








1 Department of Medicine and Surgery, University of Perugia, 06132 Perugia, Italy
2 CISM Mass Spectrometry Center, University of Florence, 50139 Firenze, Italy
†These authors contributed equally.
Abstract
Background: The recent Coronavirus Disease 2019 (COVID-19) pandemic has dramatically exposed our gap in understanding the pathogenesis of airborne infections. Within such a context, it is increasingly clear that the nasal cavity represents a critical checkpoint not only in the initial colonization phase but also in shaping any infectious sequelae. This is particularly relevant to COVID-19 in that the nasal cavity is characterized by high-level expression of the Severe Acute Respiratory Syndrome CoronaVirus 2 (SARS-CoV-2) receptor, Angiotensin-Converting Enzyme 2 (ACE2), all along the respiratory tract. As part of the nasal mucosa, commensal microbes harbored by the nasal cavity likely are far more than just innocent bystanders in the interaction between SARS-CoV-2 and the local microenvironment. Yet the role of the qualitative composition of the nasal microbiome is unclear, as is its function, whether protective or not. Methods: In this study, individuals undergoing SARS-CoV-2 molecular testing at the Hospital of Perugia (Italy) were recruited, with their residual material from the nasopharyngeal swabs being collected for microbiome composition analysis and short-chain fatty acid (SCFA) measurements (by 16S rRNA sequencing and gas chromatography-mass spectrometry), respectively. Results: After stratification by age, gender, and viral load, the composition of the nasopharyngeal microbiome appeared to be influenced by age and gender, and SARS-CoV-2 infection further determined compositional changes. Notwithstanding this variability, a restricted analysis of female subjects—once SARS-CoV-2–infected—unraveled a shared expansion of Lachnospirales-Lachnospiraceae, irrespective of the viral load and age. This was associated with a reduction in the branched SCFA isobutanoic acid, as well as in the SCFAs with longer chains. Conclusions: Our results indicate that the nasopharyngeal microbiome is influenced by age, gender, and viral load, with consistent patterns of microbiome changes being present across specific groups. This may help in designing a personalized medicine approach in COVID-19 patients with specific patterns of nasal microbial communities.
Keywords
- COVID-19
- nasopharyngeal microbiome
- short-chain fatty acids
The overall human population has been experiencing a viral pandemic caused by the Severe Acute Respiratory Syndrome CoronaVirus 2 (SARS‑CoV‑2), a strain of coronavirus that causes Coronavirus Disease 2019 (COVID-19). The numbers are impressive, with more than 6 hundred million cases and 6.67 million deaths worldwide [1]. A global effort has been made to unravel the pathogenesis of COVID-19, with the aim of developing therapeutic strategies to either prevent or cure viral infection and improve prognosis. Studies have shown that SARS‑CoV‑2 binds to the Angiotensin-Converting Enzyme 2 (ACE2) and is released inside susceptible cells upon membrane fusion, as mediated by cleaved spike protein. Genome and protein synthesis ensure viral replication and release from infected cells to complete the viral cycle. These studies have also clearly demonstrated that the host-pathogen interaction is critical in shaping the clinical features associated with SARS-CoV-2 infection, which range from an asymptomatic status to potentially fatal outcomes. From an immunological viewpoint, an initial anti-viral, interferon-based, immune response may protect from disease development, whereas a pro-inflammatory-skewed response may lead to cytokine storm and subsequent, potentially fatal multi-organ failure. Several factors have been implicated in these wide-ranging clinical pictures of COVID-19. Age and co-morbidities undoubtedly play a major role, together with genetic and biological factors of individual susceptibility. Among the latter, the communities of commensal microorganisms that colonize the surfaces exposed to the external environment, or microbiota, have been increasingly recognized as a critical factor in the host response to SARS-CoV-2 infection.
Although the gut represents a major reservoir of commensal microbes—with their metabolites exerting both local and distant immune regulatory effects [2]—emerging evidence calls for the need to consider the microbiome of the respiratory tract as a fundamental barrier against airborne pathogens [3]. The nose has particular importance in the context of COVID-19. Indeed, the nose contains the highest levels of the SARS-CoV-2 receptor ACE2 well along the respiratory tract, and consequently, infectivity progressively decreases from the upper to the lower respiratory tracts. Therefore, the nasal microbiota is involved in the early phase of SARS-CoV-2 infection, and its ability to compete against pathogen colonization—and modulate the ensuing immune response—may represent a critical checkpoint for subsequent disease outcomes [4]. The nasopharyngeal microbiome is indeed known to play a major role in the regulation of both local and systemic immunity, thus determining COVID-19 clinical outcomes [5]. This is in agreement with other studies [6, 7, 8, 9] in the search for therapeutic strategies acting at the microbiota/immune interface, including probiotic administration, modulation of nutrients, [4] and vitamin supplementation [10, 11]. Although the correlation between changes in nasopharyngeal microbiome composition and disease severity has been consistently demonstrated, it remains unclear whether the initial viral replication in the upper respiratory tract is associated with dysbiotic events. Relevant in this regard, some studies failed to detect differences in microbiome compositions between SARS-CoV-2 negative and positive individuals [12, 13]; others have nevertheless reported a significant decrease in protective, nasal commensal bacteria and a parallel expansion of opportunistic pathogens [14, 15, 16, 17, 18]. The identification of microbial signatures of SARS-CoV-2 infectivity would be relevant to the prevention of COVID-19 by allowing the identification of individuals with potentially increased susceptibility to infection and to the design of microbiome-based therapeutic approaches with a view to re-establishing a protective barrier. The achievement of this goal requires that other individual factors of microbiome variability be identified and integrated with SARS-CoV-2 infection to unravel personalized patterns of microbiome changes.
Based on these premises, we designed a cross-sectional, monocentric study whereby individuals tested for SARS-CoV-2 infection were homogenously stratified according to age, gender, and viral load, and their nasopharyngeal microbiome was characterized by 16S rRNA sequencing. As a result, we found that the nasopharyngeal microbiome is influenced by age and gender and that SARS-CoV-2 infection further determines compositional changes. Differential abundance testing and targeted metabolomics analysis could reveal consistent patterns of microbiome changes in selected groups of individuals with the potential for identifying microbial signatures of resistance vs. susceptibility to infection.
This cross-sectional and monocentric study was approved by the Ethics Committee of the Umbria Region (CER Umbria, approval No. 3990/21) with a waiver of informed consent. Nasopharyngeal swabs from individual adults presenting at the Hospital of Perugia (Umbria, Italy) for SARS-CoV-2 molecular testing were collected. The inclusion criteria consisted of collecting only specimens from the population living in Umbria, with the exclusion of people younger than 18 years or with a history of previous SARS-CoV-2 infection. Samples were collected from January to March 2021, at a time when the predominant circulating variants of concern (VOCs) were B.1.1.7 and P.1. During this interval, the vaccine campaign was in its early phase, such that the vast majority of samples were from unvaccinated individuals.
Sterile synthetic-head, plastic-shaft swabs were used to collect specimens and placed in Universal Transport Medium (UTM, Copan, Brescia, Italy). The swabs were processed for SARS-CoV-2 testing according to standard procedures/Centers for Disease Control and Prevention (CDC) guidelines. The swabs were tested by TaqPath RT-PCR COVID-19 Kit (Thermo Fisher Scientific, Waltham, MA, USA), which targets S (spike), N (nucleocapsid) and ORF1ab (open reading frame) genes, and Allplex™ SARS-CoV-2 assay (Seegene, Seoul, Korea) which targets E (envelope), N (nucleocapsid), and RdRp (RNA-dependent-RNA-polymerase) genes.
A total of 540 nasopharyngeal swabs (NPS) were collected and stratified into 18 groups based on age
(18–40, 41–60, and
The primary study end-point was the change from baseline in both diversity indexes (alpha and beta) and abundances of taxa of the microbial signatures. To define sample size, we hypothesized a taxon proportion change from baseline ranges at least equal to 5%, using a non-parametric two-sample, two-sided T-test. In order to evaluate significant differences between means of groups of interest, a minimum sample size of each group was set as equal to 30. Mann-Whitney U statistics (test power = 95%) was used with a large effect size. It follows that each of the 18 groups of interest would include at least 30 individuals in order to achieve a power of 95%.
For comparison, groups of interest were determined by age (three classes),
gender (two classes), and viral load (three classes). Study end-point objectives
required a differential analysis to be applied between pairs of groups with
constraints on the average percentage of positive individuals. Assuming a
positivity frequency of 10% (at the time of study design) with a small effect
size, the minimum sample size for each of the 6 age-gender combinations was equal
to 663, as determined by applying a binomial test to each group (test power =
95%; significance level = 5%). Power analysis was conducted with R version
3.4.4 (package pwr https://cran.r-project.org/web/packages/pwr/index.html). As a
result, the minimum number of swabs to be collected would be 663
Within positives, the study required differential comparisons between high and low viral loads for each age-gender combination. Assuming an equal probability of the high and low viral load with a medium effect size, a minimum sample size of positive viral load within each of the 6 age and gender combinations was equal to 49, as computed by applying a binomial test to each group (test power = 95%). Thus, the minimum number of positive individuals with high and low viral load was 49. This minimum sample size was already warranted by previously assumed event probabilities with a confidence level equal to 95% in that a binomial test with 49 positives out of 663 individuals provided an estimated probability equal to 7.4% with a 95% CI = (5.5%, 9.6%), and thus rejecting the null hypothesis of a 10% positives in the population. This means that sampling 663 individuals in a population with 10% positives will guarantee more than 49 positive persons with a confidence at least equal to 95%.
The 16S double-strand target gene sequencing was performed on Illumina Miseq V3
at the Bio-Fab Research company. The sequenced amplicon relates to the V4–V5
regions (515F-Y: GTGYCAGCMGCCGCGGTAA; 926R-jed: CCGYCAATTYMTTTRAGTTT) and
obtained raw fastq data was properly filtered. In particular, reads with less
than 100 nucleotides (nt) were removed, and primer sequences were detected and
then clipped. Thus, sequenced data were imported into the next-generation
microbiome bioinformatics platform Qiime2 [19] in a genomic cloud-computing
environment based on Refs. [20, 21]. Data were first denoised, dereplicated, and
filtered by both phiX reads and chimera (consensus) by using the q2-dada2 quality
control method [22] for detecting and correcting (wherever possible) Illumina
amplicon sequence data. Sequence variants (SVs) were obtained by considering the
specific sequence error profiles truncating bases at the first instance of a
quality score less than, or equal to, 2 in the reads. Strands with a number of
incorrect bases larger than 2% were discarded, and only reads with a minimum
overlap of 20 nt were retained and joined. SVs were assigned taxonomy using a
Naive Bayes classifier model trained on the Silva138 99% database trimmed to the
V4–V5 region of the 16S. A phylogenetic tree was constructed via sequence
alignment with Multiple Alignment using Fast Fourier Transform (MAFFT) [23] by filtering the alignment and applying FastTree [24]
to generate the tree. Rarefaction curves of the Shannon index emphasize a good
sequencing quality as the richness index does not increase significantly with the
sampling depth for each sample. Thus, the rarefaction strategy (to 10
The alpha-diversity of each sample was assessed using Shannon and Chao1 indexes. Corresponding statistical significances in sample group comparison were determined using a Kruskal-Wallis test [26]. Also, the between-samples beta-diversity was assessed based on the 16S rRNA, and estimates were calculated on the SVs using Jaccard and Bray-Curtis distances [27]. Permutational multivariate analysis of variance (MANOVA) [28] with 999 permutations was used to test significant differences between sample groups based on both Jaccard and Bray-Curtis distance matrices. 16S SVs were also collapsed into Phylum, Class, Order, Family, Genus, and Species taxonomic levels, and their association with the group of interest was evaluated by using LEfSe (Linear discriminant analysis Effect Size) [29]. LEfSe was applied with default alpha values for the Anova and Wilcoxon test (0.05), and the Linear Discriminant Analysis (LDA) effect size has been evaluated by setting the absolute value of the logarithmic LDA threshold equal to 3.5. Other LEfSe parameters have been set to the default.
SCFA was measured in nasopharyngeal swab solution, inactivated with guanidinium. A 300 µL volume was transferred into a 10 mL screw cap vial (Agilent Technologies, Santa Clara, CA, USA) containing 130 mg of NaCl and 5 µL of deuterated internal standard solution (an aqueous solution containing d3 acetic acid, d5 propanoic acid, d7 butanoic acid and d9 pentanoic acid); the pH of the solution was adjusted to 3.5 with 10% perchloric acid (10 µL). The solution was placed at 65 °C for 5 min, keeping the mixture under magnetic stirring (500 rpm); head space-Solid Phase MicroExtraction (HS-SPME), using a PDMS/CAR/DVB 2 cm fiber (Supelco, Sigma Aldrich Merck), was employed to extract SCFA (30 min fiber exposure at 65 °C, under magnetic stirrer agitation) and transfer the analytes in the gas chromatography (GC) injection port. Before each analysis, the fiber was conditioned in the GC injector at 260 °C for 15 min.
Samples were analyzed by gas chromatography-mass spectrometry (GC-MS) using a 6890 GC and a 5973 A MSD (Agilent Technologies, Santa Clara, CA, USA) with an electron ionization (EI) source. A Zebron ZB-waxplus GC column (Phenomenex) was used. Splitless injection (3 min valve off) was performed, and data were acquired in SIM mode. The quadrupole analyzer was calibrated before each analytical session.
For SCFA quantitation, a linear calibration curve was recorded in a blank
nasopharyngeal swab solution inactivated with guanidinium. Using six 300
µL aliquots, scalar amounts of the chemical standard aqueous
solution of SCFA and 5 µL of deuterated internal standard solution
were added and then processed as for the actual samples. The concentration range
was the following, expressed as ng/µL concentration in the
nasopharyngeal swab solution: acetic acid 4–250, propionic acid 0.1–50,
isobutanoic acid 0.08–15, butanoic, isopentanoic, and pentanoic acid 0.01–5,
hexanoic acid 0.25–5. The peak area ratio (PAR) for the specific ions of each
analyte and its corresponding deuterated IS was measured to construct the
calibration curve for each short-chain acid and calculate the sample
concentration. For isobutanoic acid, d7 butanoic acid was used as internal
standard and d9 pentanoic acid for isopentanoic and hexanoic acids. To a 300
µL volume of blank nasopharyngeal swab solution inactivated with
guanidinium, the 5 µL volume of internal standard solution was only
added to check if it was contaminated with SCFA; the measure was done in
triplicate. The presence of acetic acid was detected, and its concentration was
calculated by the standard addition method; it was estimated as being equal to
10.8
The residual nasopharyngeal swabs after SARS-CoV-2 testing were used for DNA extraction and 16S rRNA sequencing. The taxonomic composition of the nasopharyngeal microbiome revealed that Firmicutes, Bacteroidota, Proteobacteria, Actinobacteriota, and Fusobacteriota were the most abundant phyla (Fig. 1), while Prevotella, Streptococcus, and Veillonella dominated at the genus level (Supplementary Fig. 1), in line with previous studies [18, 30].
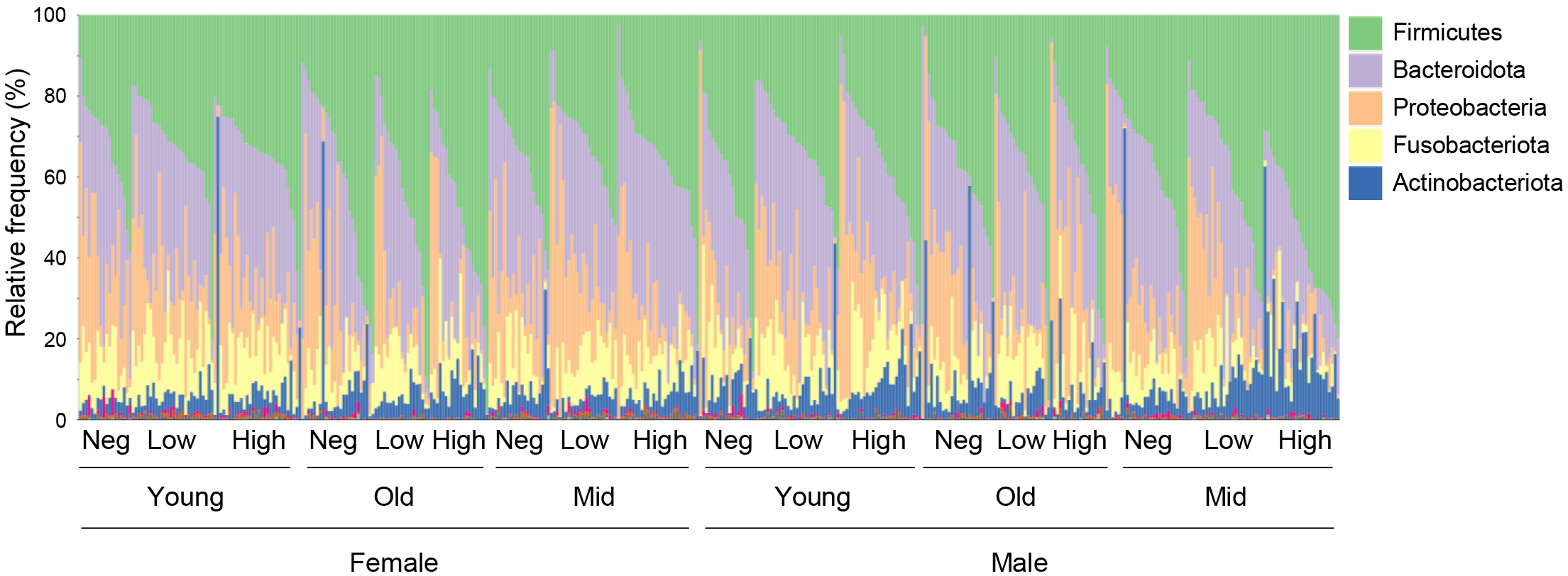 Fig. 1.
Fig. 1.Taxonomic composition of nasopharyngeal microbiome. Bar plot showing bacterial composition (abundance percentage) of each sample at the phylum level. Taxa are differentiated by colors. Samples are ranked based on the most abundant phylum (Firmicutes). Neg, Negative.
To identify the individual factors that influence microbiome composition in the context of SARS-CoV-2 infection, individuals were stratified according to age, gender, and SARS-CoV-2 diagnostic test results. In the case of positivity, samples were further stratified according to the viral load, as defined in the Materials and Methods section. The analysis of alpha diversity revealed distinct effects of age, gender, and viral load on both richness and evenness of genera, as measured by Observed Operational Taxonomic Units (OTUs) and Shannon indexes, respectively (Fig. 2).
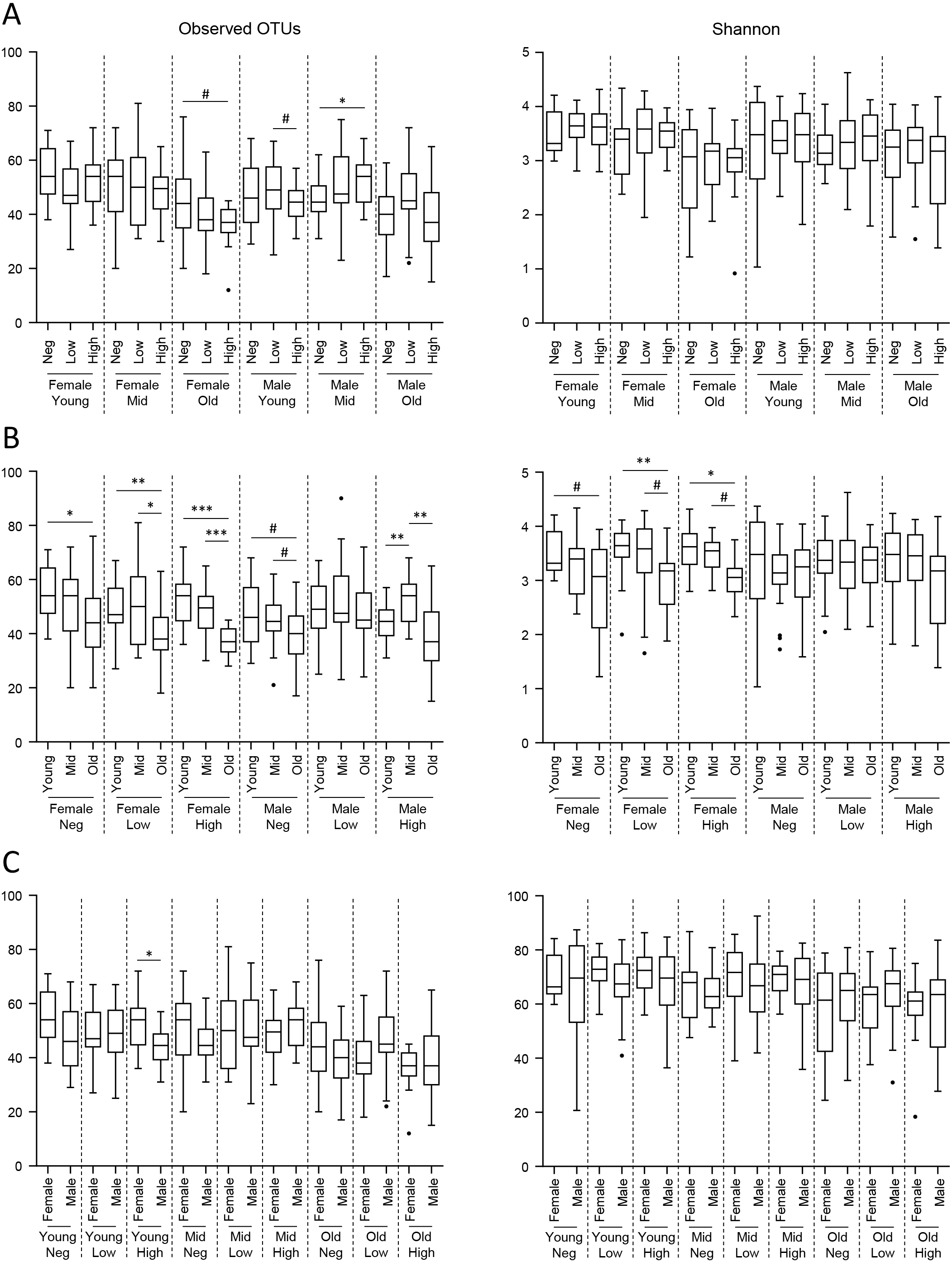 Fig. 2.
Fig. 2.Alpha diversity analysis of nasopharyngeal microbiome. Boxplots
of observed Operational Taxonomic Units (OTUs) (left panels) and Shannon (right panels) alpha diversity
indexes. Significance was evaluated by applying a pairwise Kruskal-Wallis test to
the 18 groups. Significant comparisons of interest have been explicitly
emphasized upon grouping by either viral load (A), age (B), or gender (C). #
p
More specifically, when the effect of age was considered, a decrease in the richness of genera was observed in older individuals across almost all groups, while a parallel decrease in evenness was apparently restricted to females (Fig. 2B). The effect of the SARS-CoV-2 test result (Fig. 2A) and gender (Fig. 2C) was less evident without any apparent pattern among the different groups. Age (Supplementary Fig. 2), gender (Supplementary Fig. 3), and viral load (Fig. 3) also influenced the compositional structure of the nasopharyngeal microbiome, as revealed by the Jaccard and Bray-Curtis indexes.
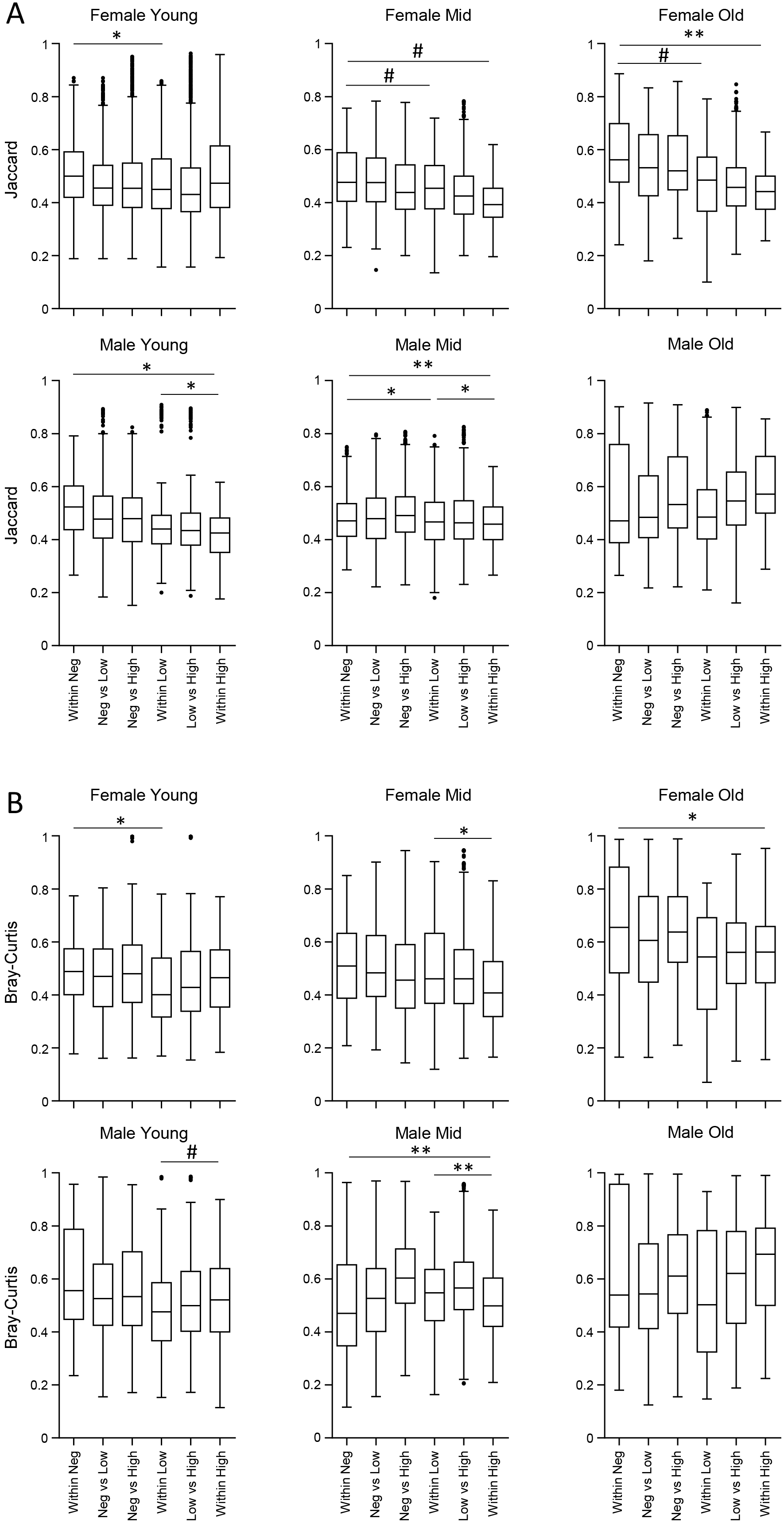 Fig. 3.
Fig. 3.Beta diversity analysis of nasopharyngeal microbiome.
Boxplots of Jaccard (A) and Bray-Curtis (B) beta diversity indexes evaluating
sample distances within or between viral load groups for each combination of age
class and gender. Significance among within-distances was evaluated by applying a
pairwise PermAnova test to the 18 groups. # p
As for alpha diversity, the compositional structure at the genus levels was mostly affected by SARS-CoV-2 viral load across all groups (Fig. 3), followed by age (Supplementary Fig. 2). The effect of gender appeared to be restricted to the high viral load across all age groups (Supplementary Fig. 3).
With regards to SARS-CoV-2 viral load, it should be noted that, in each combination of age class and gender, with the exception of the elderly male, the Jaccard and Bray-Curtis indexes are not equal for at least one viral load comparison. It follows that each population is characterized by a microbial composition capable of identifying differences among viral loads, thus justifying an associative analysis of microbial taxa to each population. It is worth mentioning, however, that the absence of significant differences in beta indexes, as observed in elderly males, does not imply the absence of significant associations and, therefore, the presence of a specific microbial composition in the same way as the other age class-gender groups.
In order to evaluate which variables contribute most and significantly to the
structure of the microbial community as an indicator of viral load, the Adonis
test was applied to the Jaccard and Bray-Curtis diversities using a
multifactorial model that also considers the interactions between gender, age,
and viral load in a sequential manner. All the variables were found to interact
significantly with each other for both metrics (Table 1) since the
p-value of the F-statistic (Pr(
All in all, the sequencing data allows us to identify a microbial compositional signature at the genus level that distinguishes the viral load in each combination of age class and gender.
| Jaccard | ||||
| Df | F.Model | R |
Pr( | |
| Sex | 1 | 2.080936 | 0.004575 | 0.015 |
| AgeClass | 2 | 7.572547 | 0.033297 | 0.001 |
| ViralLoad | 2 | 3.113238 | 0.013689 | 0.001 |
| Sex:AgeClass | 2 | 1.666735 | 0.007329 | 0.021 |
| Sex:ViralLoad | 2 | 1.607068 | 0.007066 | 0.019 |
| AgeClass:ViralLoad | 4 | 1.460523 | 0.012844 | 0.013 |
| Sex:AgeClass:ViralLoad | 4 | 1.501371 | 0.013203 | 0.008 |
| Residuals | 413 | NA | 0.907996 | NA |
| Total | 430 | NA | 1 | NA |
| BrayCurtis | ||||
| Df | F.Model | R |
Pr( | |
| Sex | 1 | 2.658643 | 0.005846 | 0.007 |
| AgeClass | 2 | 3.631918 | 0.015971 | 0.001 |
| ViralLoad | 2 | 4.115279 | 0.018097 | 0.001 |
| Sex:AgeClass | 2 | 2.480457 | 0.010908 | 0.001 |
| Sex:ViralLoad | 2 | 1.830204 | 0.008048 | 0.021 |
| AgeClass:ViralLoad | 4 | 1.761119 | 0.015489 | 0.005 |
| Sex:AgeClass:ViralLoad | 4 | 1.995595 | 0.017551 | 0.001 |
| Residuals | 413 | NA | 0.908089 | NA |
| Total | 430 | NA | 1 | NA |
Df, degrees of freedom; R
The Principal Coordinate Analysis (PCoA) plots of the beta indexes shown in Fig. 4 help in visualizing the Adonis results. The figure shows the distribution of the PCoA coordinates of the BrayCurtis and Jaccard distances evaluated over the 18 groups and organized for each of the six populations given by the combination of gender and age classes. Coordinates of each sample (dots) of the population considered and grouped by degree of infectious load, together with their distance (dashed lines) from the corresponding centroid (diamond), allow us to visualize which of the main two components explains, on average, differences in infectious load within each population. The axis labels also report the significances among viral loads explained by the corresponding PCoA component (the corresponding pairwise comparisons are shown in Supplementary Table 1). This analysis highlights that, for each of the six populations, at least one metric has a component that significantly distinguishes viral load, thus confirming that age and gender modulate, on average, microbial composition for both genera types and proportions.
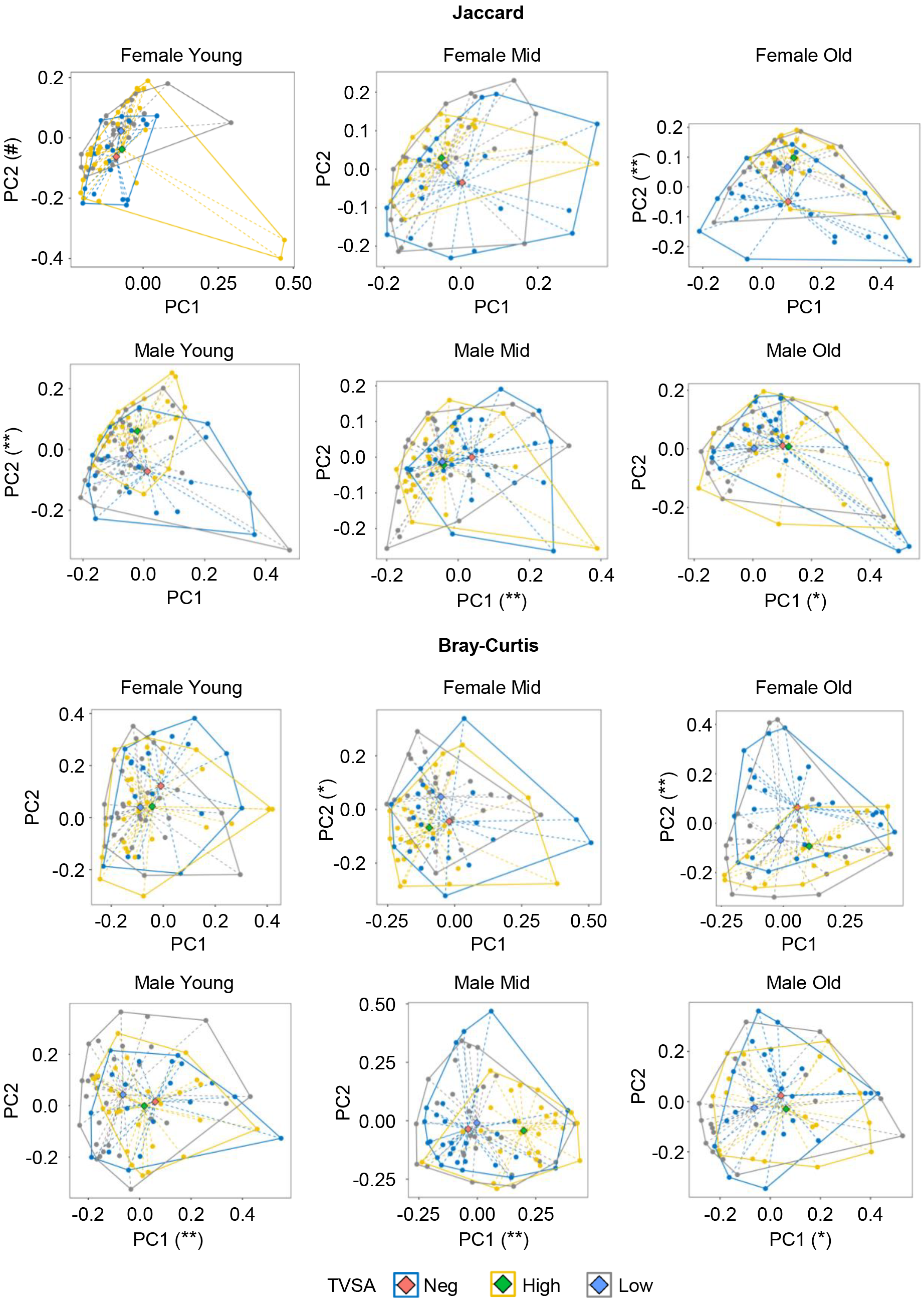 Fig. 4.
Fig. 4.Principal Coordinate Analysis (PCoA) of Jaccard and BrayCurtis diversities of
nasopharyngeal microbiome. Scatterplots (dots) of the first two PCoA components
of each beta metric are depicted in each gender and age class. Colors in each
subfigure refer to viral load groups, and the diamond represents its centroid,
according to the color legend. Dashed lines connect the centroid to each sample
of the corresponding group, while continuous lines contour the area of each
group. Each axis shows the significance of the corresponding component among
viral load groups given by the Kruskal Wallis test thus assessing that the
component can be used for evaluating a microbial signature for the viral load in
the population. #: p
Collectively, these results indicate that age, gender, and SARS-CoV-2 test result variably affect nasopharyngeal microbial composition arguing for the identification of individual microbial signatures that could predict the risk of infection.
To identify individual microbial signatures associated with SARS-CoV-2 infection, we performed high-dimensional class comparisons using linear discriminant analysis of effect size (LEfSe) [29] at the genus level. Because significant differences as a function of SARS-CoV-2 test results were detected in females across all age groups, we focused our analysis on females only. On applying the LEfSe differential abundance test, we could identify a common signature in females across all age groups (Fig. 5 and Supplementary Fig. 4). Specifically, we observed that patients positive for SARS-CoV-2 infection—and irrespective of their viral load—had a relative predominance of Lachnospirales-Lachnospiraceae over negative individuals.
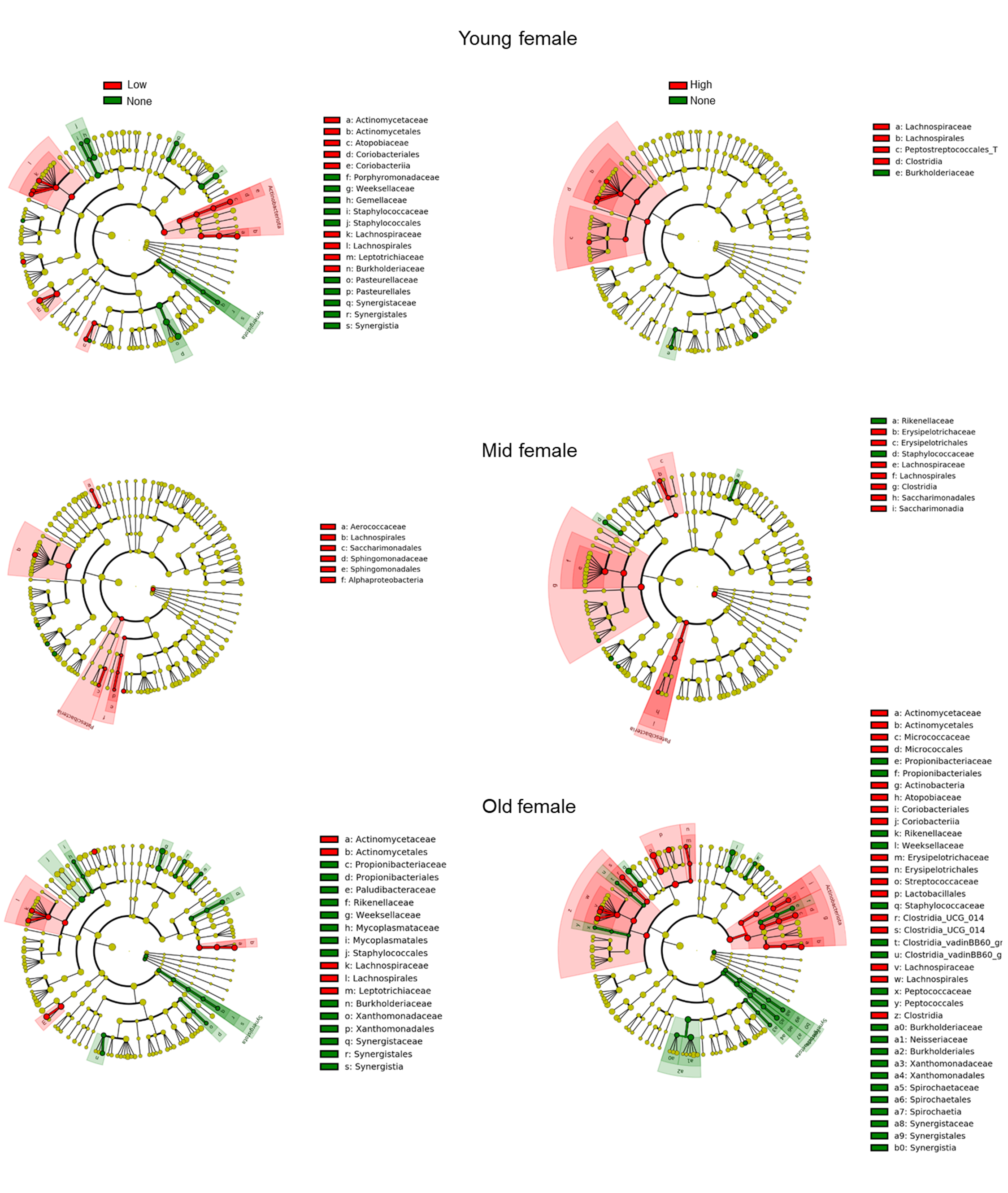 Fig. 5.
Fig. 5.Differential abundance analysis between age groups in females.
Taxonomic visualization of statistically and biologically consistent differences
between age groups in females. The cladogram simultaneously highlights high-level
taxa and specific genera. Taxa (circles) are colored red when significantly
(Linear discriminant analysis Effect Size (LEfSe), p
Collectively, these results suggest that the expansion of Lachnospirales-Lachnospiraceae may represent a microbial signature associated with SARS-CoV-2 infection in females and the consistent presence across all age groups—each characterized by a different microbiome composition—may indicate a causative role in the process.
Lachnospiraceae have been implicated in the production of metabolites able to affect human health and disease, such as short-chain fatty acids (SCFAs) [31], although with differences at the strain level [32]. We therefore performed targeted metabolomics for SCFAs in selected samples and compared their levels between positive (low and high viral loads) and negative females. While the most abundant SCFAs with shorter chains, such as acetic, propanoic, and butanoic acids, did not differ between the two groups, the branched SCFA isobutanoic acid, as well as the SCFAs with longer chains—i.e., valeric and hexanoic acids—were significantly reduced in the SARS-CoV-2-positive females (Fig. 6).
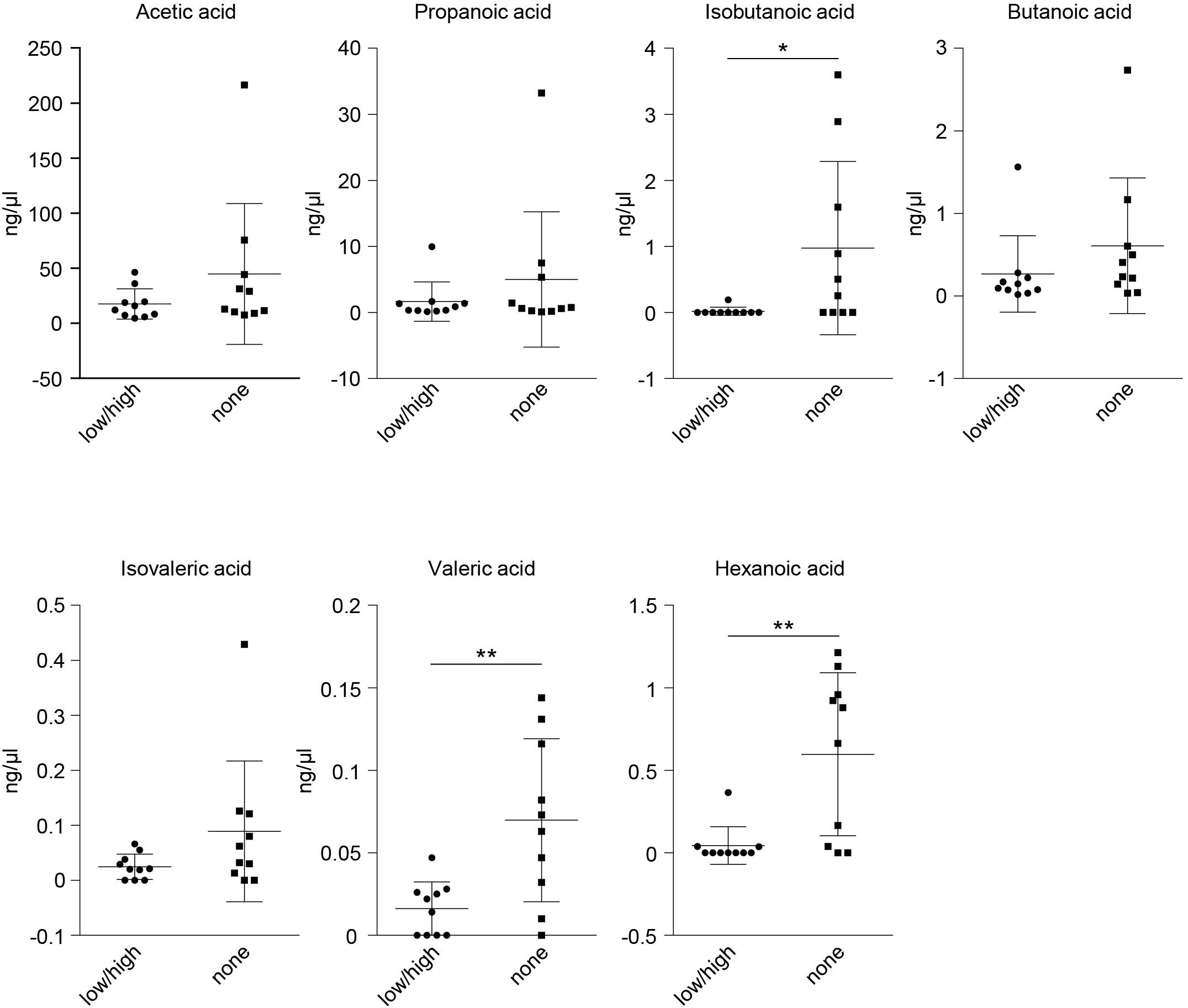 Fig. 6.
Fig. 6.SCFAs analysis in females stratified by test result. Acetic,
propanoic, isobutanoic, butanoic, isovaleric, valeric, and hexanoic acids were
measured in the nasopharyngeal swabs by gas chromatography-mass spectrometry.
Data are expressed as mean
Altogether, the targeted metabolomics analysis of SCFAs showed that the expansion of Lachnospirales-Lachnospiraceae in SARS-CoV-2-positive females is associated with a reduction in selected SCFAs, thus providing a combined compositional and functional microbial profile that characterizes females at infection.
The results presented in this study indicate that the nasopharyngeal microbiome is not a static entity but rather subject to variation as a result of demographic and microbiological parameters in the context of SARS-CoV-2 infection. Our analysis of individuals tested for SARS-CoV-2 infection and stratified into homogenous groups according to age, gender, and viral load revealed that those attributes represent important variables in the composition of the nasopharyngeal microbiome.
The impact of age was not unexpected. Several studies on the gut microbiota have consistently shown that the composition of microbial communities changes with age. In particular, aging is associated with a loss of dominant commensal taxa and expansion of pathobionts. In parallel, a second group of commensal microbes expands with age and is critical for healthy aging [33]. Age-related changes are obviously not restricted to the gut but rather affect the different body sites colonized by microbes, including the respiratory tract [34]. In the context of SARS-CoV-2 infection, it was shown that the composition of the nasopharyngeal microbiome varied as a function of age, as well as the presence or absence of respiratory symptoms, among children, adolescents and young adults less than 21 years-old [35]. In particular, the abundance of the commensal bacteria Corynebacterium and Dolosigranulum was associated with a reduced risk of respiratory symptoms [35]. Our adult cohort similarly showed an age-associated microbial composition among young, middle-aged, and old adults, with a decrease in alpha and beta diversity indexes across the different groups, although with exceptions. For instance, an age-dependent decrease in evenness was observed in females but not males, thus arguing for an important gender effect on microbial composition. Indeed, gender is known to affect diversity and species composition at several body sites, a phenomenon termed microgenderome or sex dimorphism in the microbiome [36]. However, a recent study on the nasopharyngeal microbiome of healthy subjects stratified in six groups, from 1–20 to over 70 year-old—and including 10 males and 10 females per group—did not reveal age- or sex-associated differences in alpha diversity indexes suggesting stability of bacterial diversity across the lifespan in a fashion independent of sex, although age- and sex-associated changes in the relative abundance of bacterial taxa could be identified [37].
Whereas the age range of the cohort and the criteria for stratification might explain the opposite results on the effect of age as compared to our study, we have similarly observed a lack of effect of gender on alpha and beta diversity indexes in the negative population. This would confirm that gender has a lower effect compared to age in determining the composition of the nasopharyngeal microbiome in healthy individuals. However, we observed a different compositional structure between males and females in the high viral load groups, indicating an interaction with SARS-CoV-2 infection. Interestingly, the gender-associated difference was observed in the low and middle-aged, but not older group, suggesting an intriguing hypothesis of female sex hormones influencing microbial communities in SARS-CoV-2 infection. The role of sex hormones in SARS-CoV-2 susceptibility is an interesting topic, also considering that, among others, male sex is a risk factor for severe disease and poor prognosis [38]. However, whether sex hormones are able to affect SARS-CoV-2 entry is still disputed [38], as are the potential effects on microbial composition, and further investigation is required to establish a potential role in the gender-specific response to COVID-19.
Despite the complex relationships between demographic and microbiological parameters and the composition of the nasopharyngeal microbiome, our results show that it is still possible to identify shared changes in response to SARS-CoV-2 infection. Indeed, we could identify an expansion of Lachnospirales-Lachnospiraceae in SARS-CoV-2 positive females across all age groups independently of viral load. This result is particularly important for many reasons. First, because of the expansion of Lachnospirales-Lachnospiraceae in the different groups—each characterized by a specific microbial composition—an important role for this taxon might be hypothesized in infection. In support of this hypothesis, the enrichment of the lung microbiota with gut-associated bacteria, such as Lachnospiraceae and Enterobacteriaceae, was predictive of poor ICU outcomes and the clinical diagnosis of acute respiratory distress syndrome [39]. Conversely, however, the presence of Lachnospiraceae in the nasopharyngeal swabs of COVID-19 patients was associated with a milder form of the disease, arguing for a protective role against progression to severe clinical pictures [40]. These contrasting results underlie the complexity of the role of Lachnospiraceae in health and disease [31] and argue for further characterization in specific clinical settings. Second, because Lachnospirales-Lachnospiraceae have been widely characterized within the gut microbiome for their metabolic profile, our data may provide clues as to the mechanisms underlying the initial phases of infection. For instance, Lachnospirales-Lachnospiraceae may produce metabolites that can affect human health and disease, such as SCFAs and aromatic amino acid metabolites. However, we did not observe an increase in major SCFAs in the nasopharyngeal swabs of positive patients. On the contrary, a reduction of selected SCFAs was observed. While this is in line with the role of SCFAs against bacterial and viral infections [41], it remains to be determined whether SCFAs in the nasopharynx are locally produced or reflect gut microbial metabolism via a gut-respiratory tract axis and, in both cases, how respiratory and gut Lachnospiraceae regulate their levels.
In conclusion, our results identify age, gender, and viral load-associated changes in the composition of the nasopharyngeal microbiome that, on the one hand, highlight the requirements for a proper selection of the cohort in microbiome association studies to avoid biases that could affect the interpretation of the results; on the other hand, our results may pave the way to the development of personalized medicine in COVID-19, as centered around shared changes in defined groups of individuals.
The datasets used during the current study are available from the corresponding author on reasonable request.
SB, AFM and AM were involved in the collection and processing of samples. EN and CC analyzed the data. GP, MP and GR performed the research. SB, EN, VNT, CA, PP, LR and CC designed the research study. SB, EN, LR and CC wrote the initial draft of the manuscript. All authors revised the manuscript critically. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This study was approved by the ethical committee (CER Umbria, n. 3990/21), with a waiver of informed consent.
Not applicable.
This research was funded by Fondazione Perugia, grant number 19645 (2020.0425) and HDM-FUN (European Union’s Horizon 2020 research and innovation program, number 847507).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.