




1 Department of GI Surgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 430030 Wuhan, Hubei, China
2 GI Cancer Research Institute, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 430030 Wuhan, Hubei, China
Abstract
The mitochondrial antiviral-signaling protein (MAVS), a core adaptor protein in the retinoic-acid-inducible gene-I-like receptors (RLRs)-MAVS pathway, has been demonstrated to play an important role in antiviral immune response and tumor immunology. Previous studies revealed that ubiquitylation is a key mechanism in the regulation of the RLRs-MAVS axis and immune response. Multiple E3 ubiquitin ligases and deubiquitinating enzymes control MAVS ubiquitylation and changes in MAVS function. In this review, we summarize the biological function of ubiquitylation in MAVS-related signaling and provide new insight into immunotherapy approaches that target MAVS.
Keywords
- MAVS
- ubiquitylation
- SUMOylation
- RING-finger type E3 ligase
- HECT E3 ligase
- U-box type E3 ligase
- SUMO E3 ligase
- virus-derived proteins function as E3 ligase
- de-ubiquitinase
The mitochondrial antiviral-signaling protein (MAVS), also termed virus-induced signaling adaptor (VISA), is a pivotal component in innate immune signaling [1]. After encountering viral double-stranded RNA (dsRNA), retinoic-acid-inducible gene-I-like receptors (RLRs) undergo structural alterations that activate the MAVS protein situated on the mitochondria’s outer membrane. These changes in MAVS activate biological functions that protect cells from viral attack [2]. Thus, RLRs-MAVS pathway is essential in innate immune response, and exhibits an intriguing ability to recognize and combat viral infection [3].
MAVS activity and stability are important in antiviral defense tactics. In its function as a switch in immune signal transduction, MAVS plays a dual role in antiviral signal transduction and immune homeostasis by becoming a central target of both virus and host [4]. Recently, numerous studies have investigated the regulation of MAVS signaling and a common post-translational modification (PTM) of MAVS, ubiquitylation has been found to be of increasing importance in the regulation of the RLRs-MAVS pathway [5]. Therefore, a detailed comprehension of MAVS ubiquitylation is crucial for understanding innate immunomodulation. In this review, we will briefly introduce the structure and biological function of MAVS, and summarize the ubiquitin-mediated regulation of MAVS during antiviral innate immune signaling.
The MAVS protein is located on the mitochondrial outer membrane and functions as a platform for signal transduction within the innate immune system [4]. Current evidence indicates that MAVS acts as a critical junction between the immune response and viral infections. As an adaptor molecule, MAVS transmits signals from retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5) to downstream effectors during viral infection. This signaling upregulates type I interferon and pro-inflammatory cytokine transcription, and their subsequent secretion. These cytokines may subsequently trigger an antiviral defense mechanism that inhibits viral replication and eliminates infected cells. Therefore, aberrant MAVS signaling could disrupt normal innate immune responses and promote immune-related diseases.
The MAVS protein consists of 540 amino acids and contains three primary domains:
The N-terminal caspase-recruitment domain (CARD), the central proline-rich
region, and the C-terminal transmembrane domain [6]. The CARD domain interacts
with RIG-I and MDA5 upon viral infection or the presence of exogenous nucleic
acids such as dsRNA or 5
MAVS also plays an important role in the surveillance of endogenous dsRNA. In cancer radiotherapy, DNA fragments are released into the cytoplasm and are transcribed into dsRNA through the action of endogenous retroviruses (ERVs) [14]. These ERV-associated dsRNA can also activate the innate antiviral MDA5/MAVS/TBK1 pathway and trigger downstream immune activation [14]. DNA methyltransferase inhibitors could also trigger cytosolic sensing of dsRNA via TLR3-MAVS and cause a type I interferon response and apoptosis [15]. Recently, Alu retroelements and mitochondrial transcription were reported to be alternate sources of endogenous dsRNA, and that this could be recognized by the RIG-I/MAVS signaling, and result in an interferon response [16]. A full understanding of the regulation between MAVS and endogenous dsRNA requires further investigation.
Recent studies have shown that MAVS is not only a passive sensor of viral infection, but also involved in response to bacterial and parasite infections, autoimmune diseases, and cancer progression [17, 18, 19, 20]. Appropriate inhibition of MAVS activity could avoid unnecessary or excessive deleterious inflammation [3]. Therefore, maintaining balance in MAVS signaling is necessary to maintain immune homeostasis. The function of MAVS is summarized in Fig. 1.
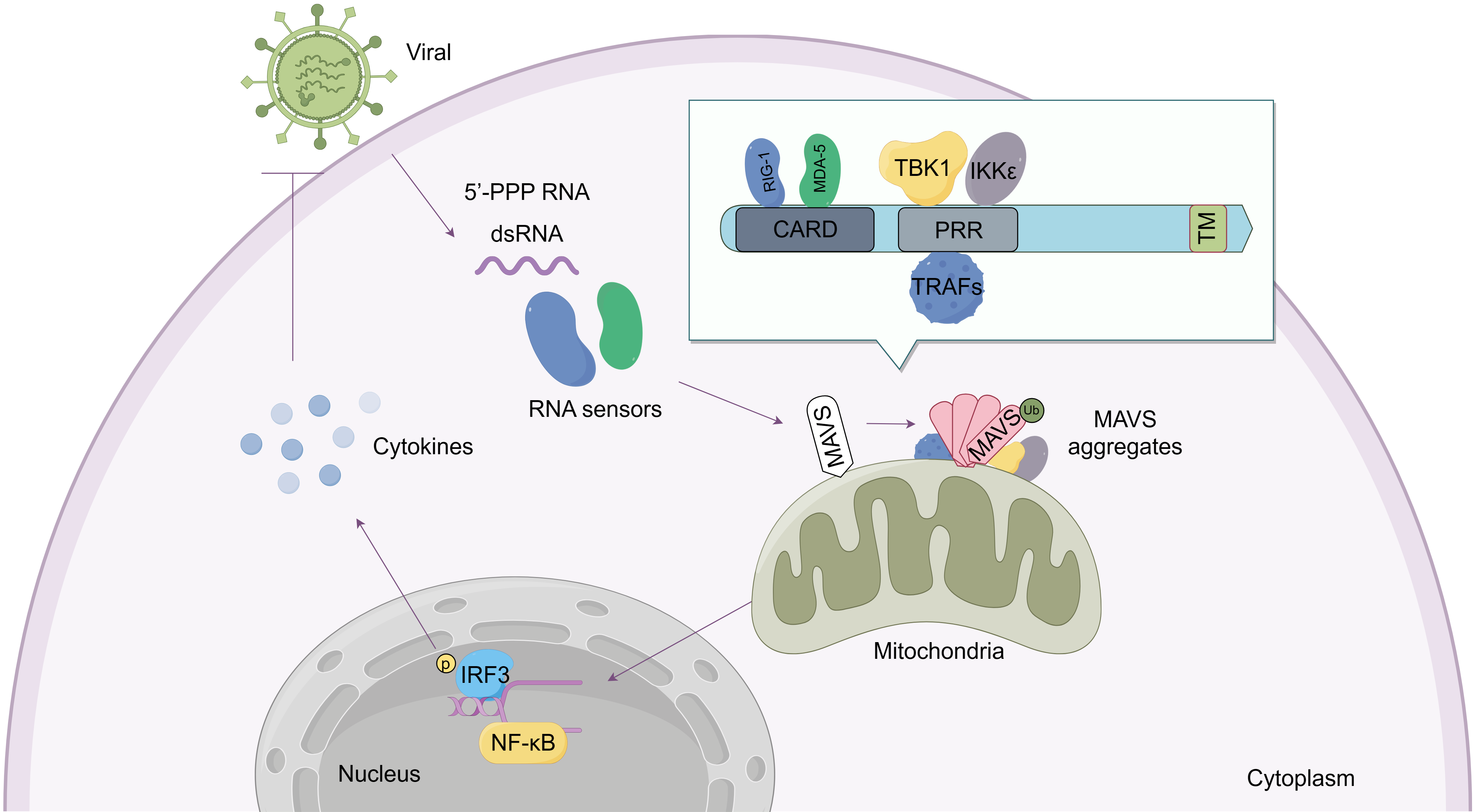 Fig. 1.
Fig. 1.Structure and function of the mitochondrial antiviral-signaling
protein (MAVS) protein. As an adaptor molecule, the MAVS protein contains three
primary domains: The N-terminal caspase-recruitment domain (CARD), Proline-rich
region (PRR), and the C-terminal transmembrane domain (TM). MAVS detects signals
from viral RNA sensors such as RIG-I and MDA5 through its CARD domain. MAVS
subsequently aggregates and recruits downstream proteins including TRAFs, TBK1,
and IKK
Four ubiquitylation patterns are observed in MAVS, these include K48-linked ubiquitylation, K63-linked ubiquitylation, K27-linked ubiquitylation, and Small Ubiquitin-like Modifier (SUMO) modification (SUMOylation). Biological functions and enzymes involved in MAVS ubiquitylation are summarized in Fig. 2.
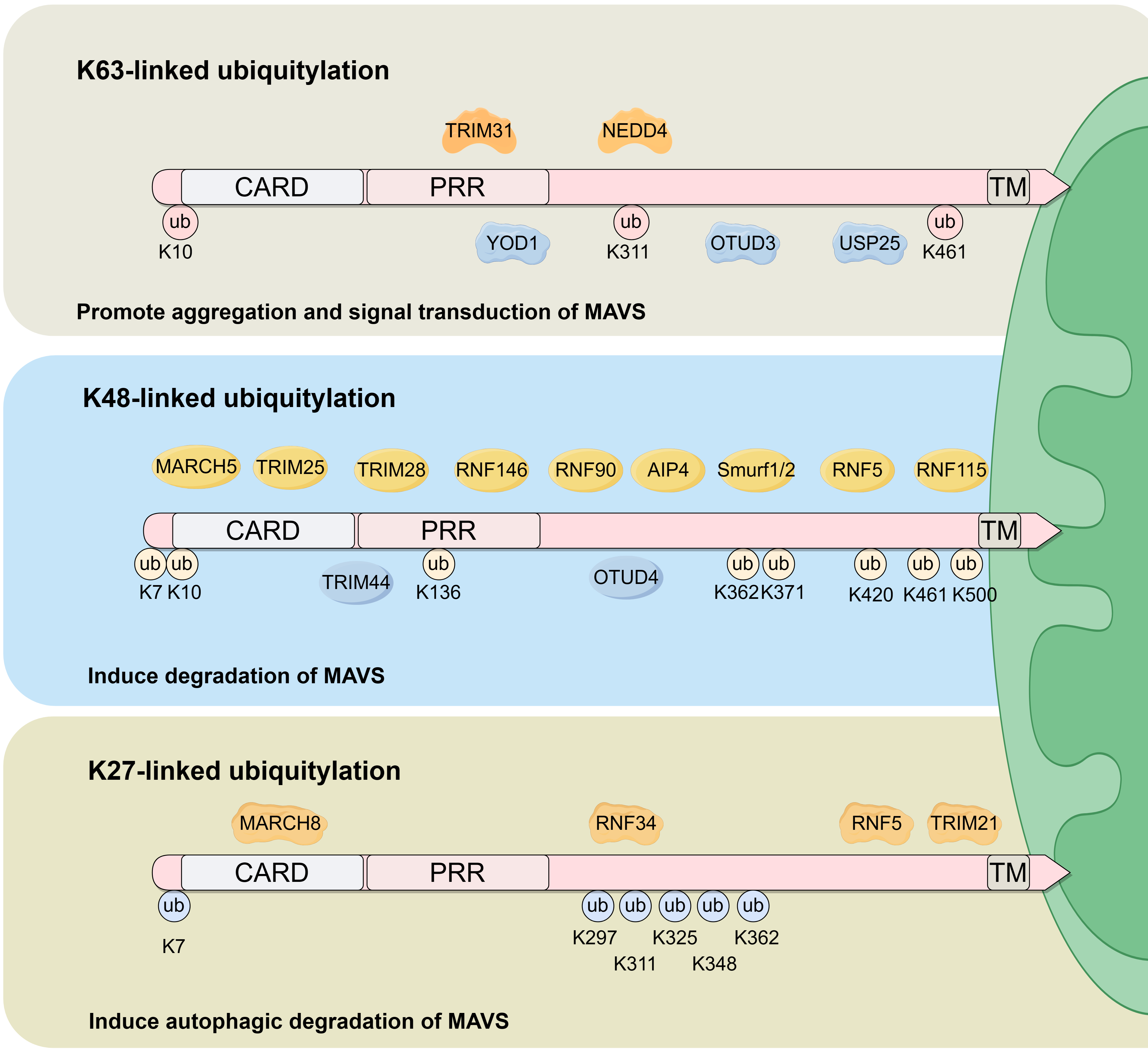 Fig. 2.
Fig. 2.Biological functions and enzymes involved in MAVS ubiquitylation. K63-linked, K48-linked and K27-linked ubiquitination are the primary ubiquitination sites within the MAVS protein. K63-linked ubiquitylation is induced by TRIM31 and NEDD4, and is inhibited by YOD1, OTUD3 and USP25. The primary biological function of K63-linked ubiquitylation is to promote MAVS aggregation and activate downstream signaling. K48-linked ubiquitylation is induced by MARCH5, TRIM25, TRIM28, RNF146, RNF90, AIP4, Smurf1/2, RNF5 and RNF115, and is inhibited by TRIM44 and OTUD4. The principal biological function of K48-linked ubiquitylation is to promote MAVS degradation. K27-linked ubiquitylation is induced by MARCH8, RNF34, RNF5 and TRIM21 and functions to promote MAVS autophagic degradation. ub, ubiquitylation.
K48-linked ubiquitylation mediates the degradation of MAVS and acts as an important negative regulator of innate antiviral immunity [21]. K48-linked ubiquitylation may limit excessive MAVS activation, which could result in imbalances in the immune system and lead to autoimmune disease. Alternatively, K-48 ubiquitination may limit anti-viral immune response and promote disease progression stemming from viral infection [22]. A series of E3 ligases, including AIP4 and Smurf1, could be transported to the mitochondria with the help of scaffolding proteins, such as PCBP2 and NDFIP1. Once present at the mitochondria, MAVS K48-linked ubiquitylation would result in MAVS degradation through the ubiquitin-proteasome pathway [23, 24]. Those factors that control K48-linked ubiquitylation and mediate MAVS degradation require further elucidation.
K63-linked ubiquitylation may enhance MAVS aggregation and induce the
interaction between MAVS and the viral infection sensor RIG-I [21, 25].
Mechanistically, K63-linked polyubiquitin chains on MAVS can be directly bound by
RIG-I and promote RIG-I-mediated MAVS aggregation through their respective CARD
interaction domains [26]. Moreover, K63-linked ubiquitylation could activate MAVS
and induce the recruitment of other downstream signal transducers, including
TANK-binding kinase 1 (TBK1) and I
K27-linked ubiquitylation of MAVS is reported to be involved in several critical biological processes including mitophagy (a form of selective autophagy), proteasomal degradation, T cell differentiation, DNA repair, innate immunity regulation, and signal transduction [25, 29, 30, 31, 32]. The function of K27-linked MAVS ubiquitylation has primarily focused on autophagy-mediated degradation [33, 34, 35]. Autophagy receptors NBR1 and NDP52 recognize MAVS K27-linked ubiquitin chains and induce autophagy-mediated degradation of MAVS [33, 34, 35]. However, a potential role for MAVS K27-linked ubiquitylation in antiviral immune response requires further elucidation.
SUMOylation, a form of post-translational modification, involves the covalent attachment of small ubiquitin-like modifier (SUMO) proteins to their substrates [36]. SUMOylation is a highly dynamic and reversible process similar to ubiquitylation, but functions differently in terms of target-specific selectivity and cellular outcomes [37]. SUMOylation is reported to regulate stability, activity, and cellular localization of target protein as well as influencing protein-protein interactions under various conditions [37].
SUMOylation plays a crucial role in allowing MAVS to receive signals from
upstream viral RNA sensors and activate downstream inflammatory response pathways
[38]. For example, MAVS SUMOylation is increased during SARS-CoV-2 infection,
thereby leading to increased levels of MAVS protein. This, in turn,
hyper-activates NF-
Ubiquitylation is a common post-translational modification of MAVS that regulates RLRs-MAVS signaling which controls a wide variety of immunological processes such as immune defense and inflammation [41]. Multiple E3 ubiquitin ligases participate in MAVS regulation through mediating different ubiquitylation patterns. For example, E3 ligases which mediate MAVS K27 or K48-linked ubiquitylation often induce MAVS degradation, thus inhibiting MAVS-related immune response [21, 33, 34, 35]. Other E3 ligases that control K63-linked ubiquitylation could enhance MAVS aggregation and activate RIG-I/MAVS signaling [26]. Generally, these E3 ligases are classified into RING-finger type E3 ligases, homologous to E6-associated protein C-terminus (HECT) type E3 ligase, U-box type E3 ligase, SUMO E3 ligase, and virally-derived proteins that function as E3 ligases. The details and biological functions of these E3 ligases in the context of MAVS signaling are summarized in Table 1 (Ref. [7, 22, 23, 24, 33, 38, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57]).
| Gene name | Ubiquitylation Type | Ubiquitylation site on MAVS | Biological function | References |
| RING-finger type E3 ligases | ||||
| TRIM31 | K63-linked ubiquitylation | K10, K311, K461 | Promote MAVS aggregation | [42] |
| TRIM25 | K48-linked ubiquitylation | K7, K10, K371, K420 | Degrade MAVS and activate type-I interferon signaling | [7] |
| TRIM28 | K48-linked ubiquitylation | K7, K10, K371, K420, K500 | Degrade MAVS | [43] |
| TRIM29 | K11-linked ubiquitylation | K371, K420, K500 | Degrade MAVS | [44] |
| TRIM21 | K27-linked ubiquitylation | K325 | Induce the recruitment of TBK1 to MAVS | [45] |
| MARCH5 | K48-linked ubiquitylation | K371, K500 | Degrade MAVS | [22] |
| MARCH8 | K27-linked ubiquitylation | K7 | Induce autophagic degradation of MAVS | [46] |
| RNF5 | K48, K27-linked ubiquitylation | K362, K461 | Induce ER-associated degradation of MAVS | [47] |
| RNF34 | K27-linked ubiquitylation | K29, K311, K348 | Induce autophagic degradation of MAVS | [33] |
| RNF90 | K48-linked ubiquitylation | Uncertain | Degrade MAVS | [48] |
| RNF115 | K48-linked ubiquitylation | K500 | Degrade MAVS | [49] |
| RNF146 | K48-linked ubiquitylation | K7 | Degrade MAVS | [50] |
| GP78 | Uncertain | Uncertain | Induce ER-associated degradation of MAVS | [51] |
| HECT E3 ligases | ||||
| AIP4 | K48-linked ubiquitylation | K371, K420 | Degrade MAVS | [23] |
| NEDD4 | K63-linked ubiquitylation | Uncertain | Induce the interaction between MAVS and other components like TRAF2 | [52] |
| SMURF1/2 | K48-linked ubiquitylation | Uncertain | Degrade MAVS | [24, 53] |
| SUMO E3 ligase | ||||
| STUB1 | Uncertain | Uncertain | Degrade MAVS | [54] |
| SYNV1 | Uncertain | Uncertain | Degrade MAVS | [55] |
| PIAS3 | SUMOylation | K461, K500 | Induce MAVS aggregation and phase separation | [38] |
| Virus-derived proteins function as E3 ligases | ||||
| NSP5 | K48-linked ubiquitylation | K136 | Degrade MAVS | [56] |
| SARS-NP | Block K63-linked ubiquitylation | Uncertain | Inhibit MAVS related antiviral signaling | [57] |
Really Interesting New Gene finger (RING-finger) type E3 ligases are the majority of the various E3 proteins characterized to date. RING-finger E3 ligases mediate direct transfer of the ubiquitin from E2 proteins to their substrate [58]. RING-finger type E3 ligases involved in MAVS ubiquitylation includes TRIM family members TRIM31, TRIM25, TRIM28, TRIM29, TRIM44, and TRIM21, MARCH family members MARCH5 and MARCH8, RNF family members RNF34, RNF90, RNF115, and RNF146, and the GP78 protein.
1.3.1.1 TRIM31
Tripartite motif containing 31 (TRIM31) is the first identified E3 ubiquitin ligase to regulates MAVS activation [42]. Liu et al. [42] found that TRIM31 could translocate into the mitochondria and interact with the proline-rich domain of MAVS protein via its C-C motif in response to viral attack. TRIM31 subsequently induces ubiquitylation on MAVS lysine residues 10, 311 and 461 [42] and promote RIG-I assembly of tetramers through the CARD domain. These modifications subsequently enhance MAVS aggregation and activate downstream signaling [42]. These findings also indicate that, similar to TAK1 and TBK1 activation [59, 60], MAVS K63-linked polyubiquitination may also recruit other adaptors with ubiquitin-binding domains that promote MAVS oligomerization. Other potential regulatory steps involved in MAVS K63-linked ubiquitylation require further elucidation [42].
1.3.1.2 TRIM25
Tripartite motif containing 25 (TRIM25) is known to mediate K48-linked
ubiquitylation at MAVS lysine residues 7, 10, 371, and 420 [7]. Interestingly,
TRIM25 mediated MAVS Lys 7 and 10 ubiquitylation could activates the type I
interferon (IFN) signaling, rather than NF-
1.3.1.3 TRIM28
Tripartite motif containing 28 (TRIM28) could induce ubiquitination at MAVS lysine residues 7, 10, 371, 420, and 500 resulting in MAVS degradation [43]. TRIM28 has various biological functions, including inhibiting type I interferon production and enhancing transcription of other pro-inflammatory cytokines [62]. Further study is needed to unveil the regulatory role of TRIM28 in MAVS-mediated RLR signaling pathway.
1.3.1.4 TRIM29
As a negative regulator in innate immunity, tripartite motif containing 29 (TRIM29) mediates the ubiquitylation at MAVS lysine residues 371, 420, and 500 and this subsequently induces MAVS degradation [44]. The C-terminal domain of TRIM29 is important for MAVS/TRIM29 interaction [44].
1.3.1.5 TRIM21
Tripartite motif containing 21 (TRIM21) is reported to mediate ubiquitylation of MAVS at lysine residue 325. This ubiquitination of MAVS induces TBK1 recruitment and activates downstream type I interferon signaling [45]. The PRY segment within the SPRY domain of TRIM21 protein binds to the transmembrane domain of MAVS, and is essential for TRIM21-mediated MAVS ubiquitylation [45]. Moreover, upon type I interferon stimulation, the interferon stimulated gene (ISG) UBL7 interacts with both MAVS and TRIM21. This association enhancing their interaction in a dose-dependent manner [63] and may form a positive feedback loop to achieve a swift and robust anti-viral response.
1.3.1.6 MARCH5
Membrane associated RING-CH protein 5 (MARCH5) is the only member of the MARCH family which located on the outer membrane of mitochondria [64]. Viral infection inhibits the autoubiquitylation of MARCH5, thus inducing its accumulation [22]. The accumulated MARCH5 protein subsequently mediates ubiquitylation of MAVS at lysine residues 7 and 500 [22]. Moreover, the CARD domain of MAVS is essential for MARCH5-MAVS interaction [22, 65]. Interestingly, MARCH5 only interacts with MAVS in the oligomeric form, rather the monomeric form [22]. This selective interaction enables MARCH5 to degrade activated MAVS in a temporally-controlled manner and thus avoid excessive immune activation. Moreover, MARCH5 induced K48-linked ubiquitylation and degradation is enhanced by PKA-mediated phosphorylation of MAVS at the threonine 54 residue [65].
1.3.1.7 MARCH8
The Membrane Associated RING-CH Protein 8 (MARCH8) catalyzes MAVS ubiquitylation at the Lys7 residue [46]. The interferon-induced bone marrow stromal cell antigen 2 (BST2) is reported to be a vital scaffolding protein regulating MAVS modification by MARCH8 [46, 66]. BST2 induces MARCH8 recruitment, thereby upregulating MAVS ubiquitylation [46]. MARCH8-mediated MAVS ubiquitylation functions as a recognition signal for NDP52-dependent autophagic degradation, and forms a negative feedback regulation in RLR-MAVS antiviral immune response to avoid excessive immune activation [46, 66].
1.3.1.8 RNF5
Ring Finger Protein 5 (RNF5), localized in endoplasmic reticulum (ER) and mitochondria, is reported to induce MAVS ubiquitylation at Lys362 and Lys461 residues. The middle domain, RING domain and transmembrane domain of RNF5, along with transmembrane domain of MAVS, are essential for RNF5/MAVS interaction [47, 67]. The function and regulation of RNF5-mediated MAVS ubiquitylation is complex. In the absence of viral infection, RNF5 can assemble with MAVS and GP78, another E3 ligase, and form a complex resulting in the degradation of MAVS within the ER [68]. In the early stages of viral infection, RNF5 could enhance the ER-associated degradation (ERAD) of MAVS protein, and could also translocate to the mitochondria to induce MAVS ubiquitylation degradation [68]. Interestingly, the V protein of the Newcastle disease virus (NDV) recruits RNF5 to induce MAVS ubiquitylation, thereby triggering an immune evasion in paramyxovirus family virus infection [69]. Moreover, the PB1 protein of the H7N9 influenza virus induces RNF5-mediated MAVS K27-linked ubiquitylation. This allows the selective autophagy receptor NBR1 to recognize ubiquitinated MAVS and inhibit MAVS-associated innate immunity [34]. Therefore, RNF5-related MAVS ubiquitylation could function, as not only a protective mechanism to prevent excessive activation of antiviral inflammatory responses, but also a viral-protein-mediated immune evasion mechanism.
1.3.1.9 RNF34
RNF34, also known as the RING finger protein 34, mediates polyubiquitylation of MAVS at lysine residues 297, 311, 348, and the Arg362 residue, thereby regulating MAVS accumulation and degradation [33]. Lys311 of MAVS maybe ubiquitinated by TRIM31 as well [42]. During viral infections, RNF34 is upregulated and translocated to the outer mitochondrial membrane, and accumulates due to MAVS K63-linked polyubiquitin chains [33]. This subsequently induces the transition of K63-linked ubiquitylation to K27-linked ubiquitylation on MAVS Lys311 residues [33]. The cargo receptor, NDP52, recognizes K27-linked polyubiquitinated MAVS aggregates and subsequently transports MAVS from the mitochondria to lysosomes for autophagic clearance [33]. Therefore, RNF34-mediated ubiquitylation may function in a negative feedback loop to control MAVS-related antiviral responses [33].
1.3.1.10 RNF90
RING finger protein 90 (RNF90) mediates ubiquitylation of MAVS and induces its degradation [48]. RNF90 expression is induced by RNA virus infection, and it may interact with residues 180–360 within the MAVS protein [48].
1.3.1.11 RNF115
RING finger protein 115 (RNF115) induces ubiquitylation on the Lys500 residue of the MAVS protein [49]. The transmembrane domain of MAVS and the N-terminal domain of RNF115 are important in promoting RNF115-MAVS interaction and this interaction is important in regulating MAVS stability. Under normal conditions, RNF115 promotes MAVS ubiquitylation and degradation [49]. Upon Sendai virus infection or cytoplasmic poly(I:C) stimulation, RNF115 disassociates from MAVS due to the likely activation of RIG-I RNA ligands. This results in stabilization of the MAVS protein [49].
1.3.1.12 RNF146
RING finger protein 146 (RNF146) mediates ubiquitylation of MAVS at the Lys7 residue. During viral infection, the poly ADP-ribose polymerase tankyrase 1/2 (TNKS1/2) is upregulated and translocated into mitochondria. This, in turn, induces MAVS ADP-ribosylation at residue Glu137, leading to the interaction of RNF146 interaction with ADP-ribosylated MAVS through its WWE domain. This interaction provokes MAVS ubiquitylation and promotes its degradation [50].
1.3.1.13 GP78
GP78, an E3 ubiquitin ligase involved in endoplasmic reticulum associated degradation (ERAD), primarily performs quality control for ER proteins by tagging misfolded proteins for proteasomal degradation [51]. GP78 is reported to localize within the mitochondria-associated endoplasmic reticulum membrane (MAM) and promotes MAVS degradation through ERAD-related ubiquitylation [51]. GP78 binds to both the N and C-terminus of MAVS through its C-terminal RING domain [51]. GP78 could also inhibit MAVS activation by competing with RIG-I for binding to the MAVS CARD domain [51].
Homologous to E6-associated protein C-terminus (HECT) domain-containing E3 ligases are the second largest group of E3 ligases, and possess the catalytic activity necessary to form a covalent thioester intermediate where the ubiquitin moiety is transferred from the E2 to a cysteine on the E3 ligase prior to its transfer to a lysine on the substrate molecule [70]. Several HECT E3s are involved in MAVS ubiquitylation, including AIP4, NEDD4, and SMURF1/2.
1.3.2.1 AIP4
AIP4, also known as ITCH, is reported to induce ubiquitylation and degradation of MAVS during viral infection [23]. During viral invasion, poly(rC) binding protein 2 (PCBP2) translocates from nucleus to mitochondria and recruits AIP4 to MAVS through its WW domain. This event subsequently mediates MAVS ubiquitylation at Lys371 and Lys420 residues and induces MAVS degradation [23].
1.3.2.2 NEDD4
The NEDD4 E3 ubiquitin ligase plays an important role in the catalysis of MAVS ubiquitylation and aggregation. N4BP3, a binding protein of NEDD4, functions as an adaptor molecule to induce interaction between MAVS and other molecules, such as TRAF2, resulting in activation of the innate antiviral immune response [52].
1.3.2.3 SMURF1/2
SMURF1/2, members of Smad ubiquitin regulatory factors, are involved in the ubiquitylation of MAVS [53, 71]. The PY motif of NEDD4 family-interacting protein 1 (NDFIP1) is critical for promoting SMURF1 and MAVS interaction [24]. Upon viral infection, OTU de-ubiquitinase 1 (OTUD1) is upregulated and subsequently increases SMURF1 stability [71]. As a result of this event, MAVS is ubiquitinated and degraded, leading to the suppression of immune and inflammatory responses [71, 72].
The U-box proteins could function as E3s in E2-dependent ubiquitylation, and are defined as a third family of E3 enzymes [73].
1.3.3.1 STUB1
STIP1 homology and U-box containing protein 1 (STUB1) has been reported to be involved in the ubiquitylation and degradation of MAVS [54]. In response to bovine ephemeral fever (BEF) infection, the interaction between STUB1 and MAVS is increased, thereby promoting MAVS ubiquitylation [54]. However, the modified amino acid sites, other potential regulators, and the detailed regulatory mechanism involved in STUB1-mediated MAVS ubiquitylation remain uncertain.
1.3.3.2 SYVN1
Synoviolin 1 (SYVN1) promotes poly-ubiquitylation and degradation of MAVS during stress and infection [55]. SYVN1-mediated ubiquitylation is induced by Mitofusin 2 (Mfn2), which is upregulated by cortisol stimulation [55]. Mfn2 enhances the interaction between SYVN1 and MAVS, thereby accelerating MAVS degradation [55]. Notably, the HR1 domain of Mfn2, the C-terminus of MAVS, and the transmembrane domain of SYVN1 are essential for Mfn2-SYVN1-MAVS signaling [55] and this signaling axis contributes to the regulation of cell stress and antiviral innate immunity [55].
During viral infection, PIAS3, identified as a SUMO E3 ligase, is reported to induce MAVS SUMOylation at Lys461 and Lys500 residues [38]. MAVS self-recognizes SUMO-ubiquitin hybrid chains through SIM, a conserved SUMO interaction motif within the MAVS CARD domain. This interaction initiating protein aggregation and facilitates phase separation [38]. Moreover, SUMO-ubiquitin hybrid chains strengthen the connection between IRF3 and MAVS, thus triggering co-phase separation and activating an antiviral immune signaling process. MAVS SUMOylation may be negatively regulated by SENP1, a protease that specifically targets SUMO modification [38]; however, the details of upstream signaling involved in MAVS SUMOylation remains uncertain.
Some virus-derived proteins function as E3 ligases and are involved in MAVS ubiquitylation [56, 57, 74]. For example, nonstructural protein 5 (NSP5), a chief cysteine protease of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) can not only cleave viral protein precursors, but also serves as an E3 ligase to mediate polyubiquitylation of MAVS at the Lys136 residue, thereby triggering immune evasion [56]. Another distinct viral protein, SARS-CoV-2 nucleocapsid protein (SARS2-NP), has been reported to block MAVS K63-linked ubiquitylation and inhibit MAVS-related antiviral signaling [57]. Further, the open reading frame 10 (ORF10) of SARS-CoV-2 could induce Nip3-like protein X (NIX) and LC3B translocation into the mitochondria, thus promoting mitophagy and MAVS degradation [74].
Ubiquitylation induces formation of prion-like aggregates and activation of MAVS [42]. To maintain appropriate levels of MAVS activation, de-ubiquitylation also plays an important role in the immunological signaling. Here we review several de-ubiquitinases which are involved in MAVS regulation (Table 2, Ref. [75, 76, 77, 78, 79]).
| Gene name | Target | Biological function | References |
| TRIM44 | K48-linked ubiquitylation | Suppress MAVS K48-linked polyubiquitylation and protecting MAVS from ubiquitin-proteasome-dependent degradation | [75] |
| USP25 | K63-linked ubiquitylation | Induce MAVS K63-linked de-ubiquitylation, thereby promoting MAVS degradation | [76] |
| OTUD3 | K63-linked ubiquitylation | Restrain MAVS K63-linked ubiquitylation, thereby inhibits MAVS aggregation and activation | [77] |
| OTUD4 | K48-linked ubiquitylation | Degrade MAVS K48-linked polyubiquitin chains, thereby maintains MAVS stability | [78] |
| YOD1 | K63-linked ubiquitylation | Remove MAVS K63-linked ubiquitin chain, thereby inhibit MAVS aggregation | [79] |
Tripartite motif containing 44 (TRIM44) is an atypical member of TRIM family due to the absence of a conventional RING finger domain, but does contain a ubiquitin hydrolase zinc finger (ZF-UBP) domain and ubiquitin specific protease (USPs) domains [80]. Unlike other TRIM family members, TRIM44 belongs to a newly evolving class of “USP-like TRIM” molecules which regulate protein de-ubiquitylation and stabilization [80, 81]. Intriguingly, TRIM44 is reported to suppress the K48-linked polyubiquitylation of MAVS following virus infection [75]. Owing to its ZF-UBP domain, TRIM44 removes ubiquitin from its substrates prior to proteasomal recognition, thus protecting MAVS from ubiquitin-proteasome-dependent degradation [75].
The ubiquitin-specific peptidase 25 (USP25) mediates MAVS de-ubiquitylation [76]. Contactin-1 (CNTN1), a member of the immunoglobulin superfamily, is reported to assist in recruiting USP25 to MAVS and inducing MAVS de-ubiquitylation. In turn, this promotes MAVS degradation through the proteasomal pathway and blocks RIG-I-MAVS-mediated interferon signaling [76].
Ovarian tumor de-ubiquitinase 3 (OTUD3), a member of the ovarian tumor de-ubiquitinase family, is reported to restrain MAVS K63-linked ubiquitylation. This inhibits MAVS aggregation and activation, and avoids excessive antiviral immune responses in uninfected cells [77]. Upon viral infection, however, OTUD3 enzymatic activity is downregulated by SIRT1-mediated acetylation, thus allowing MAVS to maintain its K63-linked ubiquitylation and activation [77].
Ovarian tumor de-ubiquitinase 4 (OTUD4), another member of the ovarian tumor de-ubiquitinase family, interacts with the transmembrane domain of MAVS via its OTU motif and induces MAVS de-ubiquitylation. This maintains MAVS stability and activates downstream innate antiviral signaling [78]. OTUD4 is upregulated by IRF3/7 signaling, and degrades K48-linked polyubiquitin chains on MAVS. This results in maintaining MAVS stability and promoting innate antiviral signaling [78]. Of note, OTUD4 also exhibits regulatory capabilities over K63-linked ubiquitylation in other proteins such as MyD88 [82, 83] and TRAF6 [84]. Whether OTUD4 regulate MAVS in K63-linked ubiquitylation dependent manner remains an open question.
YOD1, an OTU family de-ubiquitinase, translocates from the mitochondria and interacts with the proline-enriched domain of MAVS through YOD1’s UBX and ZNF domains following viral infection [79]. YOD1 removes MAVS K63-linked ubiquitin chain by virtue of its OTU domain, thereby regulating MAVS aggregation and downstream antiviral immune signaling [79].
Ubiquitylation plays a pivotal role in assuring accurate and timely regulation of MAVS-related inflammatory responses, and is affected by various upstream signals including inflammation-related feedback loops, metabolic adaptation, and cell stress [45, 55, 77]. Inflammation-associated transcription factors, such as IRF3/7, may act in a regulatory role to provide either positive, or negative, feedback regulation of MAVS ubiquitylation [78]. Moreover, some regulators, such as BST2, PCBP2, and CNTN1, are be recruited into the mitochondria through upstream signaling control and, thereby, regulating MAVS ubiquitylation and activity [23, 66, 76]. Additionally, a series of de-ubiquitinases is also involved in controlling MAVS ubiquitylation [76]. Some of them directly trigger MAVS de-ubiquitylation, while others, such as USP18, indirectly regulate MAVS through a ubiquitinase-independent mechanism [85]. The complex regulatory manner of MAVS ubiquitylation is essential in maintaining appropriate immune response against viral infection [86].
Ubiquitylation patterns are essential in controlling MAVS signaling. For instance, K27 or K48-linked ubiquitylation often induce MAVS degradation and inhibition of innate immune response [21, 33, 34, 35]. In contrast, K63-linked ubiquitylation enhances MAVS aggregation and activates RIG-I/MAVS signaling [26]. Therefore, ubiquitin “codes” either through the addition or removal of uniquitin moieties is crucial in MAVS-related immune pathway. The E3 ligases and de-ubiquitinases described above act as the “writer” of MAVS poly-ubiquitin chains and regulate MAVS activity through modulation of ubiquitylation pattern. In contrast, RIG-I could work as a “reader” by binding to the unanchored K63 ubiquitin chains and subsequently inducing signal transduction through the RIG-I pathway [87]. Furthermore, K63 polyubiquitylation of MAVS is reported to promote MAVS interaction with TBK1 and play an important role in its activation [88]. Further studies concerning other factors that control the “writing/reading” of MAVS ubiquitylation holds much promise in understanding innate immunity.
Crosstalk between other forms of MAVS post-translational modifications and ubiquitylation have been observed. For example, poly-ADP-ribosylation, glycosylation, and acetylation are reported to regulate MAVS ubiquitylation [50, 77, 89]. These protein modifications connect metabolic pathways to MAVS ubiquitylation, thereby constructing a regulatory network linking metabolism and immunity. A current focus of studies in this area include those that examine the relationship between MAVS ubiquitylation and viral infection. However, numerous studies support that MAVS signaling is also involved in diseases stemming from with autoimmune dysregulation, such as multiple sclerosis (MS), systemic lupus erythematosus (SLE), and psoriasis [90, 91, 92]. Moreover, the RLR-MAVS pathway plays a crucial role in cancer progression and tumor immunotherapy [93, 94]. Abnormal MAVS signaling is also linked to aberrant inflammatory responses resulting in immune evasion or immune-related tissue damage [95, 96].
Ubiquitylation has emerged as a central mechanism regulating the RLRs-MAVS signaling axis during immune response. Multiple E3 ubiquitin ligases and de-ubiquitinating enzymes have been characterized as controlling MAVS ubiquitylation levels and promoting changes in MAVS functional properties. Abnormal MAVS ubiquitylation could lead to immune related diseases; therefore, targeting MAVS ubiquitylation represents a potential approach for immunotherapy.
JS, Ideation; HD, literature search; JS and HD, writing the manuscript and work revision. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We would like to thank Ejear (ejearedit.com) for English language editing.
This work was supported by grants from the Hubei Provincial Natural Science Foundation of China (Grant numbers: 2023AFB118) and Science Foundation of Tongji Hospital (Grant numbers: 2022B24).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.