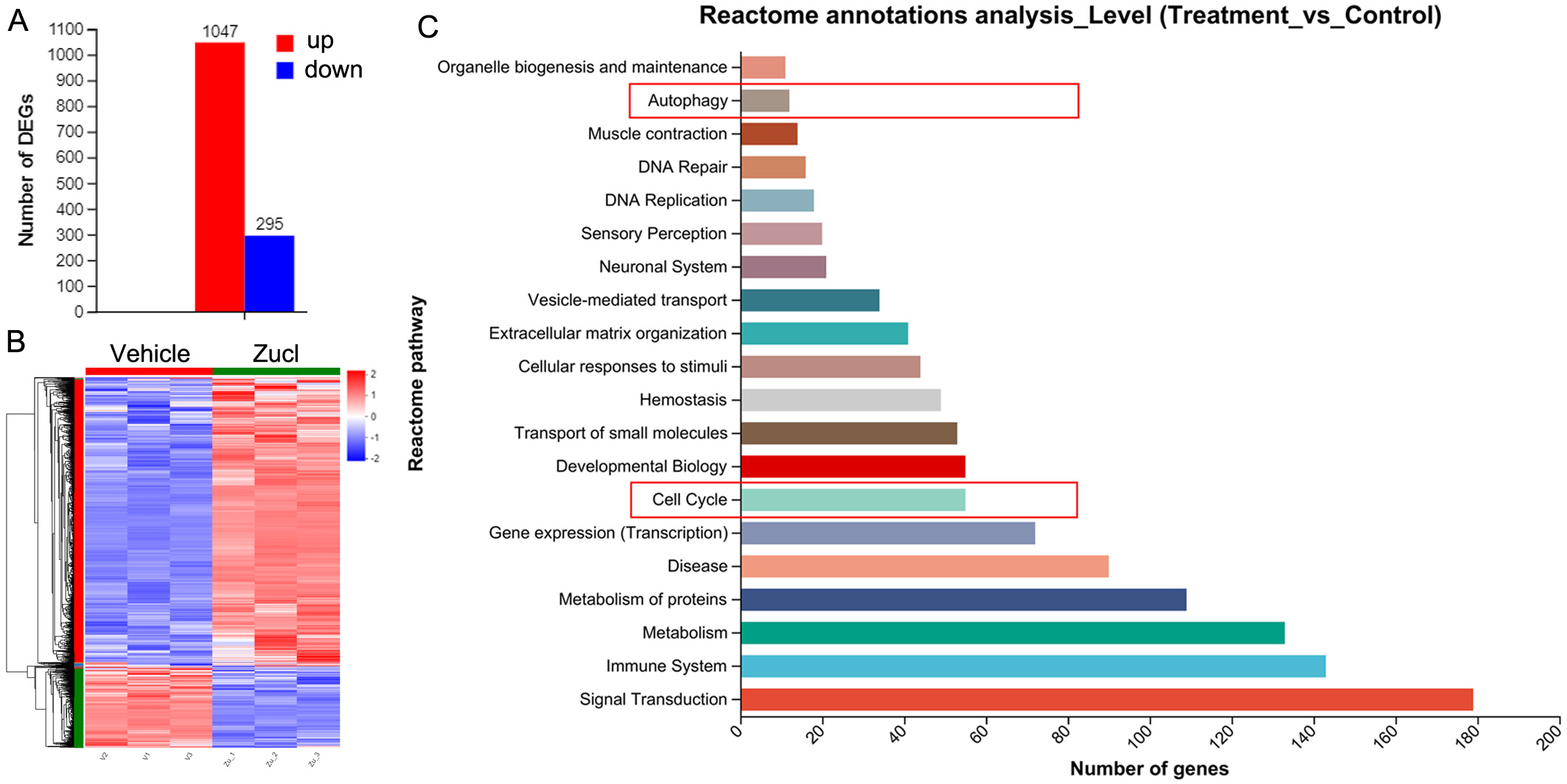
We conducted RNA-Seq analysis to study the molecular mechanism by which zuclopenthixol inhibits melanoma. Following treatment of A375 cells with zuclopenthixol, the expression of 1047 genes was found to be upregulated, while that of 295 genes was downregulated (Fig. 2A,B). Reactome annotation analysis indicated that one of the enriched pathways was the cell cycle (Fig. 2C).
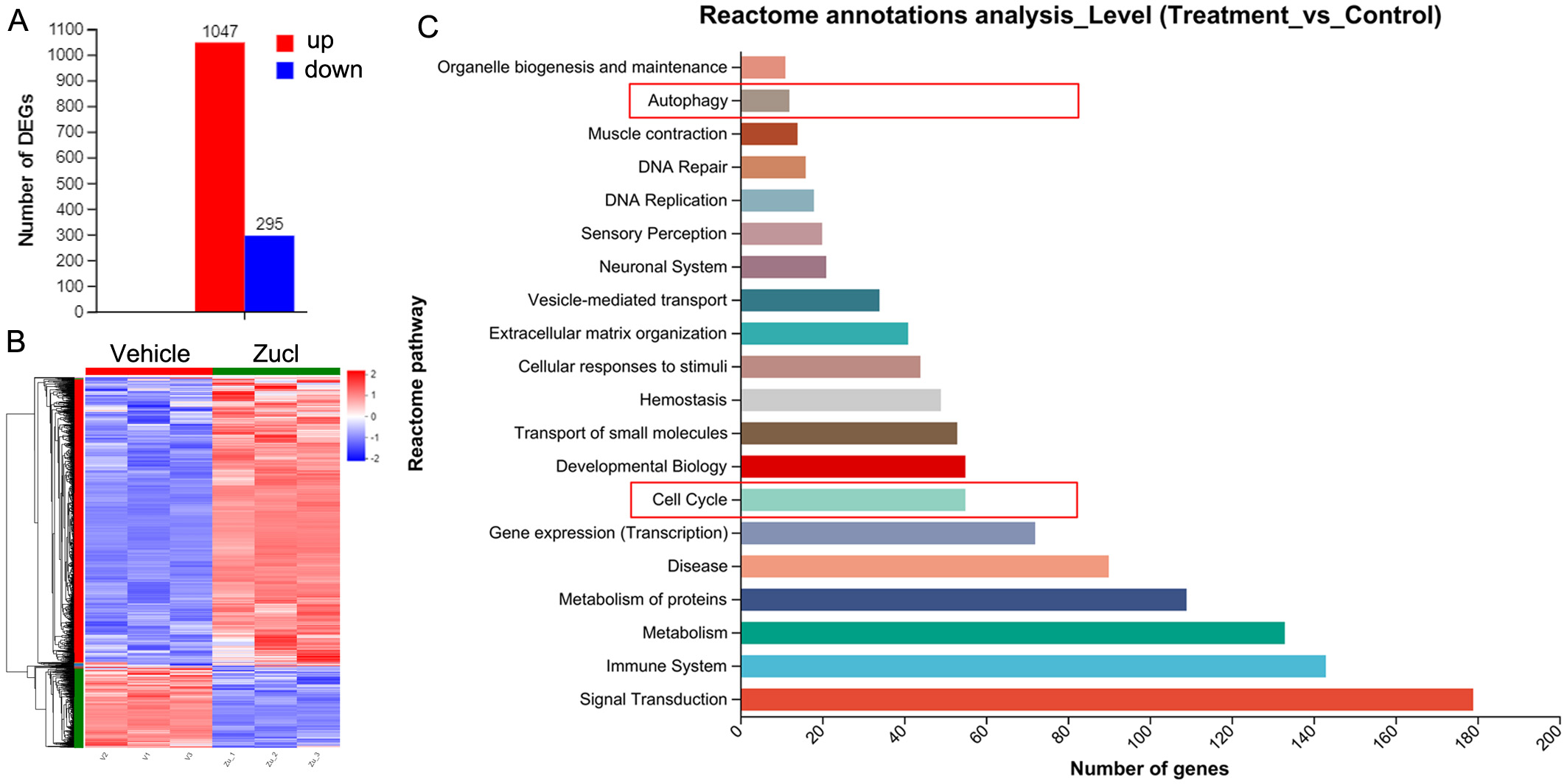 Fig. 2.
Fig. 2.The effect of zuclopenthixol on gene expression in A375 cells.
(A) The number of genes upregulated and downregulated after 12.5 µM
zuclopenthixol treatment for 24 h. Differentially expressed genes (DEGs) were
identified as genes with a log
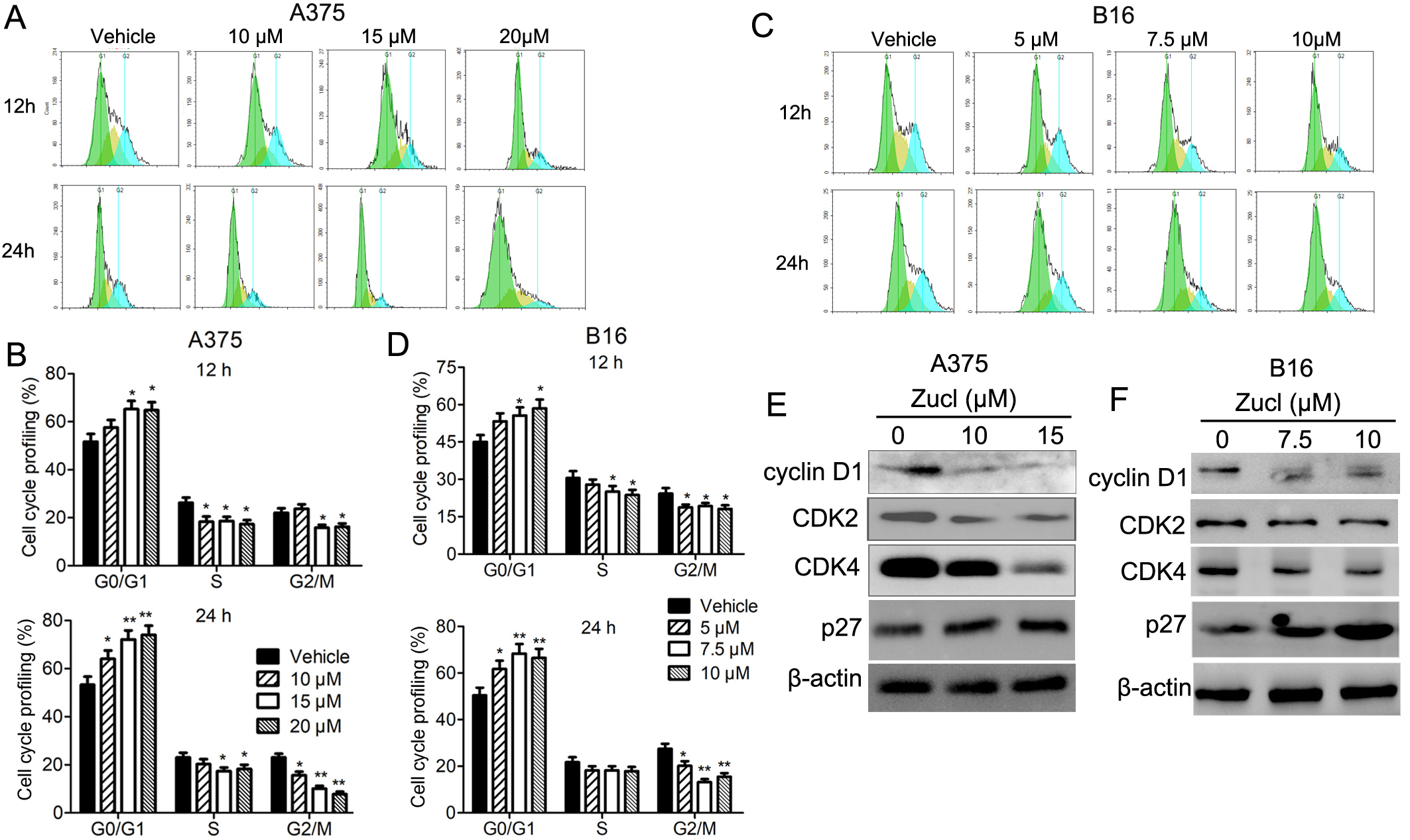
The ability of zuclopenthixol to inhibit the viability of melanoma cells may be due to its capacity to impede progression of the cell cycle and/or provoke cell demise. We therefore examined the effect of zuclopenthixol on the cell cycle of melanoma cells. In both cell lines, treatment with zuclopenthixol for 12 and 24 h led to a notable increase in the percentage of cells in the G0/G1 stage, potentially decreasing the S and G2/M phases (Fig. 3A–D). We also analyzed the expression of several important regulators of the cell cycle, including cyclin D1, CDK2 and CDK4. These proteins play crucial roles in promoting the G1-S transition, which was suppressed by zuclopenthixol treatment (Fig. 3E,F). Furthermore, zuclopenthixol increased the expression of p27. This inhibits cyclin-CDK and halts progression of the cell cycle in the G1 phase, thus preventing initiation of the normal cell cycle. These findings demonstrate that zuclopenthixol may exert its anti-tumor effects in melanoma cells in part by blocking G0/G1 phase progression.
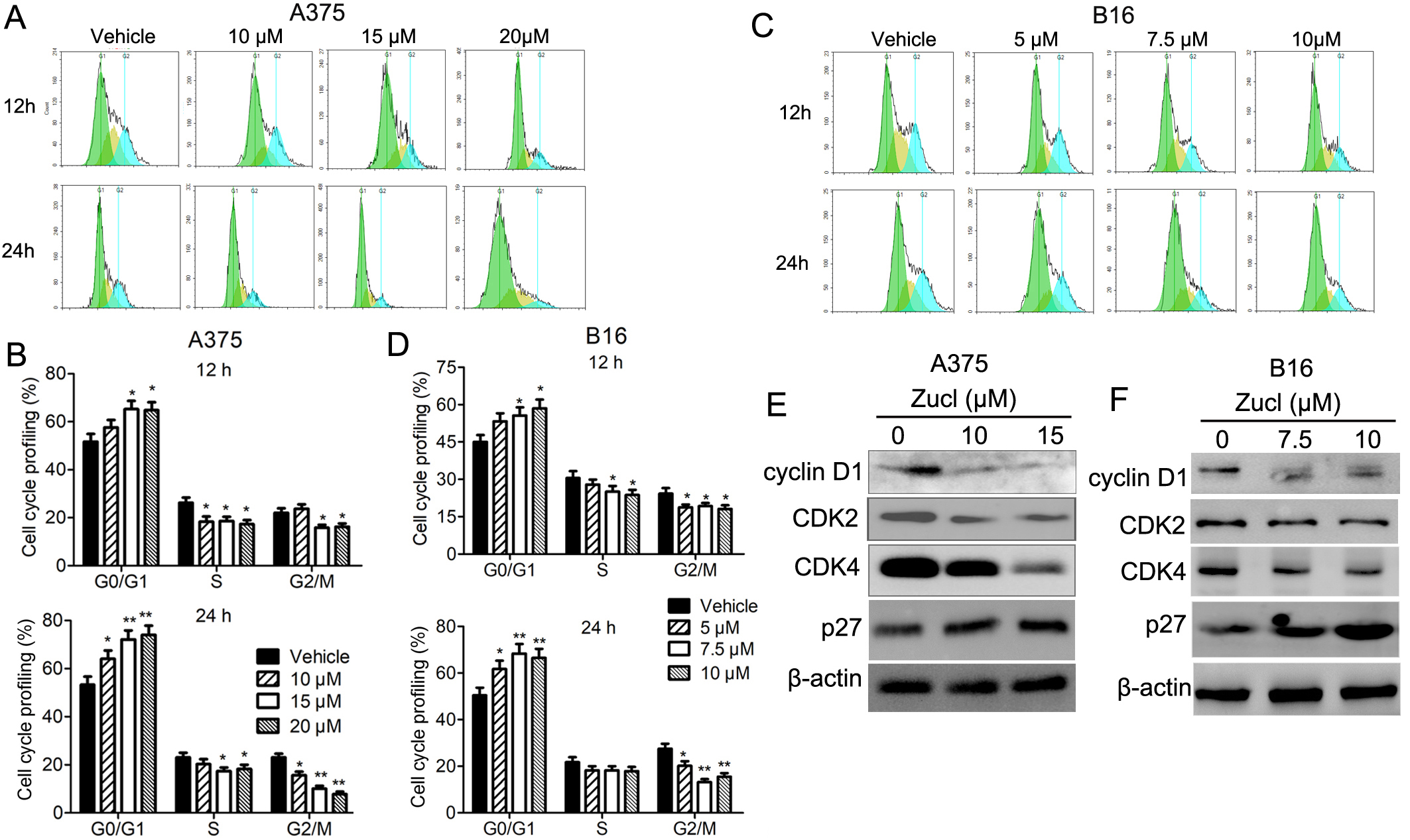 Fig. 3.
Fig. 3.Zuclopenthixol suppresses cell cycle progression in melanoma
cells. (A,C) Flow cytometry (FCM) was used to evaluate the distribution of A375
and B16 cells in distinct phases of the cell cycle following exposure to
different doses of zuclopenthixol for 12 and 24 h. (B,D) Quantitative analysis of
the results shown in A and C. (E,F) The impact of treatment with zuclopenthixol
for 24 h on the expression of proteins that regulate the G1/S transition in A375
and B16 cells. *p-values
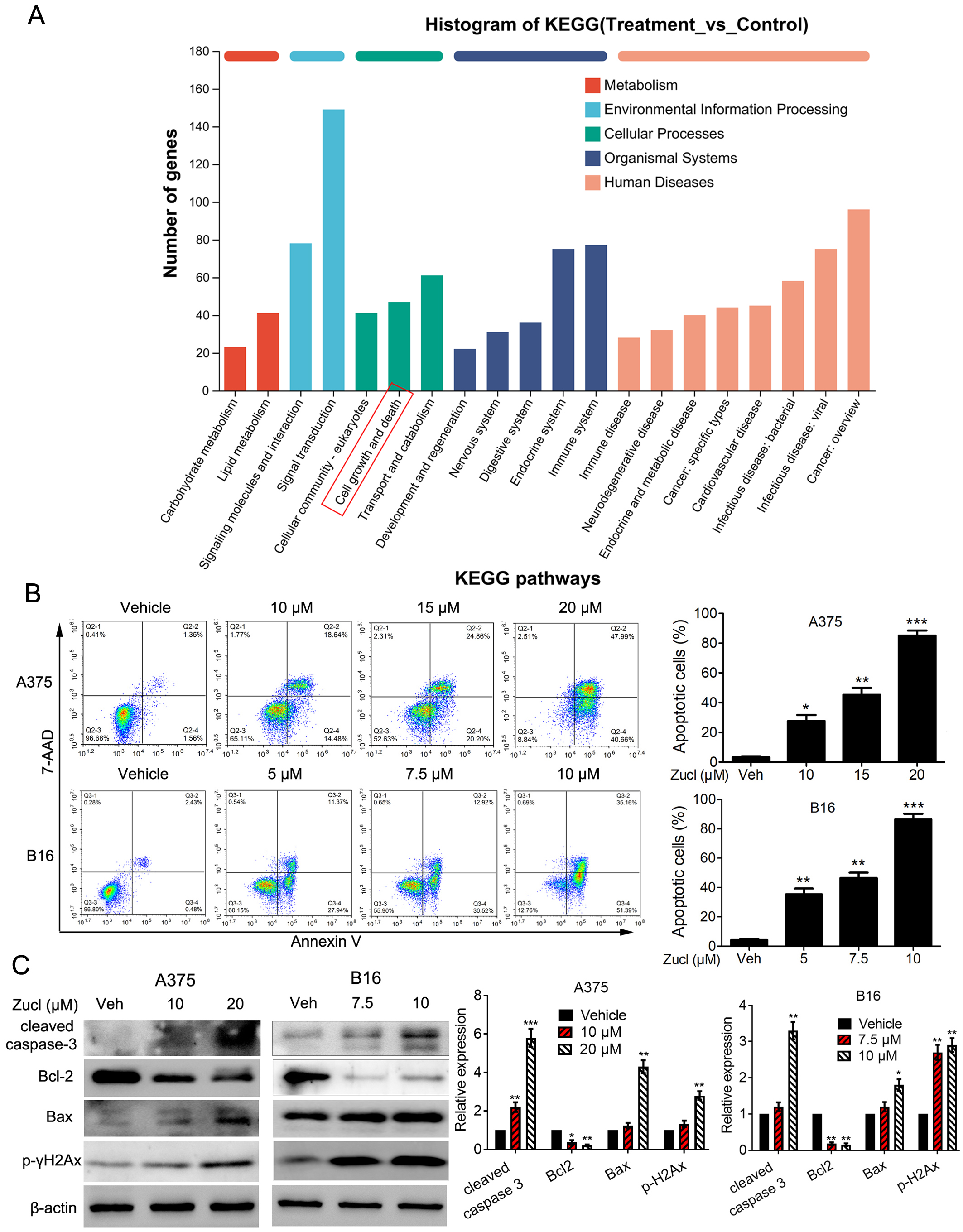
KEGG analysis of the DEGs identified
after zuclopenthixol treatment indicated enrichment for pathways related to cell
growth and death (Fig. 4A). This implies that the mechanism for zuclopenthixol
inhibition of melanoma may involve cell demise. Subsequent FCM analysis revealed
that treatment with zuclopenthixol induced significant apoptosis of melanoma
cells (Fig. 4B). Western blot analysis showed that administration of
zuclopenthixol resulted in upregulation of cleaved caspase 3, an essential factor
in apoptosis (Fig. 4C). Additionally, zuclopenthixol markedly increased the
expression of phosphorylated
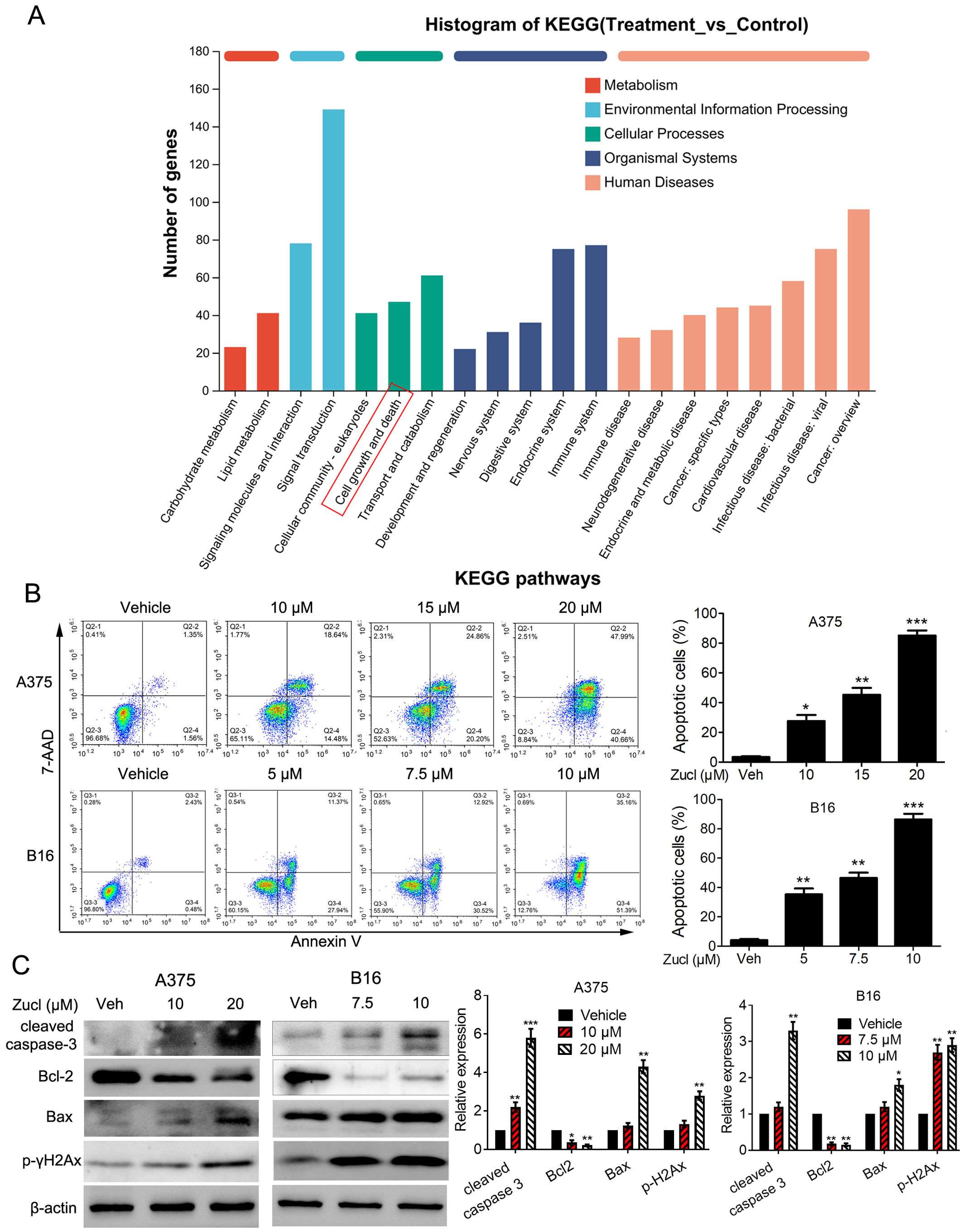 Fig. 4.
Fig. 4.Melanoma cells undergo apoptosis when exposed to
zuclopenthixol. (A) Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of
DEGs. (B) Effect of zuclopenthixol on melanoma cell apoptosis. FCM was used to
analyze the apoptosis of A375 and B16 cells after treatment with various
concentrations of zuclopenthixol for 24 h. (C) Apoptosis-associated protein
expression in cells evaluated by Western blot after 24 h treatment with
zuclopenthixol. *p-value
Two primary categories of apoptosis are currently recognized. The first is the
extrinsic apoptosis pathway mediated by cell death receptors, and the second is
the intrinsic apoptosis pathway mediated by mitochondria. Various drugs induce
apoptosis in tumor cells through the intrinsic pathway of programmed cell death.
To investigate whether this pathway is involved in zuclopenthixol-induced
melanoma apoptosis, we examined the expression of important regulatory proteins
within the BCL2 family, as these play crucial roles in maintaining mitochondrial
integrity. Treatment with zuclopenthixol significantly upregulated the expression
of Bax, while simultaneously downregulating the expression of BCL2 (Fig. 4C).
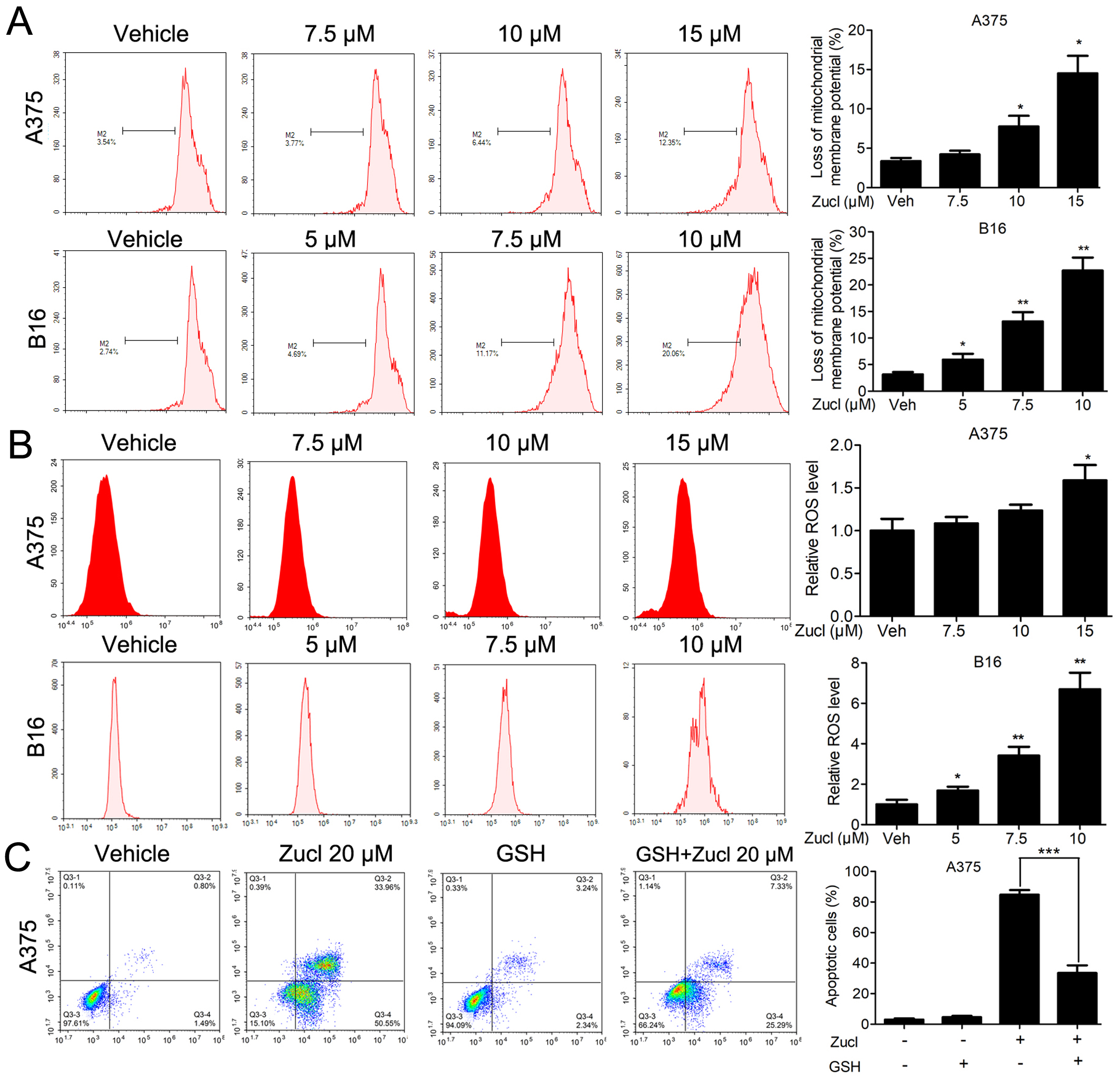
Zuclopenthixol also decreased the
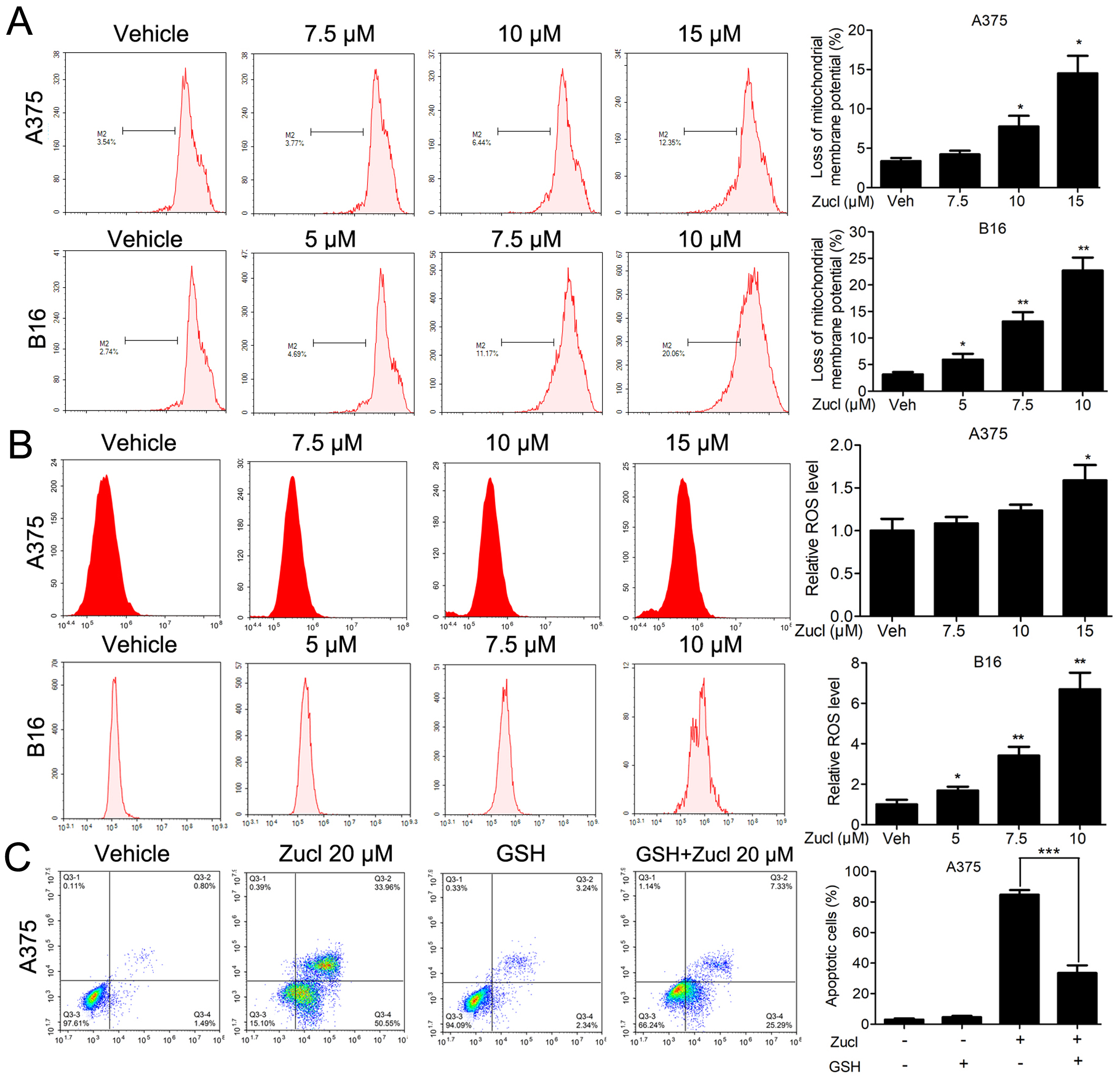 Fig. 5.
Fig. 5.Zuclopenthixol induces apoptosis of melanoma cells via the
mitochondrial-mediated intrinsic pathway. (A,B) Following treatment with various
concentrations of zuclopenthixol for 24 h, cells were stained with Rh123 or
dichloro-dihydro-fluorescein diacetate (DCFH-DA). FCM analysis was then performed
to assess the decrease in the mitochondrial membrane potential
(
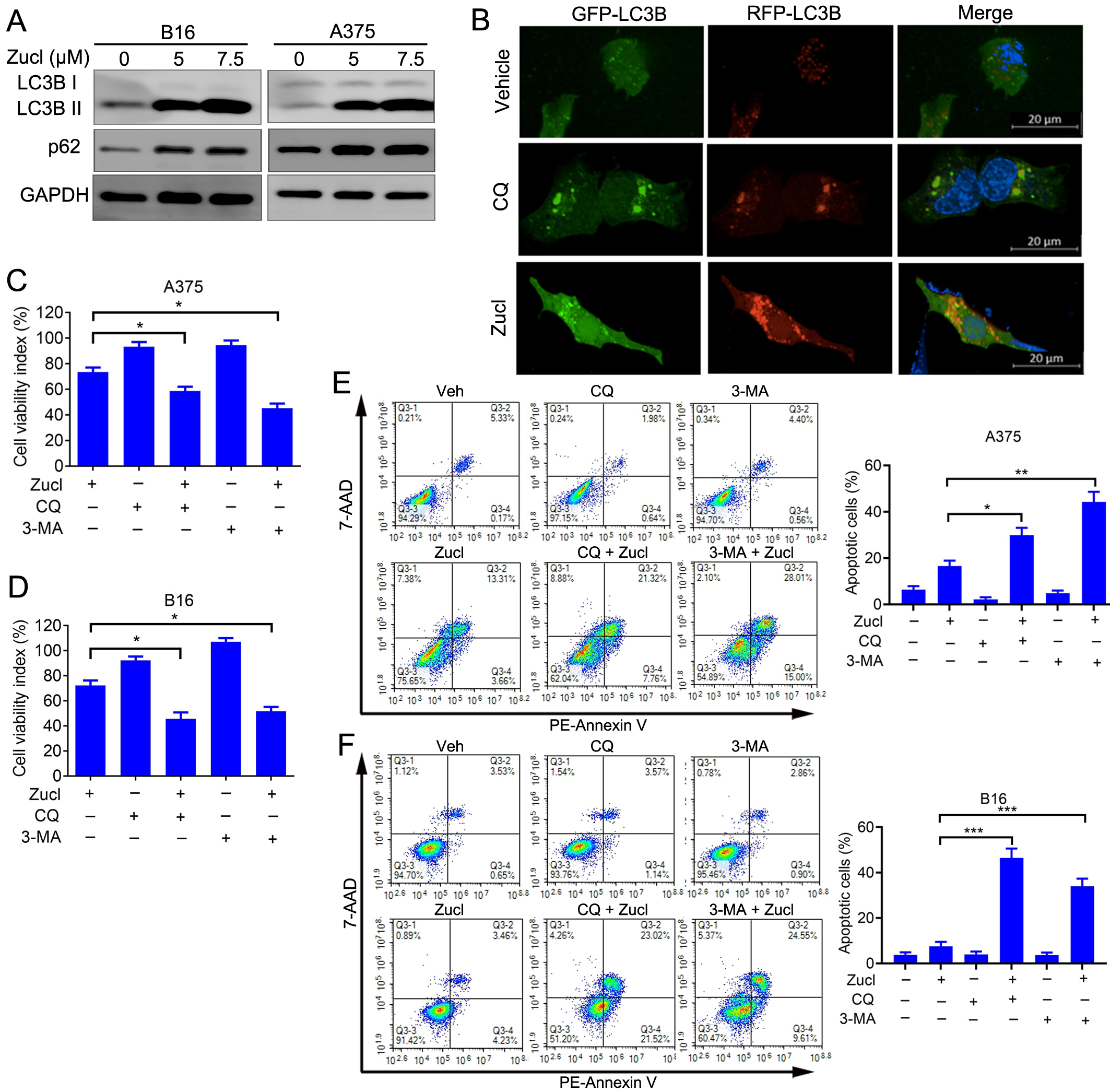
Analysis of the Reactome annotations in A375 cells revealed enrichment for autophagy following treatment with zuclopenthixol (Fig. 6A). We therefore speculated that zuclopenthixol might regulate autophagy in melanoma cells. Autophagy markers include the conversion of LC3-I to LC3-II, and the development of LC3 puncta. Treatment of melanoma cells with zuclopenthixol increased the conversion of LC3-I to LC3-II (Fig. 6A), as well as increasing the level of p62, a specific substrate for autophagy and an indicator of autophagy flux (Fig. 6A). These results indicate that zuclopenthixol can hinder the flow of autophagy. To determine whether this may be caused by the deficient fusion of autophagosomes with lysosomes, an LC3B construct labeled with tandem red fluorescent protein-green fluorescent protein (RFP-GFP) was utilized. The majority of LC3B puncta showed signal for RFP+GFP+ (autophagosomes) rather than RFP+GFP- (autolysosomes) (Fig. 6B). Therefore, treatment with zuclopenthixol results in the accumulation of autophagosomes, a reduction in the formation of autolysosomes, and the accumulation of LC3 puncta.
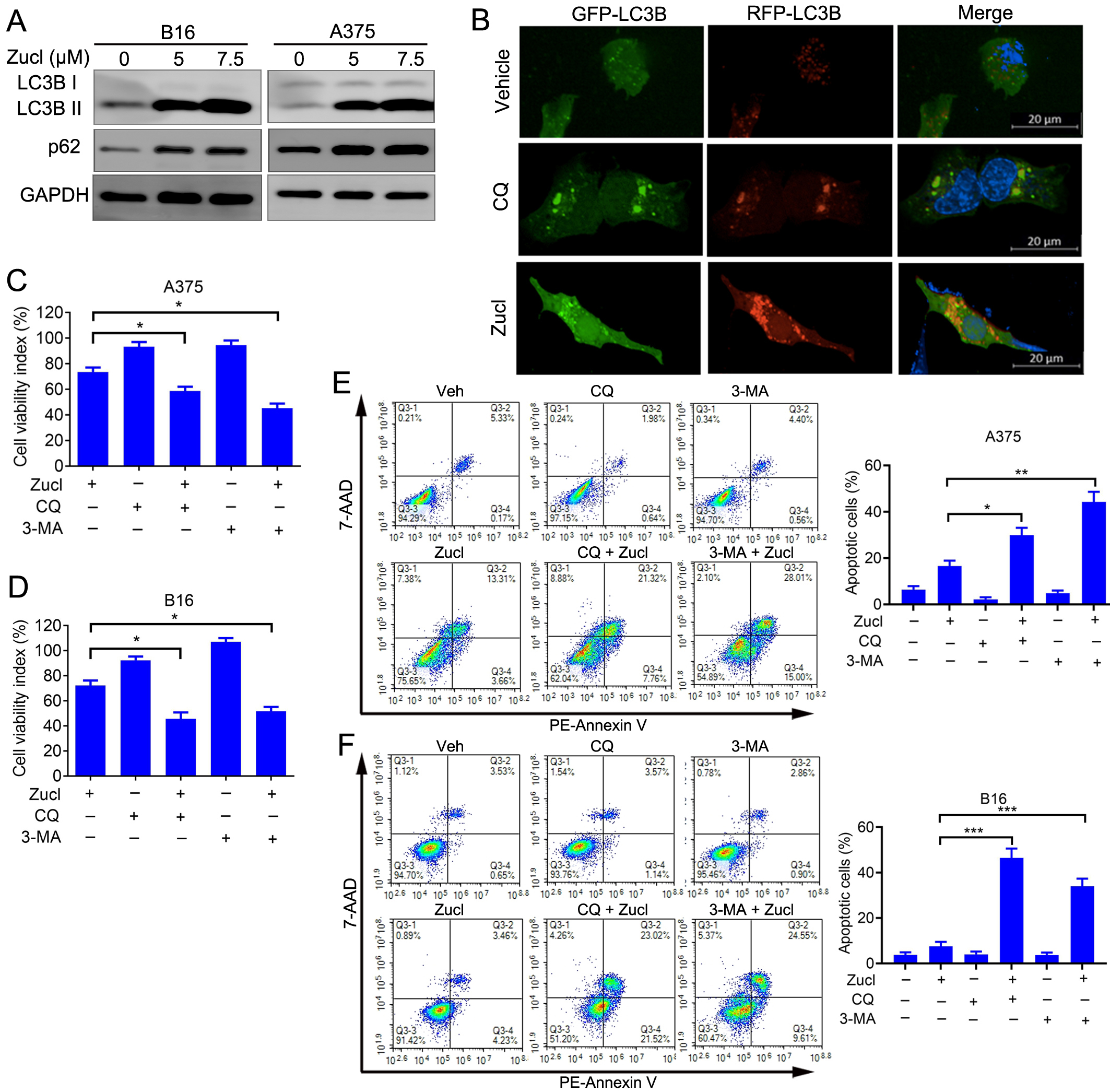 Fig. 6.
Fig. 6.Zuclopenthixol triggers protective autophagy and inhibits
autophagy flow in melanoma cells. (A) The expression of LC3 and p62 in B16 and
A375 cells was assessed after 24 h treatment with zuclopenthixol. (B)
Representative images showing A375 cells transfected with an LC3B plasmid labeled
with RFP-GFP in a tandem arrangement after being treated with zuclopenthixol
(scale bar = 20 µm). (C) Viability of A375 cells treated with
zuclopenthixol (7.5 µM) with or without chloroquine (CQ) (5 µM) or
3-methyladenine (3-MA) (1.5 mM). (D) Viability of B16 cells treated with
zuclopenthixol (5 µM) with or without CQ (7.5 µM) or 3-MA (2 mM). (E)
Rate of programmed cell death in A375 cells following exposure to zuclopenthixol
(7.5 µM) alone or in conjunction with CQ (5 µM) or 3-MA (1.5 mM) for
48 h. PE, phycoerythrin. (F) Rate of programmed cell death in B16 cells following treatment with
zuclopenthixol (5 µM) alone or in combination with CQ (7.5 µM) or
3-MA (2 mM) for 48 h. *p
The above findings indicate that zuclopenthixol may impede autophagy flux in melanoma cells. To better understand the mechanism by which zuclopenthixol exerts anti-melanoma effects, it was administered to A375 and B16 cells together with two frequently employed autophagy inhibitors. These were chloroquine (CQ), which hinders the fusion of autophagosomes with lysosomes at a late stage, and 3-methyladenine (3-MA), which is an early-stage inhibitor that impedes PI3K activity. The growth of A375 and B16 cells was significantly inhibited by the combination of zuclopenthixol and both autophagy inhibitors, as shown in FFig. 6C,D. Similarly, the combination of zuclopenthixol and autophagy inhibitors significantly increased apoptosis in these cells (Fig. 6E,F). Overall, zuclopenthixol triggers a defensive process of self-degradation in cells. Therefore, additional suppression of this process could potentially strengthen the ability of zuclopenthixol to inhibit melanoma cells.
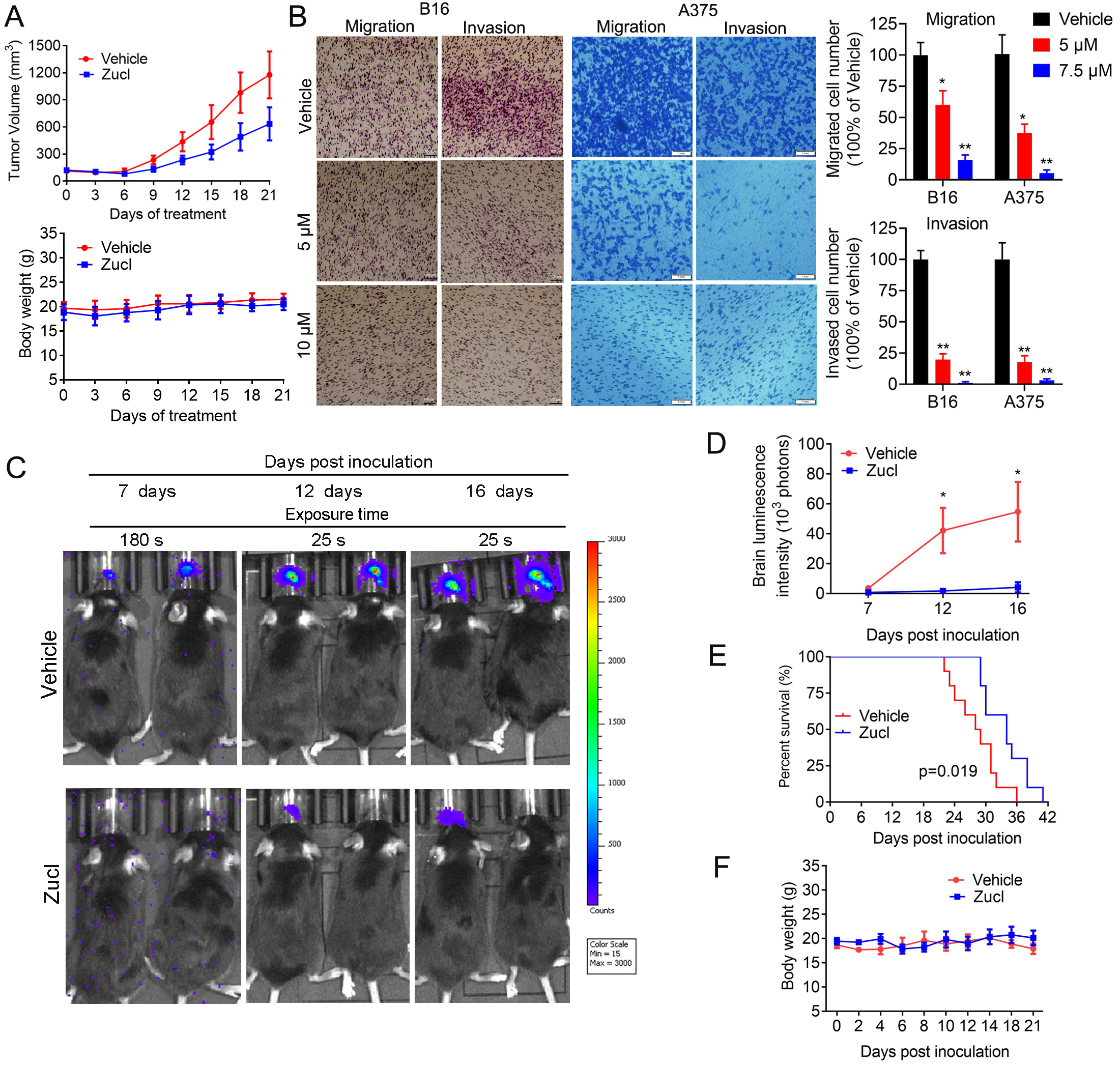
After establishing the subcutaneous melanoma model with A375 cells in BALB/c nude mice, we investigated the in vivo efficacy of zuclopenthixol for inhibiting melanoma growth. Zuclopenthixol effectively inhibited the growth of melanoma in mice, without causing a notable effect on their body weight (Fig. 7A). These findings highlight the potential of zuclopenthixol as a treatment drug for melanoma. Given its ability to penetrate the BBB, we hypothesized that zuclopenthixol may also hold promise for treating MBM. To explore this, we initially assessed its capacity to inhibit the migration and invasion of B16 and A375 cells, two critical processes involved in cancer metastasis. Zuclopenthixol significantly suppressed both processes in vitro, indicating that it could potentially suppress metastasis in vivo (Fig. 7B). In a mouse model of B16 cell brain metastasis, zuclopenthixol strongly inhibited intracranial metastatic tumor growth (Fig. 7C,D), leading to prolonged survival of mice (Fig. 7E). Of note, treatment with zuclopenthixol did not significantly decrease mouse body weight (Fig. 7F), thus providing preliminary evidence of its safety in vivo. Collectively, these findings suggest that zuclopenthixol has great potential for the effective treatment of melanoma and MBM.
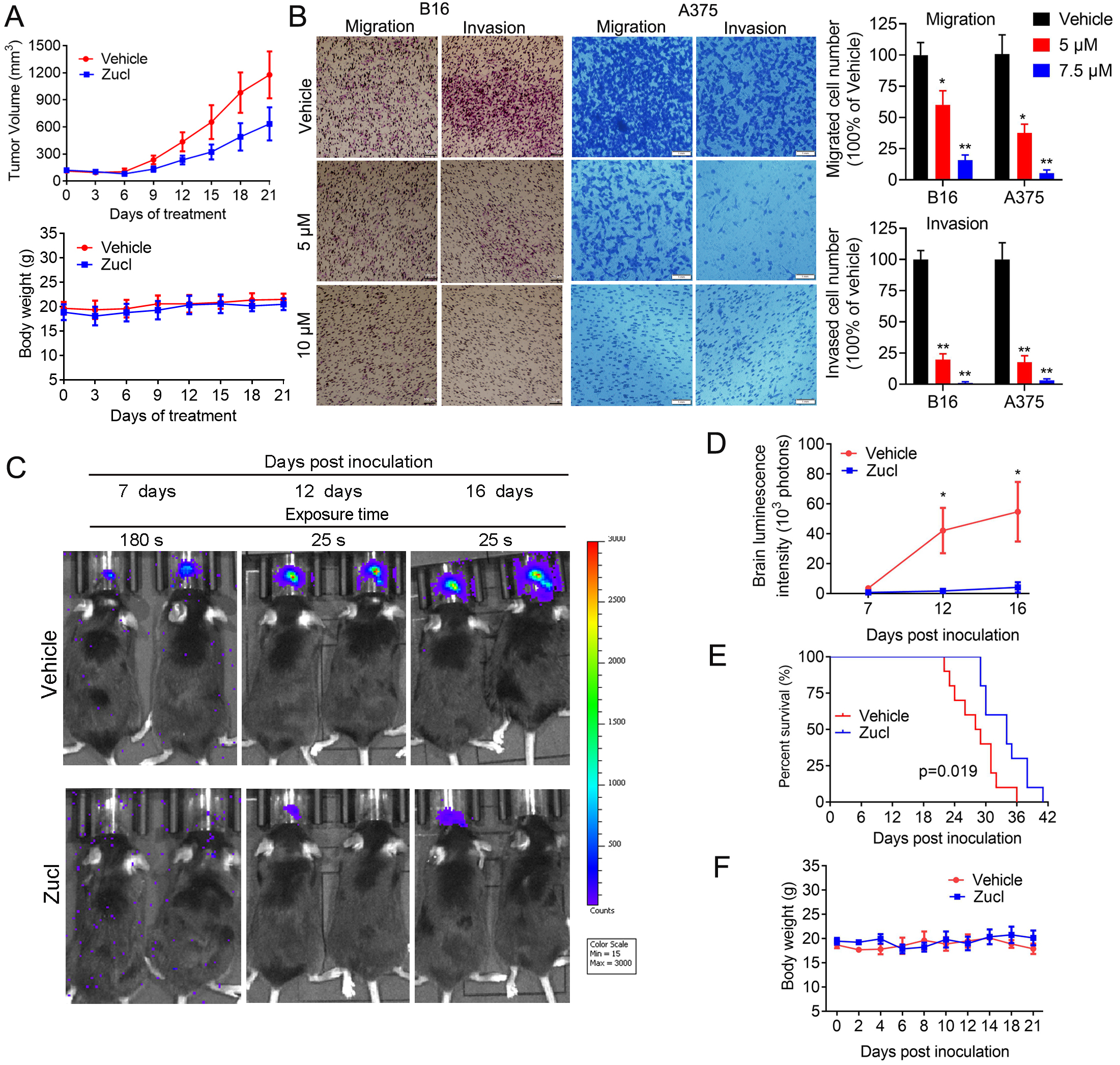 Fig. 7.
Fig. 7.In vivo efficacy of zuclopenthixol for the suppression
of melanoma growth and brain metastasis. (A) Upon reaching a mean tumor size of
approximately 100 mm
The efficacy of treatments for MBM prior to the advent of targeted therapies and immunotherapies has been poor, with patients experiencing a median overall survival of just 4–5 months [22]. While immunotherapy and targeted therapies have improved treatment outcomes, the survival of patients with MBM is still unsatisfactory [5], highlighting the urgent need for more effective treatment strategies. A major obstacle to the effectiveness of treatment for MBM is the BBB, which restricts drug penetration into the brain. Drugs that are able to cross the BBB and effectively inhibit melanoma therefore have tremendous potential for MBM treatment [7]. Anti-psychotic drugs can penetrate the BBB and have been approved and extensively studied. Moreover, some have been shown to possess anti-cancer activities, including inhibitory effects on brain tumors [19, 23, 24]. In previous studies, zuclopenthixol showed inhibitory activity against melanoma and other cancer types, prompting further exploration of its potential for the treatment of MBM [20, 21, 25]. The present results demonstrate that zuclopenthixol can effectively suppress the proliferation of various melanoma cell lines at low concentrations, resulting in G0/G1 cell cycle arrest and mitochondria-mediated intrinsic apoptosis. Furthermore, zuclopenthixol showed notable efficacy in inhibiting melanoma growth and brain metastasis in mice. These findings indicate that zuclopenthixol holds considerable promise in the treatment of MBM.
The present investigation affirmed that zuclopenthixol induces G0/G1 arrest in melanoma cells. The success of CDK4/6 inhibitors in breast cancer treatment suggests that targeting of the cell cycle may also prove effective in the treatment of melanoma. Numerous proteins participate in regulating the cell cycle, including cyclin-CDK complexes. Cyclin D binds to CDK4 and CDK6 to induce E2F-dependent transcription, DNA replication, increases in cyclin E and CDK2 levels, and subsequent phosphorylation of Rb, thereby propelling cancer cells through the G1/S checkpoint [26, 27]. p27 acts as a pivotal inhibitor during the G1/S cell cycle transition, and its elevated expression impedes the function of cyclin-CDK complexes [28]. The current results indicate that zuclopenthixol reduces the expression of CDK2, CDK4 and cyclin D1, while enhancing p27 expression. This implies that zuclopenthixol may induce G0/G1 cell cycle arrest by disrupting the relevant cyclin-CDK complexes. However, further experiments are needed to determine whether this is a direct or indirect effect.
The initiation of apoptosis is a promising strategy for cancer treatment.
Currently, apoptotic pathways are primarily classified into two types. Extrinsic
apoptosis is triggered when death ligands bind to membrane receptors, especially
death receptors. This initiates the extrinsic apoptosis pathway, which is
accompanied by activation of caspase 8 [29]. The other type is referred to as
intrinsic apoptosis and is mediated by mitochondria. In this process, the
integrity of the mitochondrial membrane is compromised, thereby reducing
Evaluation of the anti-cancer effect of drugs in experimental animals is a critical step in predicting clinical efficacy. One of the primary goals of this study was to assess the potential of zuclopenthixol in the treatment of MBM. This was investigated using an experimental brain metastasis model. Current methods for establishing brain metastasis in mice include stereotactic-guided intracranial injection, left ventricular injection, intra-carotid artery injection, and spontaneous brain metastasis models [34]. Each of these methods has advantages and disadvantages, as detailed below. Intracranial injection does not include any phase of the metastasis cascade other than secondary organ outgrowth. Left ventricular injection may also lead to extracranial metastasis and is technically challenging, while spontaneous metastasis models are time-consuming and inefficient [34]. Here, we successfully mastered the technique of intra-carotid artery injection, thereby allowing an extensive study of brain metastasis. This model allows investigation of tumor cell circulation, BBB crossing, extravasation, colonization, and outgrowth in the brain. It does have some drawbacks, however, including its technical difficulty and the inability to study the tumor cell invasion process from tissue to blood vessels. Nevertheless, we observed that zuclopenthixol inhibited MBM more strongly than it inhibited subcutaneous tumors. A previous study found the concentration of fluphenazine in the mouse brain was much higher than in the plasma [35]. Since fluphenazine and zuclopenthixol are both anti-psychotic drugs that act in the brain, we speculate that the favorable therapeutic effect of zuclopenthixol on brain metastasis may be due to its high concentration in the brain.
Despite the promising results presented in this study, further work is required to advance the use of zuclopenthixol or its derivatives as effective anti-MBM drugs. The primary focus should be on elucidating the targets and the specific mechanisms of action. Previous studies demonstrated that anti-psychotic drugs can exhibit anti-cancer effects through various mechanisms. Liu et al. [19] reported that fluphenazine and trifluoperazine could inhibit the growth and migration of breast cancer cells by suppressing DRD2. Two other studies also highlighted the significant role of DRD2 in the anti-cancer activity of anti-psychotic drugs, including its inhibition of breast cancer cell self-renewal [36], and its inhibition of stress-induced malignant tumor progression [37]. Given that zuclopenthixol is a dopamine receptor antagonist used to treat schizophrenia, DRD2 could serve as a target for the anti-cancer activity of zuclopenthixol [20]. Interestingly, in the present study we noted that DRD2 expression in A375 cells decreased following zuclopenthixol treatment, indicating a possible effect on DRD2 expression. Further studies are required to examine the effect of zuclopenthixol on DRD2 expression in diverse melanoma cells, and to unravel the functions and mechanisms of DRD2 in zuclopenthixol-induced melanoma inhibition. The pivotal role of autophagy in the anti-cancer activity of anti-psychotics has recently attracted significant attention. The current study, together with our previous work and research conducted by Lima et al. [38], collectively suggest that a viable strategy for treating melanoma may be to regulate autophagy. Lima et al. [38] found that TXA1, a thioxanthone compound, reduced the viability of melanoma cells by modulating autophagy, thus indicating its potential as a lead compound in the development of autophagy regulators with anti-cancer properties. Consistent with our present study and with prior research, zuclopenthixol is able to impede autophagy flux. However, the treatment of melanoma with zuclopenthixol and trifluoperazine gives rise to distinct outcomes with regard to autophagy. Zuclopenthixol induces protective autophagy in melanoma cells, so the suppression of autophagy could potentially amplify its inhibitory effects. On the other hand, treatment of melanoma cells with the anti-psychotic drug trifluoperazine is likely to induce cytotoxic autophagy, as evidenced by the partial reversal of its cytotoxic effect with 3-MA [21]. Moreover, another study reported that trifluoperazine could inhibit brain metastasis by blocking autophagic flux [39].
Several other studies have explored the anti-cancer activity and mechanism of zuclopenthixol. This drug was discovered as a promising compound that mimics moesin and has the ability to attach directly to the juxta-membrane area of HER2. The attachment specifically hinders the activation of HER2 in HER2-positive breast cancer, and effectively slows the progression of HER2-positive brain tumors in animal models [25]. Another study that screened a library of 3398 FDA-approved drugs found that zuclopenthixol inhibited the proliferation and migration of pancreatic stellate cells and disrupted tumor-stromal interactions, thus attenuating pancreatic cancer growth [40]. Zuclopenthixol was also reported to be a potential regulator of N6-methyladenosine in esophageal cancer [20]. However, the specific targets through which zuclopenthixol exerts its anti-cancer effects in melanoma and MBM remain unclear. Identifying these targets should contribute towards the development of novel anti-cancer therapies.
Combination therapy has become a common and effective clinical approach for treating tumors. It can enhance treatment efficacy, reduce toxicity by lowering drug doses, and also mitigate or delay the development of drug resistance. Current treatment options for MBM include immunotherapy, BRAF-targeted therapy, surgical intervention, stereotactic radiosurgery, and whole-brain radiation therapy. The use of combination therapy for the management of MBM has demonstrated promising outcomes, particularly for the combination of anti-CTLA-4 and anti-PD-1 immunotherapies, and the combination of immunotherapy with radiation therapy [41]. In the current study, autophagy inhibitors were found to synergistically enhance the anti-melanoma activity of zuclopenthixol. Therefore, combining zuclopenthixol with autophagy inhibitors, especially approved anti-tumor drugs that inhibit autophagy, may achieve better therapeutic effects against melanoma and MBM. Given the success of combination therapies in MBM treatment, future studies should investigate the efficacy of combining zuclopenthixol with existing treatment approaches.
Zuclopenthixol and trifluoperazine are potential drugs for the treatment of melanoma and MBM. They were discovered by our group using a drug repurposing strategy and during the same screening process [21]. Overall, zuclopenthixol exhibits better in vitro anti-proliferative activity than trifluoperazine, although they are both within the same order of magnitude. They also share several mechanisms of action, such as inducing G0/G1 cell cycle arrest and mitochondria-mediated intrinsic apoptosis. In addition, both can induce autophagy in melanoma cells and disrupt autophagic flux, although the effects of induced autophagy differ. Trifluoperazine induces potentially cytotoxic autophagy, whereas zuclopenthixol induces cytoprotective autophagy. The current study also employed additional experimental approaches to demonstrate the effects of zuclopenthixol. For example, we used the antioxidant GSH, whereas studies on trifluoperazine used NAC. This study also utilized the GFP-RFP-LC3 plasmid to study autophagic flux. Zuclopenthixol and trifluoperazine have different chemical structures, thus providing a better source for drug repurposing. In summary, the present study and previous research have shown that anti-psychotics are promising drugs for the treatment of melanoma and MBM. Further research is required to clarify their activities and mechanisms, thus allowing translation into clinically effective therapeutic drugs. Gaining a better understanding of the structure-activity relationships of zuclopenthixol and trifluoperazine, as well as identification of their targets, should facilitate the development of more effective drugs for the treatment of MBM.
Zuclopenthixol is a promising agent for the treatment of MBM due to its demonstrated ability to cross the BBB and exert potent anti-cancer effects. The findings in this study strongly support further research and development of zuclopenthixol as a potential therapeutic agent for MBM, with the ultimate goal of improving patient outcomes and quality of life for those affected by this devastating condition.
Data used and/or analyzed during the current study are available from the corresponding author on reasonable request.
JZ designed the research study. WL, YX and AH performed the research. WL and SC analyzed the data. WL wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The animal experiment was approved by the Ethics Committee of Sichuan University (No. 2021030544).
Not applicable.
This research was funded by the National Natural Science Foundation of China, grant number 81703083; the General Project of Chongqing Natural Science Foundation, grant number cstc20jcyj-msxmx0110.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.