






 , Peng Wang 1, Zhao Yang 2, Yue Dai 2, Sheng Wang 1,*
, Peng Wang 1, Zhao Yang 2, Yue Dai 2, Sheng Wang 1,*
1 Department of Vascular Surgery, Beijing Anzhen Hospital, Capital Medical University, 100029 Beijing, China
2 Department of Cardiac Surgery, Beijing Anzhen Hospital, Capital Medical University, 100029 Beijing, China
Abstract
Background: The endogenous
metabolism of polyunsaturated fatty acids is regulated by the fatty acid
desaturase (FADS) gene cluster and is strongly associated with diseases such as
atherosclerosis, dyslipidemia, and type 2 diabetes. However, the association
between FADS and atherosclerosis remains a subject of debate. Methods:
In this study, we specifically investigated the physiological role of
Keywords
- Δ-5 fatty acid desaturase
- atherosclerosis
- antisense oligonucleotides
- angiopoietin-like 4
Fatty acid desaturase
(FADS) is a critical enzyme in polyunsaturated fatty acid (PUFA) metabolism, and
gene polymorphisms in FADS affect the activity and function of FADS [1, 2], which
in turn affect metabolic activities in the body such as lipid concentrations,
cardiovascular disease risk, pregnancy, cognitive function, Alzheimer’s disease,
overweight, and type 2 diabetes mellitus [3, 4, 5]. Genetic studies have shown that
variants in Fads1, encoding the
Mice with Fads1 knockout or knockdown exhibited different phenotypes in atherosclerosis for unclear reasons. Regardless of the type of model mice, the phenotype may be associated with the production of specialized pro-resolving mediators or pro-inflammatory mediators regulated by FADS1 [12, 13, 14]; however, it is unknown whether Fads1 affects atherosclerosis by regulating specific signaling pathways. Studies have shown that FADS1 overexpression can promote carcinogenesis and angiogenesis by activating AKT/mammalian target of rapamycin (mTOR) signaling, meanwhile activation of phosphoinositide 3-kinase/AKT/mTOR signaling can promote the occurrence and development of atherosclerosis by aggravating the inflammatory response and causing endothelial dysfunction [15]. Given the regulatory role of Fads1 in atherosclerosis, we speculate that Fads1 can regulate endothelial cell function through AKT/mTOR signaling and experimentally explored the downstream proteins of this axis; this protein may ultimately influence atherosclerosis.
To overcome the obstacle of Fads1
Collagenase (type IV) (Cat No. C4-BIOC) used for the perfusion, Percoll (Cat No.
P1644) used for the density gradient centrifugation, and trypsin (Cat No. T4049)
were purchased from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s Modified
Eagle’s Medium (DMEM; Cat No. 11995065), Opti-Minimal Essential Medium (Opti-MEM
Cat No. 31985070), and fetal bovine serum (FBS; Cat No. A5670701) were purchased
from Gibco (Waltham, MA, USA). D-Hank’s (Hank’s Balanced Salt Solution; Cat No.
14175103) and Lipofectamine 2000 (Cat No. 11668027) were purchased from
Invitrogen (Carlsbad, CA, USA). ASOs were produced by Synbio Technologies
(Suzhou, China). The NCTC-1469 adherent cell line (Cat No. SCC-220211) and
Mycoplasma Detection Kit (Cat No. CA1080) were purchased from Solarbio Technology
Co., Ltd. (Beijing, China). Hematoxylin and eosin (H&E) staining reagent (Cat
No. G1003) and Oil Red O staining reagent (Cat No. G1015) were purchased from
Servicebio (Wuhan, China). Antibodies against cluster of differentiation 31
(CD31) (Cat Nos. GB11063-1, GB12063), alpha smooth muscle actin (
NCTC-1469 cells were cultured in DMEM containing 10% FBS. Subculturing was
performed when the cells had grown to logarithmic phase. Primary mouse
hepatocytes isolated from 8-week-old male C57BL/6J wild-type mice by collagenase
perfusion (type IV) [18] were cultured in DMEM containing 10% FBS. Primary mouse
hepatocytes were identified by cytokeratin-18 immunofluorescence, and the purity
could be more than 90%. Cells were all cultured in a humidified incubator at 37
°C and 5% CO
Fads1-ASO (GGTGCATGTTGATATCGGGG) targeting mouse FADS1 mRNA and
control-ASO (ATTCGACGGGCTTTACGTTG), which does not target any sequence in the
mouse genome, were developed and synthesized by Synbio Technologies. Each ASO
consisted of five nucleotides on the 5
| ASO | Target | Sequence | Calcd. Mass |
| Antisense-1 | Mouse Fads1 | FITC-GGTGCATGTTGATATCGGGG | 7580.0 |
| Antisense-2 | Mouse Fads1 | FITC-GGTGCATGTTGATATCGGGG-GalNAc | 9691.6 |
| Antisense-3 | Mouse Fads1 | GGTGCATGTTGATATCGGGG-GalNAc | 9154.0 |
| Control | Mouse None | FITC-ATTCGACGGGCTTTACGTTG | 7444.6 |
Note: FITC probe modification of the 5
(1) One day before transfection, 5–8
(2) For each transfection sample, ASO–Lipofectamine 2000 complexes were prepared as follows: 1 µg ASO was diluted in 50 µL Opti-MEM, and mixed gently.
(3) The stock Lipofectamine 2000 was mixed gently before use, and then 3 µL was diluted in 50 µL Opti-MEM. The solution was mixed gently and left to stand for 5 min at room temperature.
(4) Five minutes after dilution of Lipofectamine 2000, the diluted ASO was combined with the diluted lipid (total volume was 100 µL). The solution was mixed gently and left to stand for 20 min at room temperature to allow the ASO–Lipofectamine 2000 lipoplexes to form.
(5) The transfection complex (100 µL) was added to each well, followed by gentle mixing by rocking the plate back and forth.
(6) The cells were incubated at 37 °C in a humidified incubator with 5% CO
After verifying that ASO could effectively inhibit Fads1 expression,
ASO was modified by Tri-GalNAc (i.e., antisense-3) (Table 1), and its
transfection efficiency was verified in vitro. The isolated primary
hepatocytes from 8-week-old male wild-type mice in the C57BL/6J genetic
background were plated in a 12-well plate at 80–90% density, followed by the
addition of 0.8 mL Dulbecco’s Modified Eagle Medium (DMEM) (without antibiotics). Cells were cultured in an
incubator with 5% CO
To verify the effectiveness of ASO-Tri-GalNAc (i.e., antisense-3), ApoE
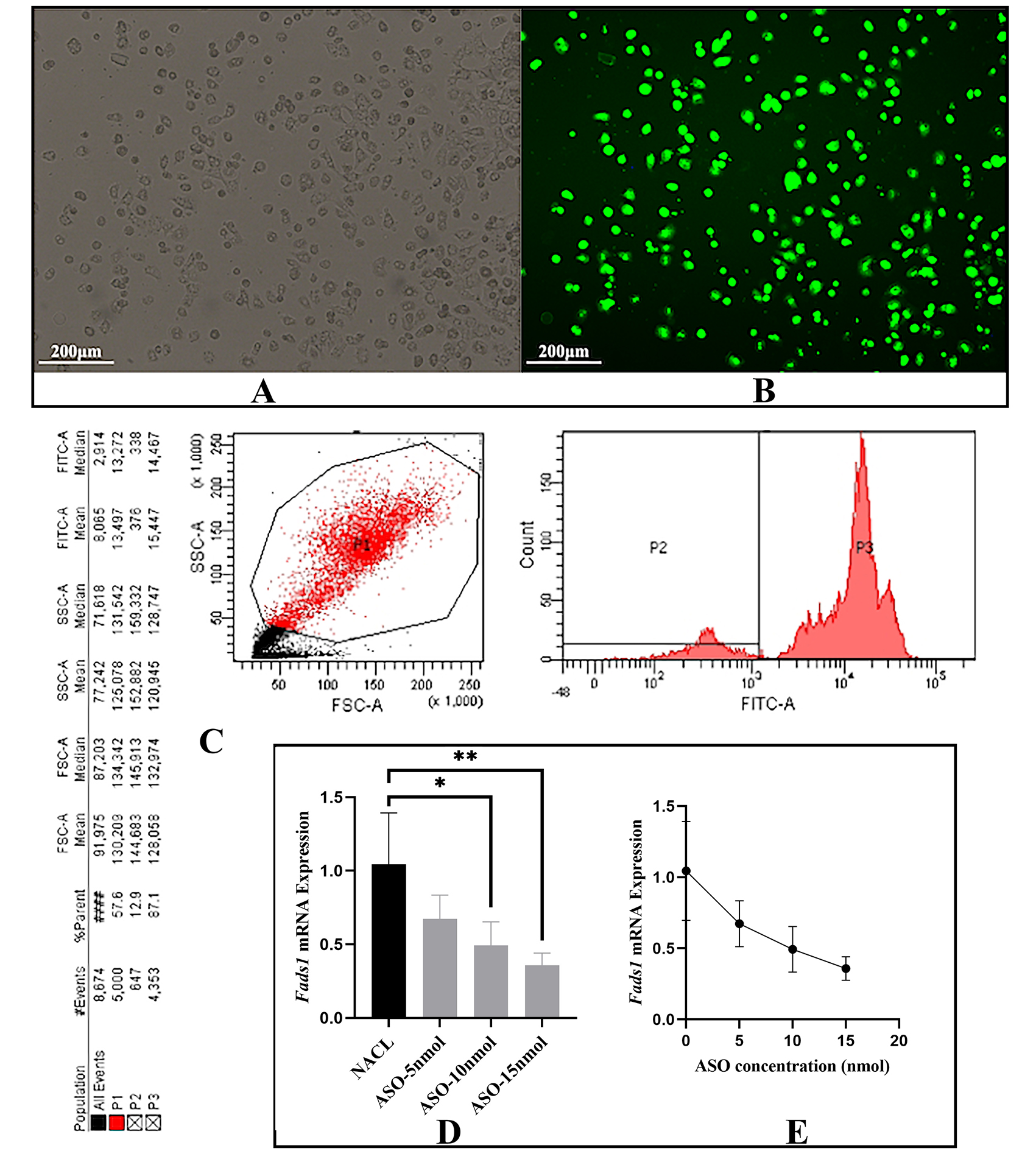 Fig. 1.
Fig. 1.In vitro transfection rate
determination and in vivo validation of effectiveness of
ASO-Tri-GalNAc. (A) Optical microscopic view of mouse
hepatocytes. (B) Fluorescence microscopic view of ASO-Tri-GalNAc transfected into
hepatocytes under the same field of view. (C) Flow cytometry to determine the
number of cells transfected with ASO-Tri-GalNAc as a percentage of total cells. (D,E) ASO-Tri-GalNAc
effectively inhibited Fads1 expression in the liver of ApoE
This study was conducted following the regulations of the Animal Ethics
Committee of Beijing Anzhen Hospital (2022174X; Beijing, China). Wild-type and
ApoE
ApoE
Mice were perfused from the left ventricle with PBS and heparin (250 U/kg). The aorta and three branches of the arch of mice were separated and fixed in 4% paraformaldehyde solution for subsequent Oil Red O staining. After complete dissection of bilateral femoral arteries, they were placed in optimal cutting temperature compound, rapidly frozen in liquid nitrogen, and stored at –20 °C for subsequent H&E, Oil Red O, and immunostaining. Part of the liver tissue was taken and treated similarly for subsequent Oil Red O staining [21, 22].
The fat on the periphery of the aorta was removed, and the blood vessel was carefully dissected longitudinally along the vessel wall with scissors, followed by staining in Oil Red O solution, differentiation in 60% isopropanol, and washing with distilled water to terminate the differentiation. Finally, photos were taken (D70; Canon, Tokyo, Japan). Multiple sections (7 µm) were prepared and stained for morphological observation with H&E, Oil Red O, and immunofluorescence. H&E and Oil Red O staining were conducted according to the instructions for the experimental reagents. For immunocytochemistry staining, sections were sealed by incubation in 3% BSA for 90 min at room temperature and then incubated with primary antibody at 4 °C overnight. Then the sections were rewashed five times with PBS (30 s each), followed by incubation with fluorescently labeled secondary antibodies for 30 min at room temperature. After washing three times with PBS (30 s each), samples were visualized with the Nikon Eclipse 80i upright microscope (Nikon, Tokyo, Japan). Quantitative analysis of histological staining and fluorescence was conducted using ImageJ software [23].
Each mouse was weighed weekly at a fixed time starting from 8 weeks old; mice were fed ad libitum before weighing. Glucose tolerance tests were performed after a 16 h fast by injecting 1 g/kg body weight glucose into the peritoneal cavity. Tail vein blood glucose levels were measured using a commercial glucometer (One Touch; Johnson & Johnson, New Brunswick, NJ, USA). Glucose tolerance tests were performed on three groups of mice at 20 weeks old.
At the end of the experiment, the carotid artery was severed to collect blood from mice. The blood was collected with a 1 mL syringe moistened with heparin beforehand. The blood was centrifuged at 10,000 g for 10 min at 4 °C to remove any remaining insoluble material and then the plasma samples were stored at –80 °C. The contents of triacylglycerol, total cholesterol, high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C) in the plasma of mice were detected with an automatic biochemical analyzer (Chemray 800; Rayto Life and Analytical Sciences, Shenzhen, China) with the test kits (Cat Nos. K068b, K074a, K075, K076; Changchun Huili Biotech Ltd., Changchun, China). The enzyme-linked immunosorbent assay (ELISA) was used to detect the inflammatory cytokines using the enzyme label detector (BioTeK, Epoch, Winooski, VT, USA) with the test kits (Cat Nos. 88-7324 and 88-7013; Thermo Fisher Scientific, Waltham, MA, USA; Cat Nos. 202/2-48, 204/2-48; Multisciences, Hangzhou, China).
RNA was isolated from cells and tissues using the Quick-RNA Purification Kit
(Cat No. RN001; ESscience Biotech, Beijing, China) according to the
manufacturer’s instructions. For mRNA expression analysis, 2 µg total RNA
was reverse transcribed using the GoScript™ Reverse Transcription
System (Cat No. A5001; Promega, Beijing, China). Quantitative PCR (qPCR) was
performed using the BlazeTaq™ Probe qPCR Mix (GeneCopoeia,
Rockville, MD, USA) on the CFX Connect Real-Time PCR Detection System (Bio-Rad,
Hercules, CA, USA). The expression values were normalized to the housekeeping
genes
Samples were homogenized in ice-cold lysis buffer consisting of 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA-2Na, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate (SDS). A total of 30 µg protein per sample was resolved by SDS-polyacrylamide gel electrophoresis (5%), electotransferred to polyvinylidene fluoride membrane, and imaged using the ChemiScope 6100 Imaging System (Clinx Science Instruments Co. Ltd., Shanghai, China). Primary antibodies for Western blotting were ANGPTL4 (Cat No. 18374-1-AP; Proteintech), GAPDH (Cat No. GB15004; Servicebio) raised in rabbit, and Prestained Protein Marker (Cat No. G2083; Servicebio) as internal standards. The secondary antibody for Western blotting was anti-rabbit conjugated to horseradish peroxidase (Cat No. GB23303; Servicebio).
Results are expressed as the mean
ASO-Tri-GalNAc (i.e., antisense-3) can enter mouse hepatocytes without
transfection reagents, and the transfection efficiency was about 85% (Fig. 1A–C). The expression of hepatic Fads1 was effectively inhibited by IP
injection in ApoE
There were no deaths in the three groups of high-fat diet ApoE
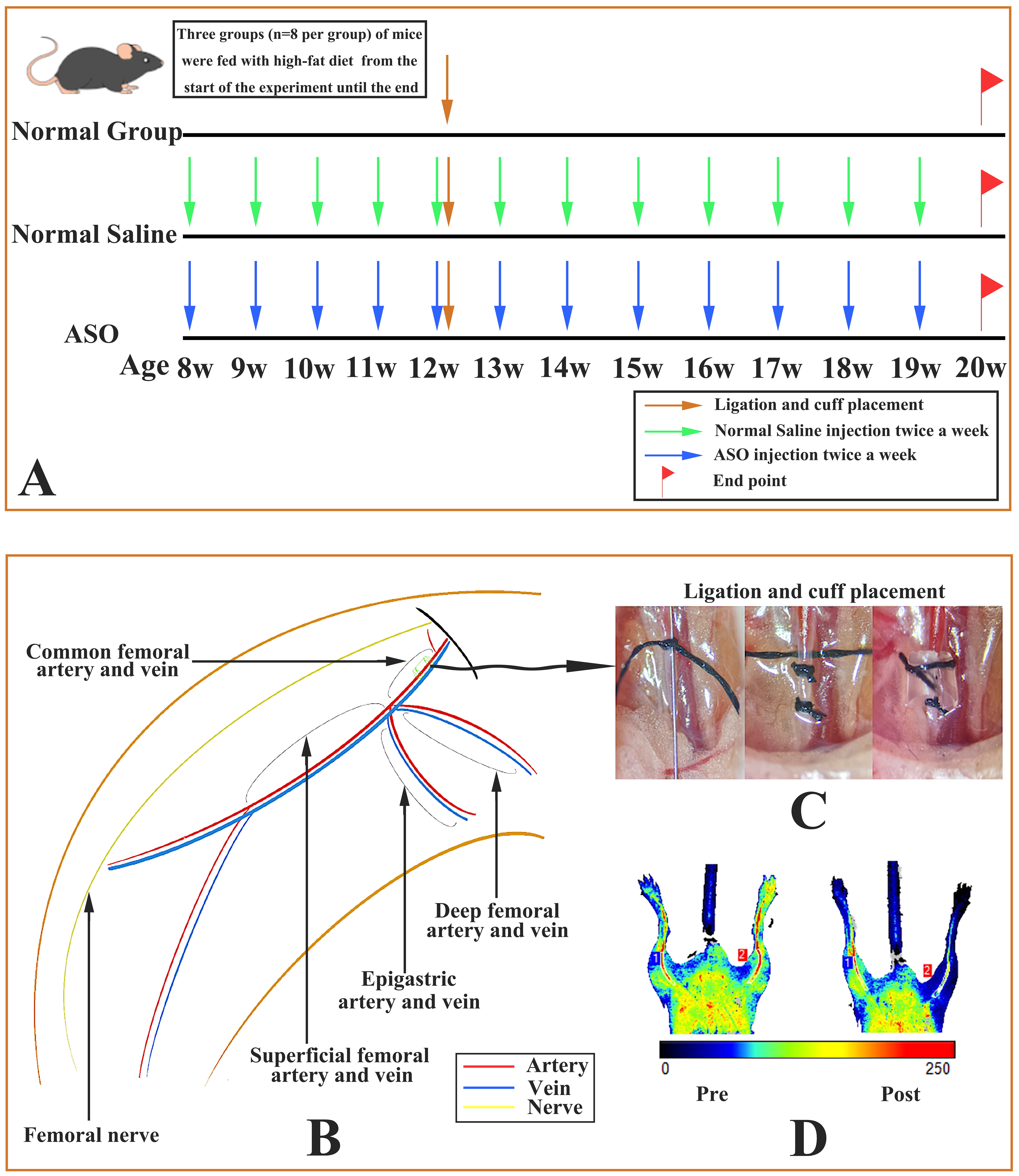 Fig. 2.
Fig. 2.Different interventions in the three groups of mice and modeling process of atherosclerosis in mouse femoral arteries. (A) During the experimental cycle, the three groups of mice were subjected to different interventions. (B) Vascular anatomy of the mouse right lower limb. (C) Right femoral artery cuff placement combined with tandem stenosis. (D) Preoperative (left) and postoperative (right) laser Doppler perfusion imaging of mice showed postoperative stenosis of the right femoral artery and decreased perfusion of the distal femoral artery.
We found that ASO-Tri-GalNAc-mediated knockdown of liver Fads1significantly aggravated atherosclerosis (Fig. 3A,B). The aortic root lesion area in the ASO Group statistically differed from the Normal Group but did not statistically differ from the Normal Saline Group; the area ratio of plaque/vessel in the ASO Group statistically differed from the Normal Group and Normal Saline Group (Fig. 3C,D). We also found that ANGPTL4 expression was upregulated in the aorta of the ASO Group, whereas ANGPTL4 expression was not found in the Normal Group or Normal Saline Group (Fig. 3E,F). Interestingly, FADS1 mRNA expression in the ASO Group was upregulated in the aorta, inconsistent with the downregulation of hepatic FADS1 mRNA expression (Fig. 3G). Further analysis of the expression levels of signaling pathways in the aorta revealed the upregulation mRNA expression of mTOR complex 1 (mTORC1), AKT1, and ANGPTL4 in the ASO Group compared to the Normal Saline Group (Fig. 3H–J).
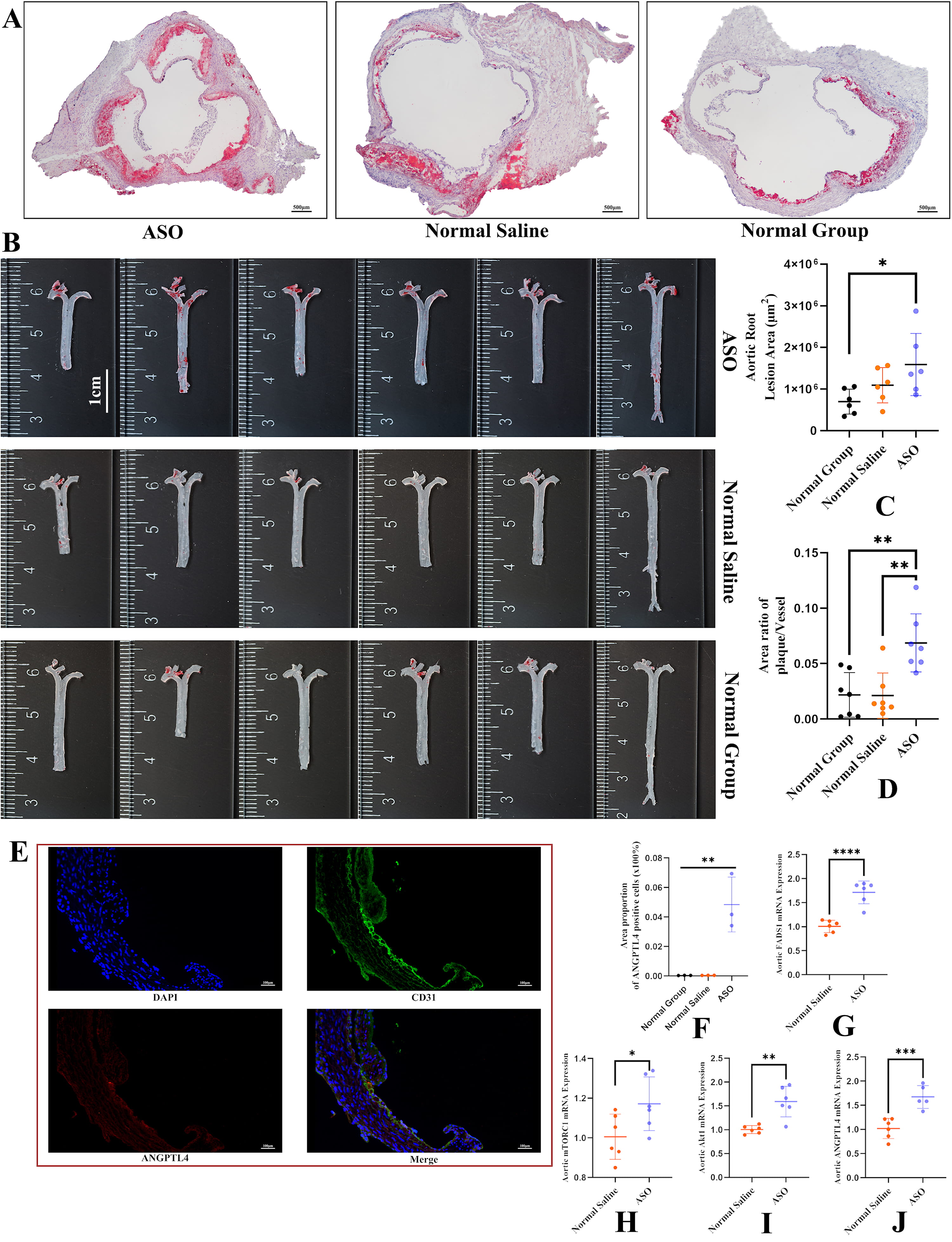 Fig. 3.
Fig. 3.Quantitative analysis of aortic plaque and upregulated
expression of angiopoietin-like 4 (ANGPTL4) in the aorta with more severe
atherosclerosis. (A) Representative Oil Red O staining of aortic roots in the
three groups. Scale bar, 500 µm. (B) Aortic Oil Red O staining in the three
groups. Scale bar, 1 cm. (C) Quantitative analysis of aortic root plaque area (n =
6). (D) Quantitative analysis of the ratio of plaque area to aortic area (n = 6).
(E) Representative images of aortic root from mice treated with
Fads1-ASO. Sections were immunostained with CD31, in green. Sections
were immunostained with ANGPTL4, in red. Scale bar, 100 µm. (F)
Quantitative analysis of the area proportion of ANGPTL4-positive cells (n = 3).
(G–J) FADS1, mTOCR1, AKT1, and ANGPTL4 mRNA expression in the aorta (abdominal
aorta to iliac artery); n = 6 per group. *, p
To explore the effects of hepatic Fads1 knockdown on the peripheral arteries, by establishing a model of femoral atherosclerosis and the femoral artery between the proximal and distal ligation sites was selected for Oil Red O staining, we found that selective knockdown of liver Fads1 had no significant effect on femoral atherosclerosis in mice (Fig. 4A). Three groups of mice both developed severe atherosclerosis in the femoral artery; however, there was no difference in the ratio of plaque area to the area under the internal elastic plate (Fig. 4B). The percentage of plaque area in the femoral artery was higher than that in the aortic root and aorta in all three groups of mice (Fig. 4C). Femoral artery immunofluorescence showed prominent infiltration of macrophages, proliferation of smooth muscle cells, incomplete arrangement of endothelial cells, and the upregulated expression of VCAM-1 in the femoral atherosclerotic sites of mice in the three groups (Fig. 4D). However, there was no difference in the area proportion of positive cells of the three groups (Fig. 4E–G).
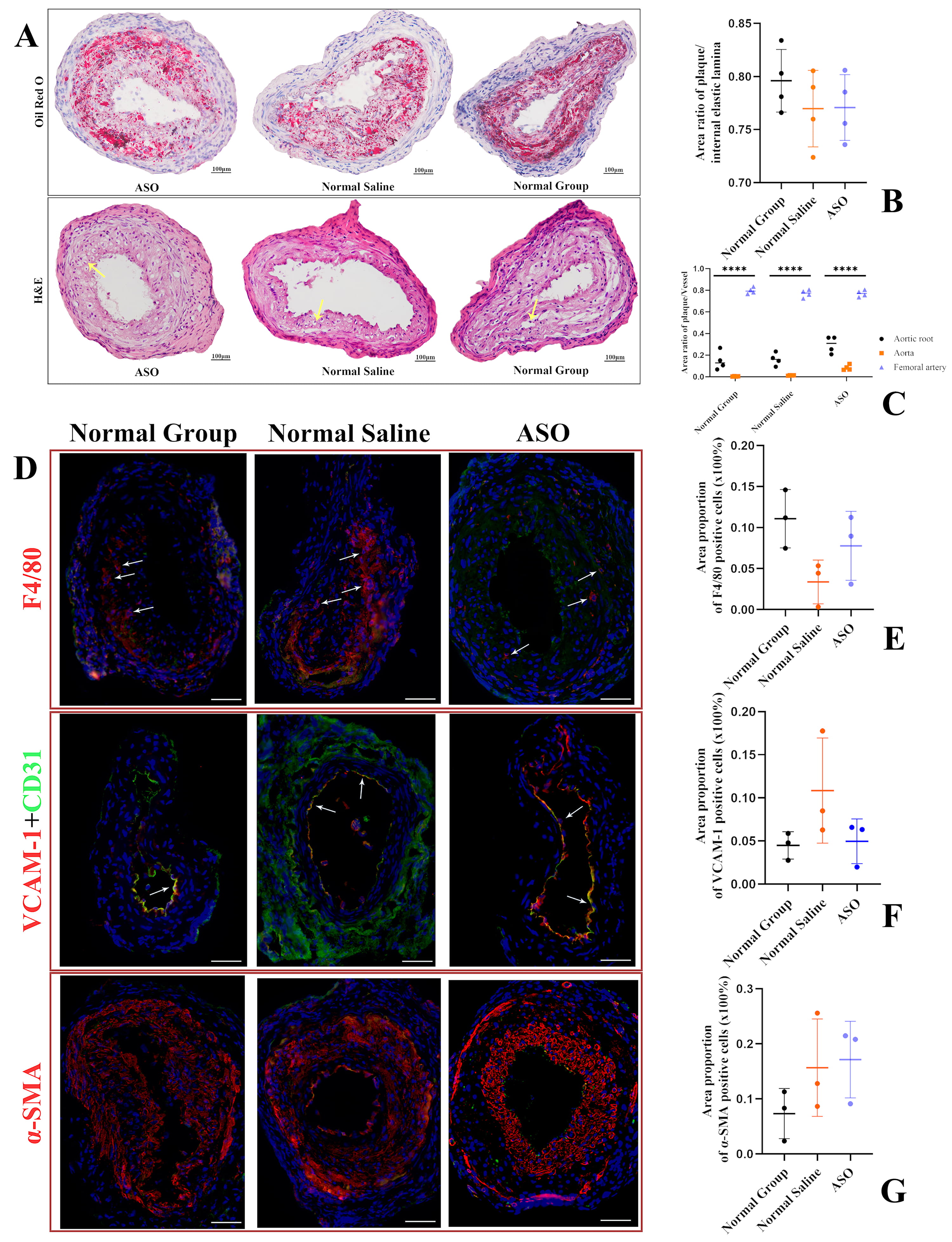 Fig. 4.
Fig. 4.Quantitative analysis of femoral artery plaque and
representative images of femoral artery from three groups. (A) Vessels between
the ligation sites of the two ends of the femoral artery were stained with Oil
Red O and H&E. Oil Red O staining showed atherosclerotic plaque formation and a
larger lipid core in all three groups. Hematoxylin-Eosin staining H&E staining showed disturbed endothelial
cell arrangement and foam cell aggregation. The arrow in the figure shows typical
foam cells. Scale bar, 100 µm. (B) The ratio of plaque
area to the area under the internal elastic plate (n = 4). (C) The ratio of
plaque area to the vessel or area under the internal elastic plate (n = 4). (D)
Sections were immunostained with F4/80, VCAM-1, and
We found that selective knockdown of liver Fads1 did not affect plasma lipid and inflammatory cytokine levels (Fig. 5A–G). There was no difference in the mRNA expression of pro-inflammatory interleukin 6 (IL-6) and anti-inflammatory IL-10 in the aorta between the Normal Saline Group and ASO Group (Fig. 5H,I). Previous studies have shown that Fads1 knockout mice generated by a gene-trapping strategy are lean relative to wild-type littermates [24]. To confirm whether selective knockdown of hepatic Fads1 would affect body weight, we monitored the body weight of mice and found a significant reduction in body weight at 19–20 weeks of age (Fig. 5J). Moreover, there was a trend of improved glycemic control in the ASO Group mice at 20 weeks of age (Fig. 5K).
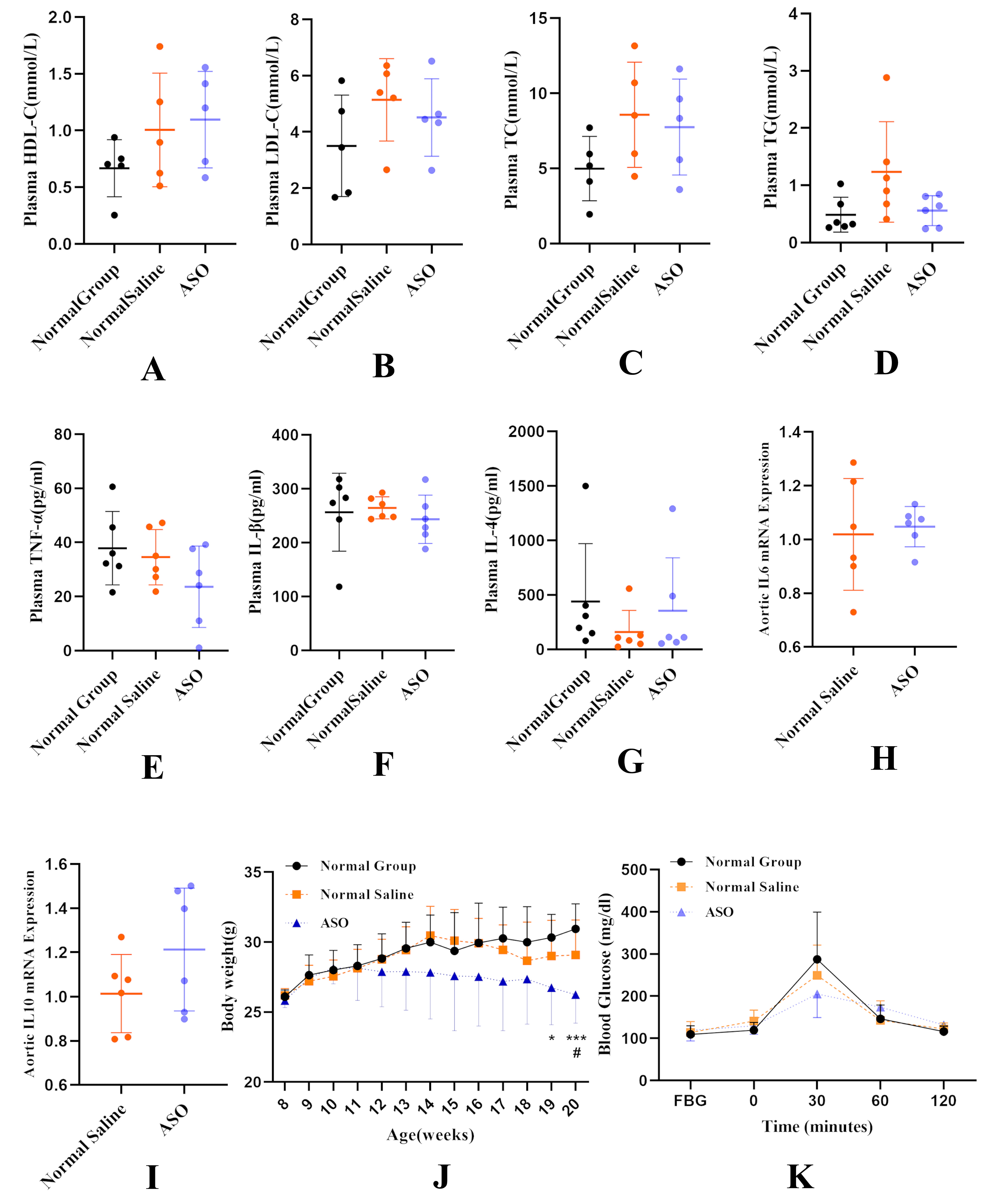 Fig. 5.
Fig. 5.Knockdown of liver Fads1 aggravated atherosclerosis
without significantly changing plasma lipid and inflammatory cytokine levels,
reduced body weight, and improved glycemic control. (A,B) High-density
lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C)
levels after 12 weeks of high-fat diet in the three groups; n = 6 per group.
(C,D) Plasma total cholesterol and total triglyceride; n = 6 per group. (E–G)
Plasma tumor necrosis factor
Liver Oil Red O staining showed that selective knockdown of liver Fads1 significantly aggravated non-alcoholic fatty liver disease (NAFLD) (Fig. 6A). Given that ANGPTL4 was highly expressed in liver and adipose tissue, we explored whether downregulation of hepatic Fads1 would affect ANGPTL4 production. Western blot analysis showed no significant correlation between them (Fig. 6B,C). ASO-Tri-GalNAc treatment resulted in the selective knockdown of hepatic Fads1 without altering FADS2 mRNA expression (Fig. 6D,E), showing that ASO-Tri-GalNAc had a specific effect and did not affect the expression of Fads2. In addition, selective knockdown of hepatic Fads1 did not significantly affect the weight of liver or white fat (epididymal fat) (Fig. 6F,G).
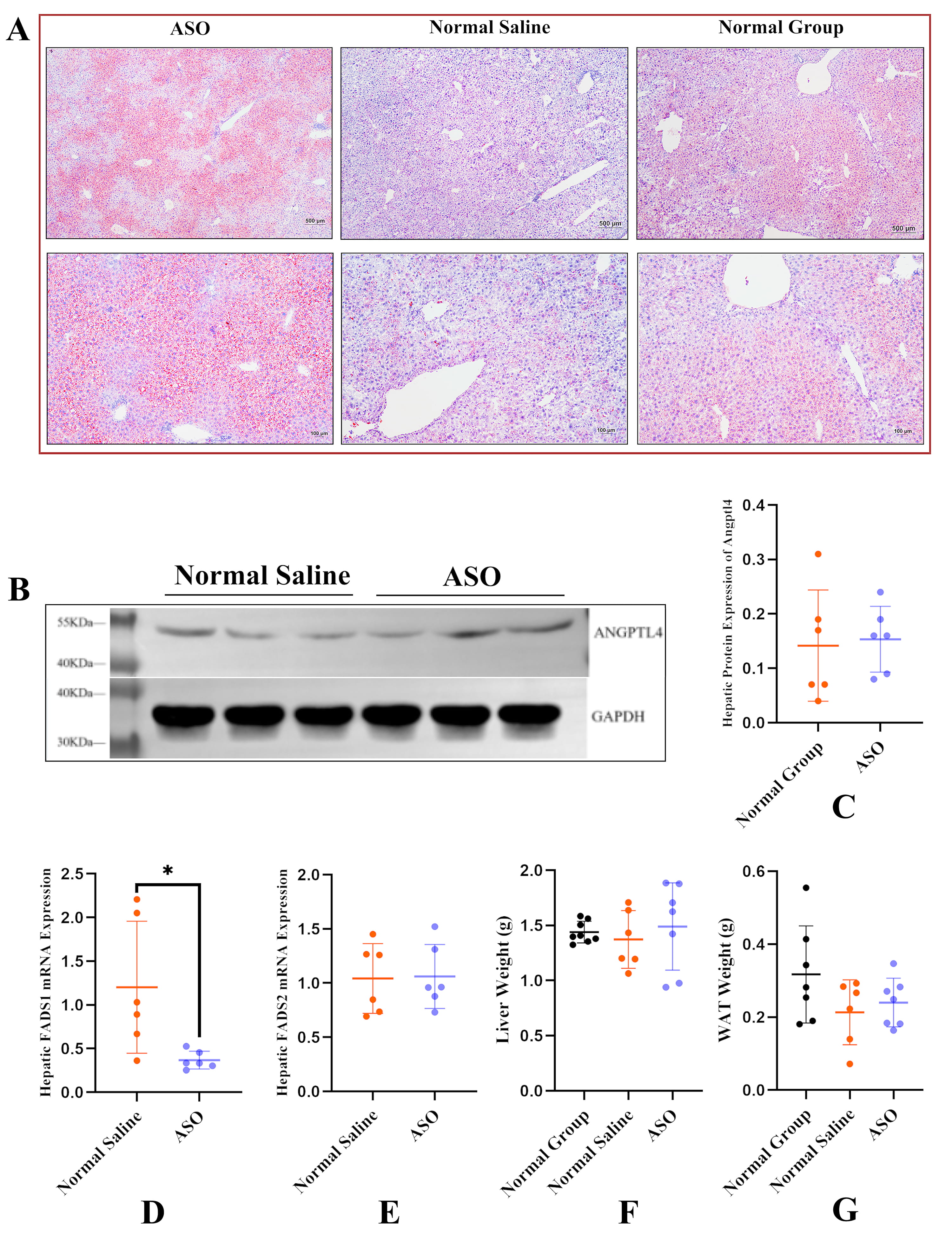 Fig. 6.
Fig. 6.Knockdown of liver Fads1 aggravated non-alcoholic fatty
liver disease (NAFLD). (A) Representative images of liver Oil Red O staining in
three groups. View at 40
Multiple factors cause atherosclerosis, indicating its complex pathogenesis,
which has not been fully elucidated. Given that Fads1 encodes
Powell et al. [12] showed that ApoE
After Fads1 knockdown in the liver of ApoE
Endothelial cells are the main component of the endothelial barrier, and destruction of the endothelial barrier leads to changes in vascular permeability. Endothelial dysfunction is responsible for the initiation of atherosclerosis, which contributes to the occurrence of atherosclerosis [26, 27]. Although the effect of ANGPTL4 on atherosclerosis remains a subject of debate [28, 29, 30], multiple studies have shown that ANGPTL4 promotes vascular inflammation and increases vascular permeability [31, 32, 33, 34, 35, 36, 37]. Adipose tissue and liver-derived ANGPTL4 might promote atherogenesis by regulating lipid uptake and inflammation in other tissues [38, 39, 40]. Furthermore, ANGPTL4 might promote atherogenesis through its vascular effects independently of the regulation of circulating lipids. ANGPTL4 mediates the inhibition of lipoprotein lipase (LPL) activity under different circumstances, and accumulating evidence has shown a direct correlation between ANGPTL4 and the risk of atherosclerosis. Although ANGPTL4 is a secreted protein, ANGPTL4 also controls lipoprotein metabolism and energy homeostasis as well as LPL-independent functions in the tissues where it is expressed, the relative contributions of cell-intrinsic and endothelial-specific versus circulating ANGPTL4 from other sources in regulating these processes are unclear [38]. Li et al. [41] indicated that FADS1 could activate AKT/mTOR signaling, and this hypothesis was further validated by both in vivo and in vitro assays. In addition, RNA sequencing revealed ANGPTL4 as a candidate downstream gene of the mTOR pathway [42]. In light of these conclusions, we speculated that FADS1 could affect atherosclerosis through AKT/mTOR signaling. In this study, we quantitatively analyzed the expression of ANGPTL4 in arterial endothelial cells, aorta, and liver. We found that ANGPTL4 expression was upregulated in the mouse aorta with more severe atherosclerosis, consistent with the upregulated mRNA expression of FADS1 and AKT1/mTORC1 (Fig. 3E–J). Furthermore, we inhibited hepatic FADS1 mRNA expression; however, the expression of ANGPTL4 was not affected (Fig. 6B–D), which may be related to the expression of ANGPTL4 in the liver being also regulated by nutritional and metabolic states [38]. The above results suggest that liver-derived ANGPTL4 is not responsible for the exacerbation of atherosclerosis, but the arterial endothelium-derived ANGPTL4 may exacerbate atherosclerosis. Therefore, we speculated that the upregulation of FADS1 expression could exacerbate atherosclerosis by upregulating ANGPTL4 expression in the aorta through AKT1/mTORC1 signaling. However, this study had some limitations. We did not analyze the expression of AKT1/phospho-AKT1 or mTORC1/phospho-mTORc1 by Western blotting. In the future, further validation of whether FADS1 in arterial endothelial cells can affect ANGPTL4 expression via the mTORC1/AKT1 signaling pathway and whether it further affects LPL activity is required.
Interestingly, the mRNA expression of FADS1 in the liver and vessel was
inconsistent. ASO inhibited hepatic FADS1 mRNA expression, but FADS1 mRNA
expression was upregulated in the aorta. We suggest that the upregulation of
FADS1 mRNA expression in the vessel was not significantly associated with ASO:
(1) ASO underwent Tri-GalNAc modification by targeting hepatocytes and is
unlikely to have the ability to bind to vascular tissues; (2) the metabolic sites
and accumulation sites of ASO, even without modification, are mainly concentrated
in the liver, adipose, kidney, and reticuloendothelial cell systems, lacking
selectivity for vascular tissues; and (3) ASO inhibits the expression rather than
promotes the expression of target genes. We speculated that selective knockdown
of hepatic FADS1, a key enzyme in the metabolism of
In the current study, we demonstrated that knockdown of liver Fads1 aggravated aortic atherosclerosis without significantly changing plasma LDL-C, HDL-C secretion rates, and inflammatory cytokine levels. We also found that ANGPTL4 expression was upregulated in the mouse aorta with more severe atherosclerosis, consistent with the upregulated mRNA expression of FADS1 and AKT1/mTORC1. Interestingly, selective knockdown of liver Fads1 had no significant effect on femoral atherosclerosis caused by surgical intervention. ASO-Tri-GalNAc-treated mice exhibited more severe NAFLD. In addition, knockdown of liver Fads1 reduced body weight and improved glycemic control. We also emphasize the need for an in-depth exploration of the regulation of FADS1-AKT1/mTORC1-ANGPTL4-LPL signaling in arterial endothelial cells for atherosclerosis to more clearly elucidate the molecular mechanism underlying FADS1 regulation of atherosclerosis.
The primer sequence is available in the supplementary material. The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
QL, PW and SW planned the project; QL designed diets, designed experiments, analyzed data, and wrote the article; QL, ZY, YD conducted mouse experiments, performed biochemical workup of mouse tissues, and aided in article preparation; all authors were involved in the editing of the final article. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All mouse experiments were performed according to the local relevant guidelines. All mice were kept under specific pathogen-free conditions in the Beijing Institute of Heart Lung and Blood Vessel Diseases and given free access to food and water. Our study was approved by the Ethics Committee of Beijing Anzhen Hospital (ethics number: 2022174X). The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, 1996, revised 2011; available from www.nap.edu/catalog/5140.html).
Not applicable.
This study was supported by grants from the Director’s Fund of Beijing Anzhen Hospital (2022409422 to S.W.).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.