






 , Jiantao Wang 1,*
, Jiantao Wang 1,*1 Shenzhen Eye Hospital, Jinan University, Shenzhen Eye Institute, 518040 Shenzhen, Guangdong, China
2 Center for Genome Analysis, Ruixing Biotechnology Co., Ltd., 430075 Wuhan, Hubei, China
Abstract
Objective: The morphology and functions of the human trabecular meshwork (HTM) are dysregulated in glaucoma, and the molecular mechanisms of this dysregulation remain unknown. According to an established in vitro model, whose function was to study the regulatory networks sustaining the response of HTM cells to the increased substrate stiffness, we systematically analyzed the expression pattern of long noncoding RNAs (lncRNAs), the important regulatory RNAs in cells. Methods: Bioinformatics analysis was performed to identify the dysregulated lncRNAs in response to increased substrate stiffness using transcriptome sequencing data (RNA-seq). Then we interfered with the expression of several dysregulated lncRNAs in HTM cells to explore their molecular targets. The cross-linking immunoprecipitation and sequencing method (CLIP-seq) was used to identify enhancer of zeste homolog 2 (EZH2)-targeted RNAs in HTM cells. The chromatin IP and sequencing method (ChIP-seq) was used to identify the targets of EZH2 and histone H3 at lysine 27 (H3K27me3). Results: The response of thousands of dysregulated lncRNAs to increased substrate stiffness was identified through RNA-seq. Functional prediction of these lncRNAs revealed that they potentially regulated key biological processes, including extracellular matrix (ECM) organization. By interfering with the expression of lncRNA SHNG8, ZFHX4-AS1, and RP11-552M11.4, the results demonstrated that those lncRNAs extensively regulated the expression levels of ECM-associated genes. Moreover, we found that EZH2 expression was significantly decreased at high substrate stiffness. Using CLIP-seq to identify EZH2-targeted RNAs in HTM cells, we found that SNHG8 was bound by EZH2. According to the CLIP-seq data of EZH2, we found that EZH2 binding sites were observed in the transcripts of SNHG8-regulated genes, but not in the ChIP-seq results of EZH2 and H3K27me3. Conclusion: Our results suggest that SNHG8 and EZH2 may cooperate to regulate the expression of a subset of genes by influencing their RNA abundance, explaining how they support HTM cell morphology and high density. This study contributes to the understanding of the alteration of HTM during the progression of glaucoma by identifying functional lncRNAs, especially SNHG8, and suggests novel therapeutic targets to treat glaucoma.
Keywords
- glaucoma
- human trabecular meshwork cells
- lncRNAs
- EZH2
- SNHG8
Glaucoma is the leading cause of irreversible blindness worldwide, with elevated
intraocular pressure (IOP) being the major risk factor, especially for primary
open-angle glaucoma (POAG) [1, 2]. Short-term IOP fluctuation is a signature risk
of glaucoma [3]. Thus, IOP has been considered a therapeutic target by modulating
its level in glaucoma patients [4]. At the molecular level, several molecules
reportedly elevate IOP in glaucoma, including FN-EDA/TLR4 axis [5],
Src/TGF-
An in vitro HTM cellular model simulating the in vivo HTM cell environment is necessary to investigate the underlying mechanisms of increased IOP in glaucoma patients. A previous study demonstrated that HTM cells cultured on polyacrylamide hydrogels with altered compliance values can mimic the in vivo glaucomatous HTM cells [13]. High meshwork compliance mimicking glaucoma can affect HTM cell physiology and subsequent responses. Alterations in substratum compliance simulating the glaucoma-related environment can also regulate the compliance of HTM cells [14]. Our previous study also demonstrated the highly dynamic expression patterns of protein-coding genes and noncoding RNAs of HTM cells by changing the polyacrylamide hydrogel stiffness, and validated ECM gene expression changes using clinical HTM tissues [15, 16]. However, how those HTM cells respond to normal IOP fluctuation and mechanical stresses, and when HTM cells become dysfunctional under overload substratum stiffness are unknown.
In ophthalmology, the high-throughput sequencing method has been employed to explore disease biomarkers [17]. Meanwhile, long noncoding RNAs (lncRNAs) also participate in the pathogenesis of glaucoma on a genetic basis [18, 19]. In rats with glaucoma, lncRNA-MALAT1 mediates neurodegeneration through the continuous CREB signaling activation [20]. However, most studies on the role of lncRNAs in glaucoma have focused on the genetic variation [21]. LncRNAs can perform their functions with diverse mechanisms, including their primarily studied function of cis/trans-acting transcriptional regulation helped by other proteins, such as polycomb repressive complex 2 (PRC2) [22, 23]. Thus, the underlying mechanisms of lncRNAs in glaucoma need to be further investigated.
According to our previously established in vitro HTM cell model mimicking in vivo IOP increasing in glaucoma and high-throughput transcriptome sequencing (RNA-seq) data [15], we investigated the dysregulation of lncRNAs under high substratum compliance and predicted their potential targets. Additionally, we selected several highly dysregulated lncRNAs and altered their expression levels to validate their downstream targets using the RNA-seq method. Our study provides insights into how lncRNA expression profiles are dysregulated in pressure-increasing HTM cells and reveals the complicated lncRNA-mRNA regulatory mechanisms.
The human SNHG8, ZFHX4-AS1 and RP11-552M11.4 genes were cloned by reverse transcription and polymerase chain reaction (PCR) amplification methods. The purified DNA fragment from a gel (28004, MinElute PCR Purification Kit, Qiagen, MD, USA) was cloned into the pIRES-hrGFP-1a vector (240031, Agilent Technologies, Savage, MD, USA) using the hot fusion method by CE Design V1.04 (Vazyme Biotech Co., Ltd., Nanjing, China). Plasmids were introduced into Escherichia coli strain by chemical transformation. Then E. coli strains were plated onto LB agar plates with 1 µL/mL ampicillin (7177-48-2; Sigma, St. Louis, MO, USA), and incubated overnight at 37 °C. Colony PCR (28 cycles) was used to screen lncRNA colonies with universal primers (located on the backbone vector). Finally, the inserted sequences of lncRNAs were verified by Sanger sequencing.
Primary HTM cells (YS905C, Yaji Biological, Shanghai, China) were donated by
anonymous deceased individuals without detailed information, cultured in DMEM/F12
(PM150310, Procell, Wuhan, China), and were validated by STR profiling, and
tested negative for mycoplasma. Cells were cultured in a humidified incubator at
37 °C and 5% CO
We used glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal
control for assessing the effects of SNHG8, ZFHX4-AS1, and
RP11-552M11.4 overexpression, as well as the ECM-related gene
expression. The cDNA synthesis and following RT-qPCR were performed on the
Bio-Rad S1000 Thermal Cycler with Hieff™ qPCR
SYBR® Green Master Mix (Low Rox Plus; YEASEN, Shanghai, China),
followed by denaturing at 95 °C for 10 min, 40 cycles of denaturing at
95 °C for 15 sec and annealing and extension at 60 °C for 1
min. The expression level of each transcript was normalized to GAPDH by
the 2
HTM cells were prepared for ultraviolet (UV) cross-linking with UV irradiation
type C (254 nm) at 400 mJ per cm
HTM cells were cross-linked with 1% formaldehyde for 10 min at room temperature and then lysed to obtain a chromatin-protein complex. Soluble sheared chromatin was obtained by sonication (average DNA length of 200–500 base pairs). The 10 µg anti-EZH2 antibody (5246, Cell Signaling Technology, Boston, MA, USA) and anti-H3K27me3 antibody (12-565, Millipore, Burlington, MA, USA) were used for IP with 100 µL chromatin as input at 4 °C overnight with two biological replicates. The next day, 30 µL protein beads were added and the samples were further incubated for 3 h. Immunoprecipitated DNA was used to construct the sequencing libraries following the protocol of ChIP-seq Library Prep Kit (NOVA-5143-02, Bioo Scientific, Austin, TX, USA) and the libraries were applied to the Illumina HiSeq X Ten system (Illumina, Inc., San Diego, CA, USA) for 150 nt paired-end sequencing.
The RNA-seq data files of GSE123100 from our previous study [15] were re-analyzed. The raw files were converted to fastq file by the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) Tool fastq-dump. Low-quality bases were removed from the raw reads using the FASTX-Toolkit (version 0.0.13, The Hannon Lab, New York, NY, USA; http://hannonlab.cshl.edu/fastx_toolkit/). The short reads less than 16 nt were also dropped. Then the quality filtered reads were evaluated using FastQC (version0.12.1, Babraham Institute, Cambridge, MA, USA, http://www.bioinformatics.babraham.ac.uk/projects/fastqc).
Raw sequenced reads were filtered to remove low quality reads using the above methods. Then, clean reads were aligned onto the human GRCH38 genome using the HISAT2 (version2.2.1, University of Texas Southwestern Medical Center, Dallas, TX, USA) alignment program [26] with no more than four mismatches. Uniquely mapped reads were used for gene expression calculation by FPKM values (fragments per kilobase of transcript per million fragments mapped) [27]. The statistical power of this experimental design (one biological replicates), calculated by online implementation (https://rodrigo-arcoverde.shinyapps.io/rnaseq_power_calc/) was 0.93.
StringTie (version2.1.6, Johns Hopkins University, Baltimore, MD, USA) [28] was used to assemble and
predict transcripts. Final transcripts were screen out by eliminating the
transcripts with FPKM
We used edgeR (version3.32, The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia) [34] to identify
DEGs with p-value
After alignment, uniquely mapped reads were used to predict EZH2-bound regions
(peaks) by the “ABLIRC” strategy (version1.0, ABLife Inc., Wuhan, China) [35] and CIMS
(version1.0, The Rockefeller University, New York, NY, USA) [36]. After stimulation and calculation, all the
observed peaks with p-value
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways
were identified to sort out the functional categories of DEGs with the KOBAS
server (version2.0, Peking University, Beijing, China) [40]. Principal component analysis (PCA) was
performed using R package factoextra
(https://cloud.r-project.org/package=factoextra). The pheatmap package
(https://cran.r-project.org/web/packages/pheatmap/index.html) in R software
(version4.2, https://www.r-project.org) was used to perform clustering based on
Euclidean distance. Student’s t-test was used to compare the two groups
with p-value
Our previous study systematically investigated the mRNA expression profiles in
response to the tunable stiffness and found extensive dysregulation of
ECM-associated genes [15]. Here, we explored the expression profile of lncRNAs,
which are important regulatory RNAs that may influence the global transcriptome
profile. We re-analyzed the GSE123100 dataset and performed lncRNA prediction and
expression analyses. The investigation pipeline used in this study is shown in
Fig. 1A. Biological replicates were HTM cells cultured for 3 days (D3) and 5 days
(D5) with the same Young’s modulus. After predicting novel lncRNAs and
calculating their expression levels, we divided the expressed genes into mRNA and
lncRNA groups. The PCA results demonstrated that D3 and D5 samples were separated
by the first component, and that obvious distances for D5 samples with high
Young’s modulus were observed (Fig. 1B). Then we assessed the expression patterns
of several glaucoma-associated lncRNAs, including
ANRIL/CDKN2B-AS1, MALAT1, and LOXL1-AS1 [18].
ANRIL/CDKN2B-AS1 showed lower expression levels at 1.1 kPa and
50 kPa; the expression level of LOXL1-AS1 was decreased at 50 kPa;
whereas MALAT1 showed specifically higher expression level at 50 kPa
(Fig. 1C). These results demonstrated that their expression patterns were
dynamically changed with the increase of Young’s modulus. Together, these results
indicated that in HTM cells, lncRNA expression levels are extensively regulated
by increased substrate stiffness and that lncRNAs may have important regulatory
functions. We then analyzed the differentially expressed lncRNAs (DElncRs) using
the same method for mRNAs [15]. By analyzing DElncRs between adjacent samples
with increased stress, we found that DElncR number was the largest in 34.3 kPa
vs. 11.9 kPa, following by 11.9 kPa vs. 2.5 kPa, and that
DelncR number was very small in samples with lowest or highest stress (Fig. 1D).
According to a previous study, the average Young’s modulus of HTM was 80.0
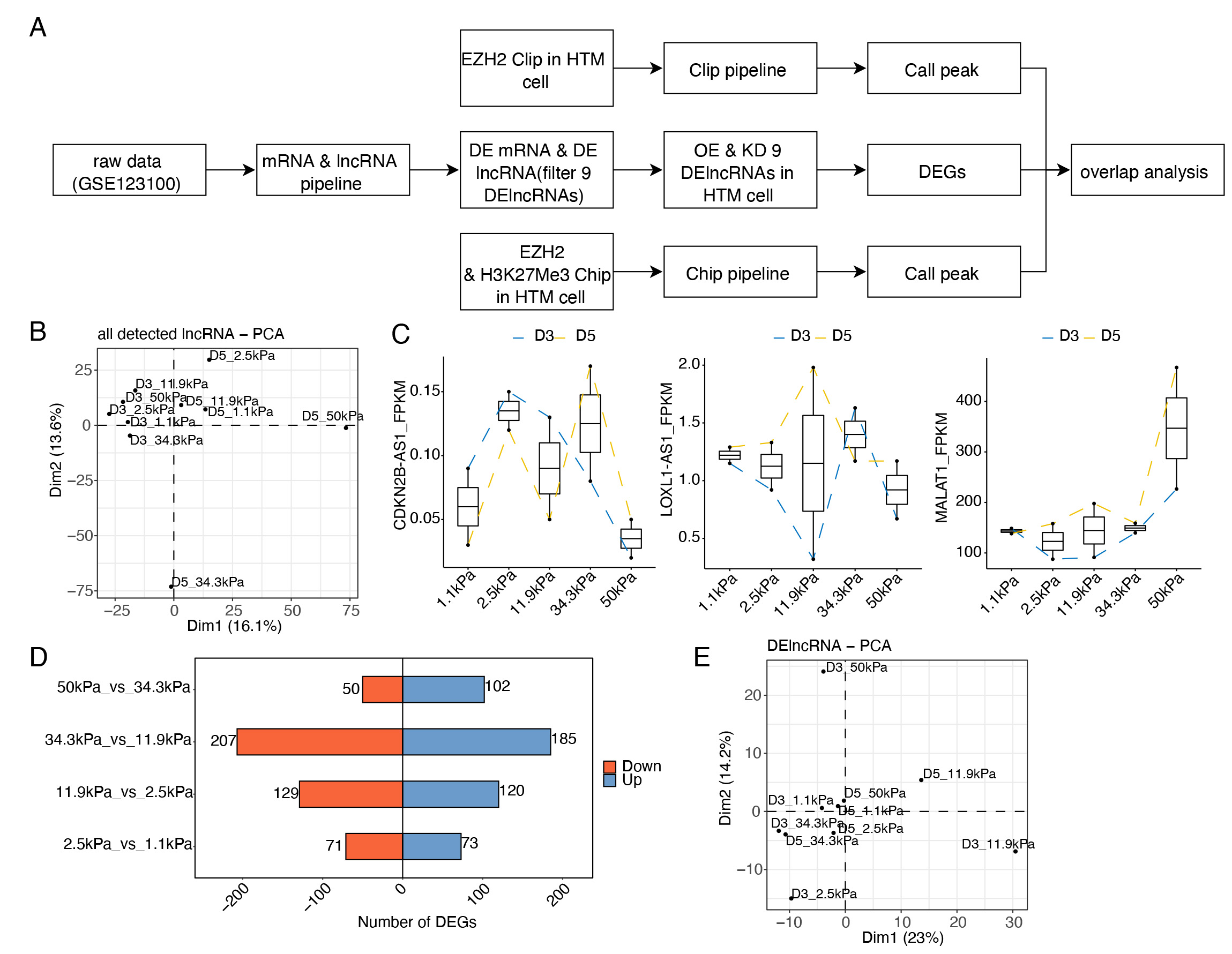 Fig. 1.
Fig. 1.Global analysis of long noncoding RNAs (lncRNAs) from human trabecular meshwork (HTM) cells. (A) The work flow of this study. (B) Principal component analysis (PCA) results showing the diversity of expressed lncRNAs in the HTM cell model. (C) Line plot showing the expression patterns of CDKN2B-AS1, MALAT1, and LOXL1-AS1 in HTM cells in response to stiffness. (D) Bar plot showing the numbers of differentially expressed lncRNAs (DElncRs) in different groups. (E) PCA result showing the diversity of DElncRs in the HTM cell model. mRNA, messenger RNA; CLIP, cross-linking immunoprecipitation; EZH2, enhancer of zeste homolog 2; DE, differentially expressed; Chip, chromatin immunoprecipitation; DEGs, differentially expressed genes; OE, over expression.
Based on the above results, we explored the potential functions of DElncRs in
HTM cells. Focusing on the two groups with the most DElncRs (Fig. 1D), we found
that 97 DElncRs were detected in both groups (Fig. 2A, p-value = 3.43347
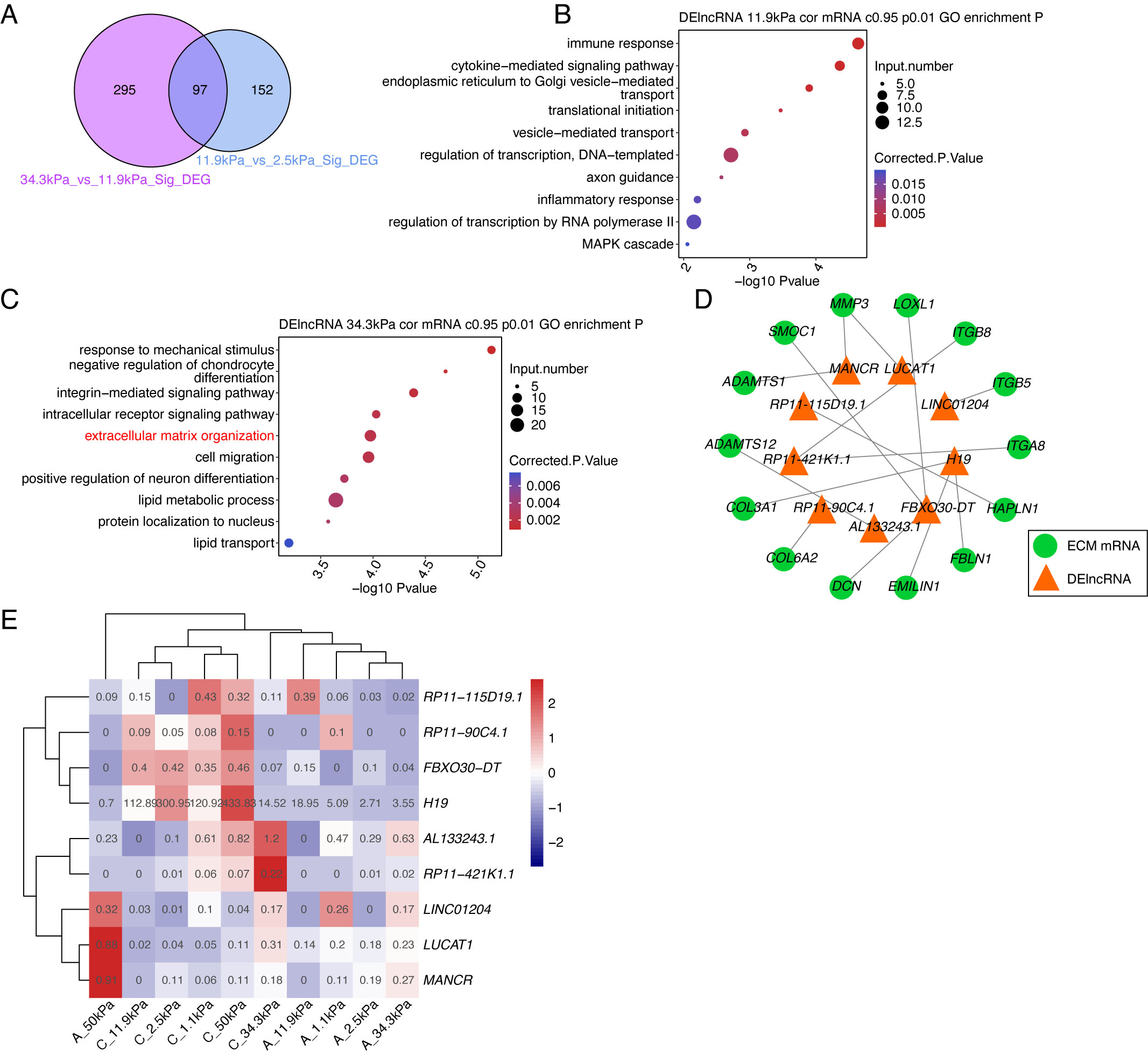 Fig. 2.
Fig. 2.DElncRs are tightly associated with extracellular matrix (ECM) genes by co-expression analysis. (A) Venn diagram showing the overlapped DElncRs in the 11.9 kPa vs. 2.5 kPa and 34.4 kPa vs. 11.9 kPa group. (B) Bubble plot showing the top enriched Gene Ontology (GO) Biological Process (BP) pathways of co-expressed DElncRs-DEGs in the 11.9 kPa vs. 2.5 kPa group. (C) Bubble plot showing the top enriched GO BP pathways of co-expressed DElncRs-DEGs in the 34.4 kPa vs. 11.9 kPa group. (D) Network presentation of DElncR-DEG pairs enriched in ECM-related pathways. (E) Heatmap presentation of DElncRs co-expressed with ECM-related DEGs. DEG, Differentially Expressed Gene.
According to these results, we proposed that lncRNA expression levels were extensively changed between 11.9 kPa vs. 34.4 kPa, and were associated with ECM organization. Next, we selected several lncRNA-mRNA pairs with a negative correlation to further explore their relationships. The following 10 lncRNAs were selected to explore their potential functions: ZFHX4-AS1, NNF-AS1, CRNDE, TP73-AS1, SNHG8, DLEU2, CTD-3252C9.4, LINC00707, RP11-552M11.4, and MEG8. Most of these lncRNAs were located in intergenic regions, and may regulate other genes or RNAs in a cis or trans manner. Three lncRNAs, ZFHX4-AS1, NNF-AS1, and TP73-AS1, were at the antisense strand of protein coding genes, functioning in a cis manner. These known lncRNAs were classified into two clusters: increased expression levels by elevated stiffness, and the contrary cluster. The RT-qPCR experiment was performed to validate their expression patterns during substrate stiffness elevation. The RT-qPCR results of the eight lncRNAs showed a consistent pattern with the RNA-seq data (Fig. 3, Supplementary Table 1). These results demonstrated that the dysregulated lncRNAs may have important functions in the HTM cells of glaucoma pathogenesis.
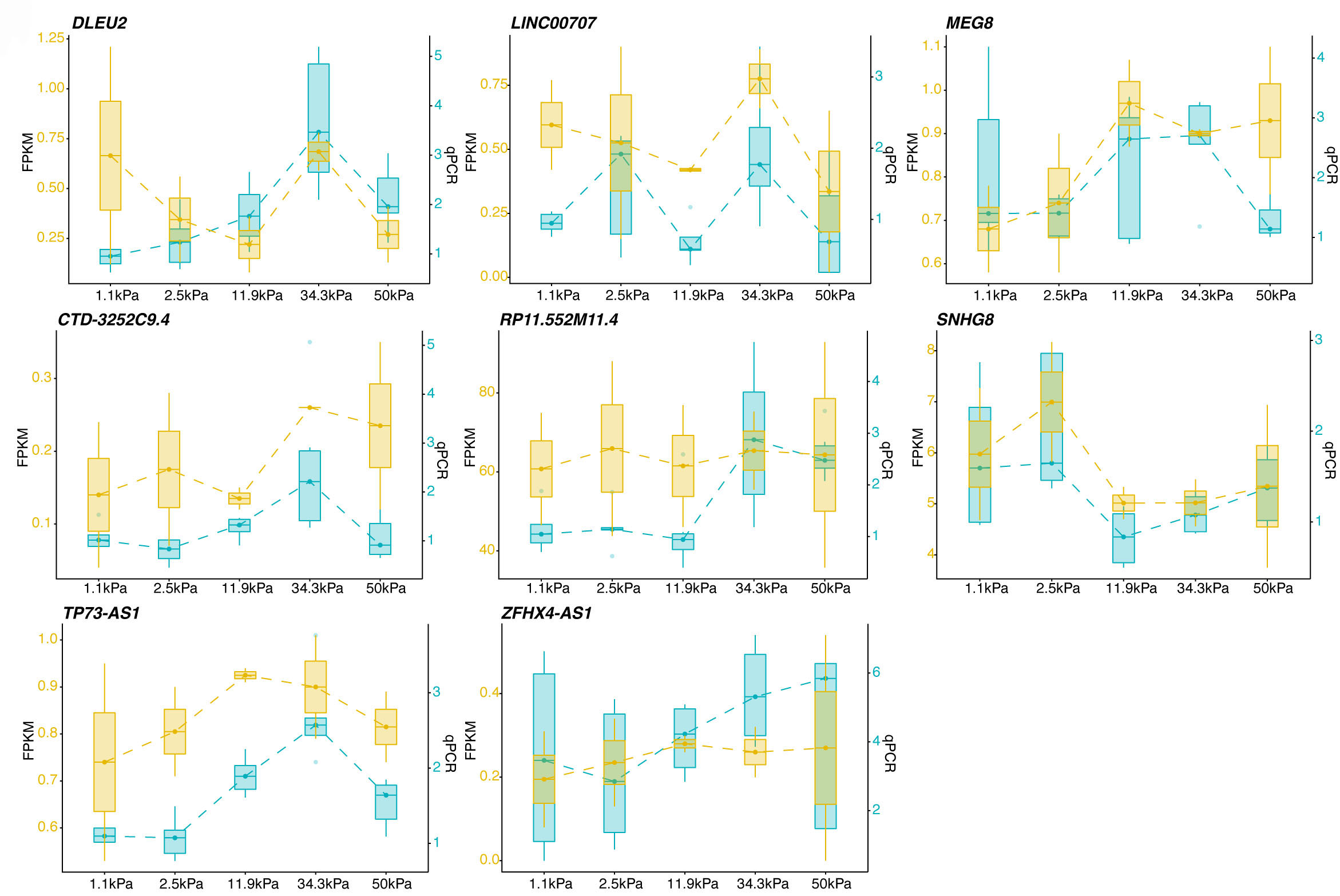 Fig. 3.
Fig. 3.Reverse Transcription and Quantitative Polymerase Chain Reaction (RT-qPCR) validation of lncRNAs under the effect of substrate stiffness in HTM cells. Box plot showing the RT-qPCR validation of selected lncRNAs for D3 and D5 samples. RNA-seq results (brown box) and RT-qPCR results (light green box) showed the consistency of these two methods. RT-qPCR analysis was performed with three biological replicates.
To further explore the functions of these selected lncRNAs, we attempted to
construct lncRNA overexpression cell models by transfecting overexpression
plasmids into normal HTM cells. Plasmid without lncRNA sequences was used as the
common negative control (NC). RT-qPCR has validated the overexpressed
transcriptional levels of several lncRNAs (Fig. 4A). Finally, three lncRNAs with
significant expression changes were selected for overexpression experiments,
including ZFHX4-AS1, SNHG8, and RP11-552M11.4.
Meanwhile, ZFHX4-AS1 itself had control (NC_ZFHX4-AS1_OE) because the
experiment was done in another batch, and SNHG8 and RP11-552M11.4 had
the same control (NC_OE). Then RNA-seq experiments were performed to explore the
transcriptional influence of these three lncRNAs. The expression analyses showed
that they showed distinct higher levels in the corresponding overexpression
samples, and were not changed in other samples (Fig. 4B). Sample correlation
analysis revealed that overall gene expression levels were highly correlated with
PCCs
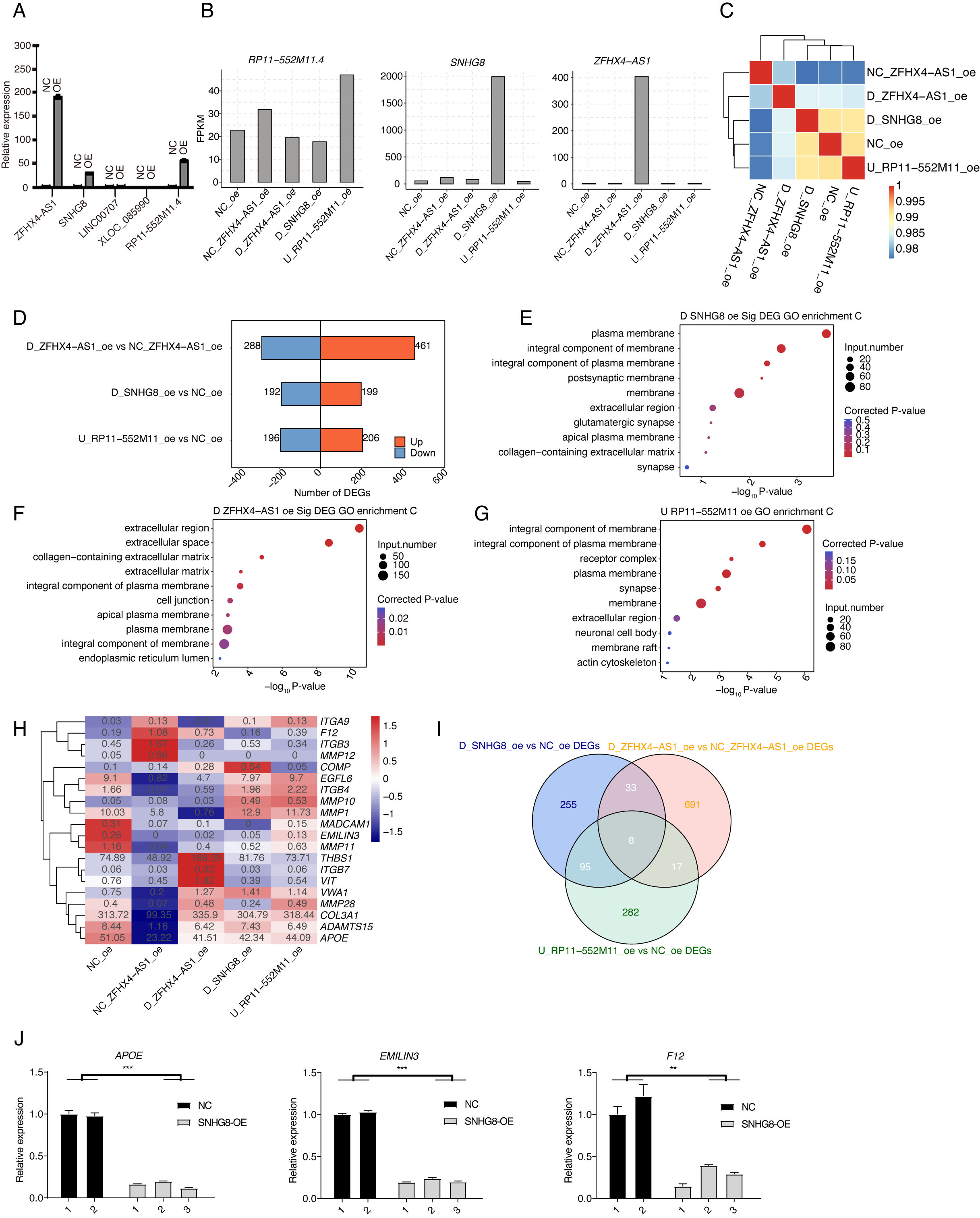 Fig. 4.
Fig. 4.LncRNA overexpression extensively regulates the expression of
ECM remodeling-related genes. (A) Bar plot showing the overexpressed lncRNA
levels in HTM cells using RT-qPCR method. (B) Bar plot showing the expression
levels of three overexpressed lncRNAs and two negative controls. (C) Sample
correlation heatmap of RNA-seq samples. (D) Volcano plot showing the DEGs between overexpressed lncRNAs and negative
controls. (E) Bubble plot showing the enriched GO Cellular Component (CC) terms
of DEGs from SNHG8-OE samples compared with negative control. (F) The same with
(D) but for the DEGs from ZFHX4-AS1-OE. (G) The same with (D) but for the DEGs
from RP11-552M11.4-OE. (H) Hierarchical clustering heatmap showing the expression
patterns of ECM-associated and ZFHX4-AS1-regulated DEGs. (I) Venn diagram showing
the overlapped and specific DEGs among these three DElncRs. (J) Bar plot showing
the changed expression levels of APOE, EMILIN3, and F12 between SNHG8-OE
vs. NC samples using RT-qPCR. One NC sample was removed due to its
outlier feature. ** p-value
LncRNAs regulate their targets in multiple functional types with the help of RNA
binding proteins (RBPs), including cis- and trans-acting [42]. A well-studied
partner of lncRNAs is EZH2, the core component of the PRC2 complex [43]. We
performed CLIP-seq analyses for EZH2 to explore the interacting RNA partners of
EZH2 in HTM cells. After alignment of sequenced tags, genomic region distribution
analysis showed that compared to IgG, EZH2 preferred to bind to coding sequence
(CDS) and 3
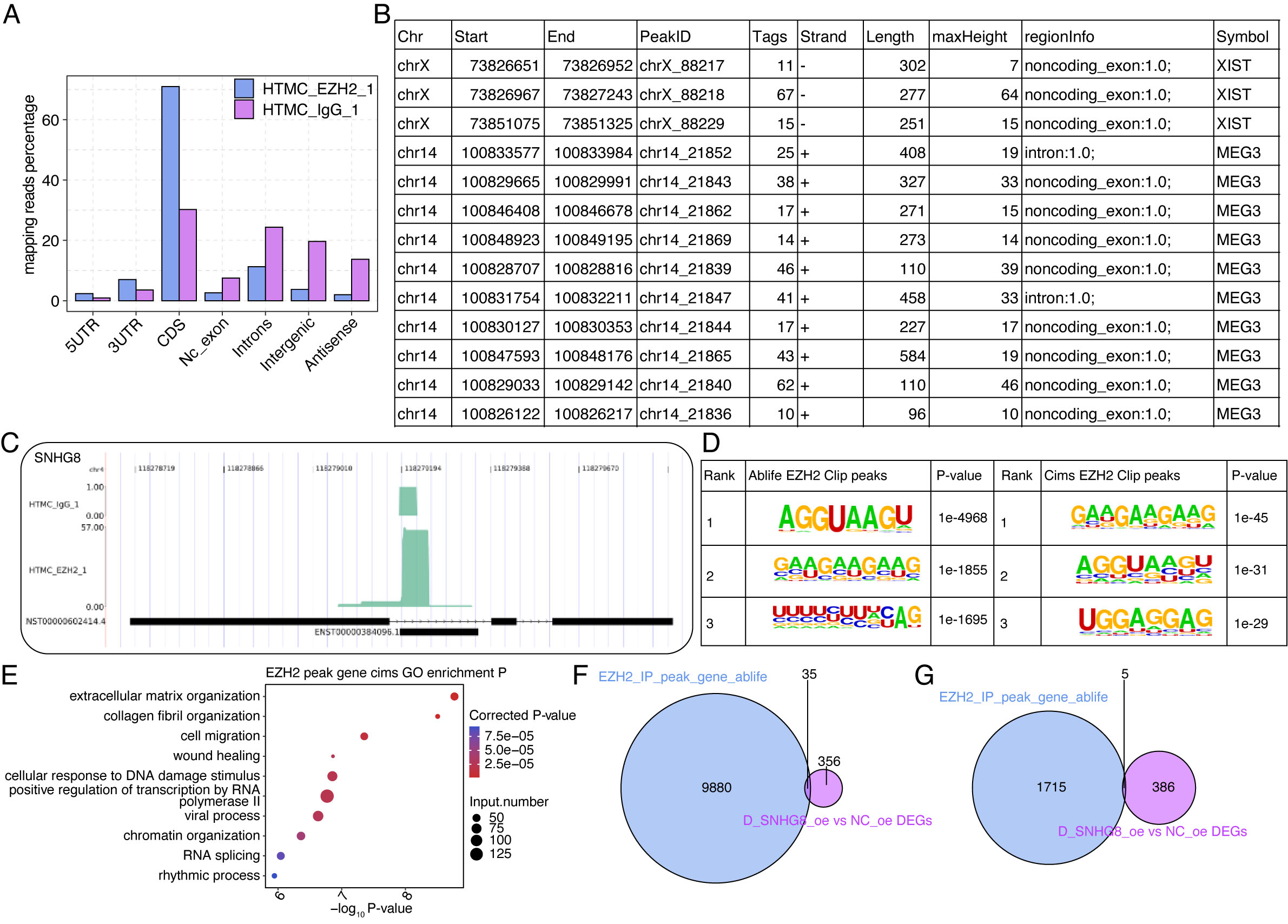 Fig. 5.
Fig. 5.EZH2 interacts with mRNAs and lncRNAs in HTM cells. (A) Bar plot showing the genomic distribution of mapped reads in EZH2-IP and IgG samples. (B) Table showing the detailed information of EZH2-bound peaks from XIST and MEG3 lncRNAs using ABLIRC method. (C) Reads density plot for lncRNA SNHG8 in EZH2-IP and IgG samples. (D) Motif analysis showing the top three enriched motifs from EZH2-bound peaks using ABLIRC and CIMS methods. (E) Bubble plot showing the top enriched GO BP pathways of EZH2-bound gene transcripts using CIMS method. (F) Venn diagram showing the overlapped genes between EZH2-bound transcripts (replicate 1) and SNHG8-OE-DEGs. (G) Venn diagram showing the overlapped genes between EZH2-bound transcripts (replicate 2) and SNHG8-OE-DEGs.
The physical interactions between EZH2 and SNHG8 have been
demonstrated, but it remains unclear how this protein-RNA complex plays a role in
HTM cells remains unclear. Increasing evidence has shown that EZH2-RNA
interactions prevent EZH2 from catalyzing H3K27 methylation to regulate gene
expression [46, 47, 48]. Thus, we performed ChIP-seq analyses of EZH2 and H3K27me3,
with two biological replicates for each. After MACS2 software was used to call
peaks [38], significantly enriched binding peaks of EZH2 and H3K27me3 were
obtained (p-value
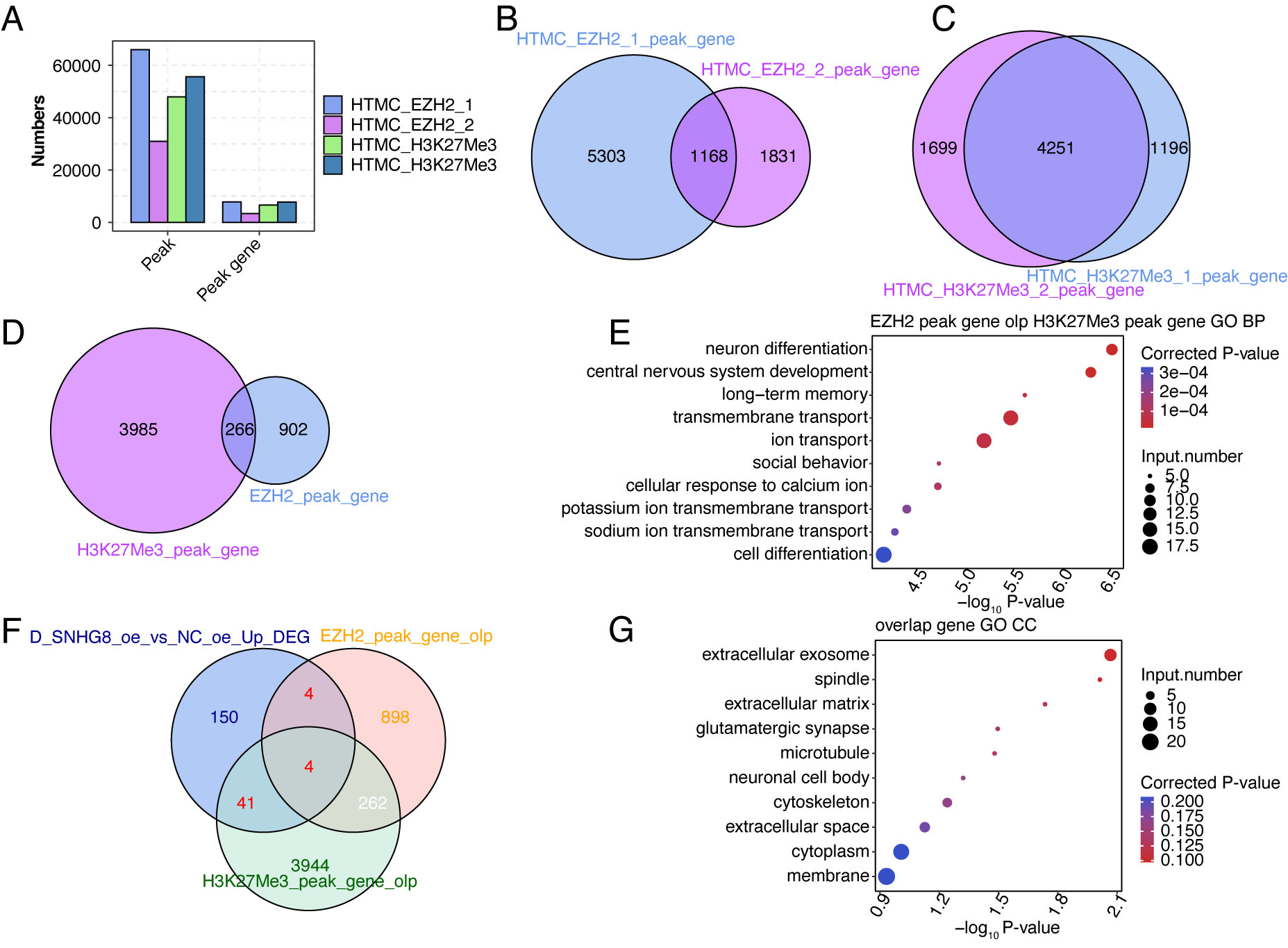 Fig. 6.
Fig. 6.ChIP-seq analysis reveals the DNA interaction profiles of EZH2 and H3K27me3 in HTM cells. (A) Peaks and genes identified by EZH2 and H3K27me3 ChIP-seq data are shown in the bar plot. (B) Venn diagram showing the overlapped genes between the two replicates of EZH2 ChIP-seq- associated genes. (C) Venn diagram showing the overlapped genes between the two replicates of H3K27me3 ChIP-seq-associated genes. (D) Venn diagram showing the overlapped genes between EZH2 and H3K27me3 ChIP-seq- associated genes. (E) Bubble plot showing the enriched GO BP pathways of overlapped genes in (D). (F) Venn diagram showing the overlapped genes among SNHG8-OE-DEGs, EZH2-associated genes, and H3K27me3-associated genes. (G) Bubble plot showing the enriched GO CC pathways of 49 overlapped genes in (F).
Glaucoma-induced irreversible blindness affected millions of people worldwide. The primary risk factors of glaucoma are increased outflow resistance and elevated IOP mediated by dysregulated TM and ECM [12]. In this study, next generation sequencing and gene interfering technologies were used to explore the transcriptome and epigenetic profiles of altered HTM cells by mimicking the in vivo glaucomatous HTM tissue. We focused on the dysregulated lncRNAs and explored their potential functions and targets. Our study revealed that many lncRNAs were dysregulated due to the increased substrate stiffness and that their dysregulation induced the expression changes of genes with important biological functions, including ECM organization and assembly pathways. The identified lncRNA SNHG8 has potential functions in ECM modulation and may fulfill these functions by interacting with EZH2 or other RNA binding proteins to modulate the transcriptional program of associated genes.
Cells in the HTM tissue play a key role in maintaining IOP homeostasis [49]. A
better understanding of this process will greatly improve the therapeutic
approaches to glaucoma, a disease whose IOP homeostasis was compromised or lost
[50]. Our previous in vitro experiment on substrate stiffness-increased
HTM cell model revealed the global ECM gene expression changes of HTM cells [15].
The influence of noncoding RNAs on ECM proteins, including several important
lncRNAs, has been previously discussed [51]. A total of 248 lncRNAs and 4 hub
lncRNAs were identified through competing endogenous RNA network analysis in POAG
[52]. Global transcriptome exploration on how HTM cells responded to mechanical
stretch also revealed hundreds of dysregulated mRNAs, metabolism- and ECM
interaction-associated lncRNAs and several master regulatory microRNAs [53].
Other studies have mainly focused on single lncRNAs and linked them to the
genetic basis of glaucoma [18]. In HTM cells, the antisense RNA of TGF
In this study, several lncRNAs were finally identified to modulate ECM gene expression, including SNHG8, ZFHX4-AS1, and RP11-552M11.4. These lncRNAs were mainly analyzed in the tumor area based on searching previous studies. Down-regulation of ZFHX4-AS1 can suppress the invasion and migration of breast cancer cells via the Hippo signaling pathway [58], whereas the other functions of ZFHX4-AS1 still remain largely unknown. In cervical cancer, RP11-552M11.4 promotes tumorigenesis and development by targeting ATF1 [59], and in ovarian cancer, by targeting BRCA2 [60]. SNHG8 has many functions, for example, SNHG8 can regulate EBV-associated gastric cancer [45], and promote the development of hepatocellular carcinoma (HCC) by sponging miR-149-5p [61]. In this study, we investigated how these three lncRNAs transcriptionally regulated ECM genes in HTM cells and found ECM associated genes were extensively regulated by these lnRNAs. Based on the complex functional mechanisms of lncRNAs, we propose that these dysregulated lncRNAs due to substrate stiffness may form a regulatory network with their target molecules to influence the ECM patterns of HTM cells, extending their important regulatory roles and underlying mechanisms in the glaucoma pathogenesis. Meanwhile, due to the lack of solid validation experiments and less biological replicates, further studies should be performed to validate the regulatory mechanisms of these dysregulated lncRNAs.
Furthermore, we found that SNHG8 could modulate ECM gene expression by interacting with EZH2 through post-transcriptional regulation. A previous study summarized the epigenetic regulatory functions of lncRNAs with the PRC2 complex, which contains the core methyltransferase EZH2 [62]. By interacting with genetic and environmental factors, epigenetic regulation could promote TM fibrosis generation through several pathways, resulting in elevated IOP [63]. A recently published study validated that SNHG8, a mechanical force-sensitive lncRNA, can modulate the osteogenic differentiation of periodontal ligament stem cells through epigenetic pathways by regulating EZH2 expression [64]. Another study also reported that SNHG8 and EZH2 coordinately regulate proliferation and apoptosis in human papillomavirus (HPV)-induced cervical cancer [65]. Using the CLIP-seq data of EZH2, we identified the direct interaction between SNHG8 and EZH2 in HTM cells. Meanwhile, we also found that EZH2-bound mRNA genes were significantly enriched in ECM-related pathways, which indicated us that SNHG8 cooperates with EZH2 to regulate the fate of RNAs interacting with them. Meanwhile, we also performed ChIP-seq on EZH2 and H3K27me3 to identify their targeting genes. However, few genes were found to be overlapped between SNHG8-regulated DEGs and genes with both EZH2 and H3K27me3 binding signals, suggesting that the two gene sets function normally without being mutually influenced and other mechanisms may exist for the regulatory program of SNHG8 in HTM cells.
In conclusion, we systematically investigated the lncRNA profiles in HTM cells responding to the increased substrate stiffness and found that the dysregulated lncRNAs were tightly associated with the altered ECM organization, suggesting their important roles in ECM organization and glaucoma pathogenesis. Meanwhile, we explored the direct association between SNHG8 and EZH2, and discussed how SHNG8 and EZH2 coordinately modulated the expression of the downstream targets. These results provide an understanding of the important roles and molecular functions of lncRNAs in glaucoma progression. The identified lncRNAs, especially SNHG8, and their potential targets could provide potential therapeutic targets in glaucoma treatment in future investigation.
The RNA-seq data sets analyzed during the current study are available in the NCBI Gene Expression Omnibus (GEO) repository with ID GSE123100. The other data sets were used under license for the current study and so are not publicly available. Data are however available from the authors upon reasonable request.
JW and ZY designed the study and revised the manuscript. JG partially designed the study, performed the majority of the experiments, and drafted the manuscript. YW, YS, and DC performed part of the experiments and analyzed the data. YH and XS performed part of the experiments. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by the National Natural Science Foundation of China (82070961), the Basic Research Project of Shenzhen (JCYJ20230807114605010), Shenzhen Science and Technology Program (No.KCXFZ20230731093359004), and Shenzhen Key Medical Discipline Construction Fund (No. SZXK037).
The authors declare no conflict of interest. Yue Sun, Yunfei Wu, and Dong Chen are employed by Wuhan Ruixing Biotechnology Co., Ltd.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.