

 , Chun-fen Mo 1,2,*
, Chun-fen Mo 1,2,*1 Department of Immunology, School of Basic Medical Sciences, Chengdu Medical College, 610500 Chengdu, Sichuan, China
2 Department of General Surgery, Second Affiliated Hospital of Chengdu Medical College (China National Nuclear Corporation 416 Hospital), 610083 Chengdu, Sichuan, China
3 Department of Gastroenterology, First Affiliated Hospital of Chengdu Medical College, 610500 Chengdu, Sichuan, China
†These authors contributed equally.
Abstract
Primary liver cancer is one of the most common malignant tumors with high mortality and increasing incidence worldwide. Currently, chemotherapy is an important comprehensive treatment for moderate or advanced liver cancer. Despite the effective therapeutic effects initially achieved by chemotherapy, the high phenotypic and molecular heterogeneity of liver cancer cells facilitates resistance to conventional chemotherapy or targeted therapy and even leads to multidrug resistance (MDR), which is one of the major obstacles for clinical chemotherapy. Drug resistance exhibits multiple and complex molecular mechanisms to antagonize therapy under pharmacological pressure, including overexpression of drug efflux transporters, downstream adaptive response (such as apoptosis, autophagy, and endoplasmic reticulum stress), dysfunction of DNA damage repair (DDR), epigenetic modification, tumor microenvironment (TME) as well as extracellular matrix (ECM). In this paper, we summarize the recent research progress and intervention strategies for drug resistance in hepatocellular carcinoma (HCC), which will provide a promising therapeutic strategy for overcoming MDR in liver cancer.
Keywords
- chemotherapy
- hepatocellular carcinoma
- multidrug resistance
- tumor microenvironment
- targeted therapy
- immunotherapy
The main pathological types of primary liver cancer are hepatocellular carcinoma (HCC), intrahepatic cholangiocarcinoma (ICC), and combined HCC-cholangiocarcinoma (cHCC-CC). Of these, HCC is the most common and fatal cancer, accounting for approximately 80%–85% of primary liver cancer cases [1]. The initiation and progression of HCC is a complex process involving various modifications to multiple molecular pathways, as well as altered gene expression [2]. Ethnic and regional differences also have a major impact on the incidence and mortality of HCC. Inadequate screening may result in the late diagnosis of HCC and hence poor prognosis [3].
HCC is an innately drug-resistant tumour and HCC patients are generally insensitive to chemotherapy drugs. They are prone to develop multidrug resistance (MDR) during chemotherapy, leading to reduced survival and poor prognosis [4, 5, 6]. The two main types of drug-resistance in cancer patients are caused by intrinsic factors and exogenous factors. Intrinsic resistance, also called primary resistance, is the consequence of genetic alterations present in tumour cells before treatment. This includes drug resistance attributed to cancer stem cells (CSCs) [7], and increased expression of drug efflux transporters that recognize and remove chemotherapeutic compounds [8]. In exogenous resistance, also known as acquired resistance, the cancer cells are initially sensitive but then develop drug resistance after a period of chemotherapy [8, 9]. Recently, the emergence of targeted therapy such as sorafenib has become an important treatment strategy for HCC. Although targeted drugs have led to significantly improved overall survival of HCC patients, most will eventually develop drug resistance through secondary mutations or via constitutive activation of bypass signal pathways. In the present review, we systematically summarize the underlying mechanisms and intervention strategies for drug resistance in liver cancer, and provide insights into possible future therapeutic targets for HCC.
Currently, the main drug therapies for liver cancer are chemotherapy, targeted therapy and immunotherapy. The conventional chemotherapeutic drugs used in the clinic are 5-fluorouracil (5-FU), cisplatin, and adriamycin (ADM). Sorafenib, lenvatinib, regorafenib, tivantinib and cabozantinib are the commonly used molecular targeted drugs. Immunotherapy for HCC is a relatively new management option that mainly involves immune checkpoint inhibitors/monoclonal antibodies against programmed cell death protein 1 (PD-1), PD-1 ligand (PD-L1) and receptor cytotoxic T lymphocyte antigen-4 (CTLA-4). These include drugs such as nivolumab, MED14736, pembrolizumab, tremelimumab and ipilimumab. However, acquisition of MDR to the above agents may cause therapeutic failure during HCC treatment.
Adriamycin/Doxorubicin (ADM) is a DNA topoisomerase II inhibitor from the
anthracycline family of anticancer drugs. The targets of ADM are human type IIA
topoisomerases (Top2
5-FU is a heterocyclic aromatic chemotherapeutic agent that is widely used for
HCC treatment. 5-FU targets thymidylate synthase (TS), leading to inhibition of
thymidine formation required for DNA synthesis and hence the inhibition of cell
proliferation [18]. In clinical practice for the treatment of gastrointestinal
cancers, 5-FU is often combined with oxaliplatin and calcium folinate as FOLFOX
therapy [19]. Clinical trial data showed that the median survival of HCC patients
after FOLFOX4 therapy was 8.2 months [19]. Another clinical trial using modified
FOLFOX6 (mFOLFOX6) showed that the median survival of patients with metastatic
colorectal cancer (mCRC) treated with this regimen was 8.7 months [20]. Combined
treatment with 400 mg per day of sorafenib and mFOLFOX6 was not effective [20].
However, drug resistance to 5-FU is readily achieved during the treatment of
liver cancer. Hong and colleagues found that GLIPR1 promoted the resistance of
HCC to 5-FU by activating the PI3K/PDK1/ROCK1 pathway [21]. In addition, Li
et al. [22] reported that ALCL3 attenuated 5-FU-induced apoptosis by
activating ERK signalling. On the other hand, ALCL3 triggered cell cycle arrest
through downregulation of p-Chk2
Cisplatin is the first metal-based anticancer drug used for liver cancer treatment [23]. This drug interferes with the DNA repair mechanism, thereby resulting in DNA damage and inducing apoptosis [24]. An objective response rate (ORR) of 73.2% was achieved using the newly developed technique of balloon-occluded, alternate infusion of cisplatin [25], which also proved to be a safe and effective method. However, the long-term use of cisplatin can predispose to complications such as deafness. A clinical trial showed that sodium thiosulfate given 6 hours after cisplatin chemotherapy could reduce the incidence of cisplatin-related hearing loss in children with hepatoblastoma without affecting their overall survival [26]. However, a previous study found that chemotherapeutic agents can induce the expression of a peptide (circMRPS35-168AA) that promotes cisplatin resistance in HCC [27], indicating that HCC patients are at risk of developing resistance. Moreover, downregulation of miR-33a-5p attenuated cisplatin sensitivity in MHCC97L and Hep3B cells, suggesting this miRNA may be a potential target for reversing resistance during HCC treatment [28].
Targeted drugs specifically target tumour cells, rather than killing cells indiscriminately. In addition, such drugs have less harmful side-effects on normal tissues. Compared to chemotherapy drugs, targeted drugs are therefore safer and have fewer side-effects. In addition, the targeted drug delivery system (TDDS) developed in recent years also helps to specifically transport targeted drugs to cancer sites, thus reducing toxicity while killing cancer cells at the same time. Targeted drugs therefore have the advantages of specificity, safety and fewer side-effects, as well as being suitable for combined treatment with other types of drugs. The resistance of targeted drugs that are commonly used in liver cancer are summarized below.
Sorafenib in an oral multi-kinase inhibitor that can facilitate apoptosis,
mitigate angiogenesis, and suppress tumour cell proliferation. It does this by
targeting BRAF, Raf-1, Flt3, VEGFR-2/3 and PDGFR-
Lenvatinib is an oral small-molecule inhibitor of multiple receptor tyrosine kinases (TKIs) and has been approved for first-line therapy of HCC. Compared to sorafenib, lenvatinib shows a higher remission rate and prolongs the survival of HCC patients with advanced stage disease. Long-term survival can be achieved when lenvatinib is combined with surgery [42]. Lenvatinib may also be an appropriate second-line therapy for unresectable HCC patients that express fibroblast growth factor receptor 4 (FGFR4) and are therefore resistant to sorafenib [43]. A phase II clinical study showed that 12 mg lenvatinib taken once daily is the recommended dose for HCC patients [44], with acceptable toxicity and all safety standards met. Another clinical trial in HCC patients suggested that lenvatinib may not have a survival advantage compared with sorafenib [45]. However, the time to progression (TTP) for patients treated with lenvatinib was much longer than with sorafenib, suggesting it is more suitable for HCC treatment when used in combination with other drugs [45]. Unfortunately, although there is a good initial response to lenvatinib in sorafenib-resistant HCC, the cells eventually develop resistance to this drug. Long non-coding RNAs are important regulators of lenvatinib resistance. Cao et al. [46] reported that LCC-ZEB2-19 inhibits HCC progression and attenuates resistance to lenvatinib. Furthermore, low expression of LCC-ZEB2-19 correlated with poor prognosis of HCC. In contrast, the long non-coding RNA AC026401.3 is upregulated in HCC tissues and was associated with poor prognosis. Knockdown or deletion of AC026401.3 enhanced the sensitivity of HCC cells to lenvatinib [47].
Regorafenib is a diphenylurea multi-kinase inhibitor that targets angiogenesis
(VEGFR1-3, TIE2), PDGFR-
Tivantinib (ARQ 197) is a novel mesenchymal-epithelial transition (MET) inhibitor with an ATP-independent binding mechanism that stabilizes the inactive conformation of MET receptor tyrosine kinase and disrupts ligand-mediated activation [52]. A clinical trial from Japan found that HCC patients treated with tivantinib had a median survival of 10.3 months, compared to 8.5 months for those taking placebo [53]. There was an study reported that Tivantinib had the effect of attenuating HCC resistance in vitro, but must be used in advance [54]. According to Kobayashi et al. [54], the pre-treatment of HepG2 cells with tivantinib can reduce the resistance of these cells to hepatocyte growth factor. Wu et al. [55] demonstrated that increased activity of ABCG2 caused by tivantinib can increase resistance to drugs such as mitoxantrone, thus leading to MDR. This observation may be related to the timing of tivantinib use.
Cabozantinib (XL184) is a potent, oral pan-tyrosine kinase inhibitor that inhibits VEGFR2, c-Met, Kit, Axl, and Flt3. This drug was approved by the U.S. FDA for the treatment of liver cancer in 2019 [56, 57]. It is used for patients with advanced liver cancer, often after they develop resistance to sorafenib [58]. In clinical trials, 7% of patients showed a partial response, 64% had stable or progressive disease, and 59% died during follow-up [58]. The median survival for the entire cohort after the start of cabozantinib treatment was 7.0 months. Of the patients enrolled in these trials, 92.0% had been treated with sorafenib, and 42% had undergone liver transplantation and liver resection. The incidence of side-effects with cabozantinib was only 17%, and was mainly diarrhea [58]. Therefore, canbozantinib shows good efficacy and high safety, and is an ideal drug for patients with mid- to advanced HCC. However, there are still reports of resistance to canbozantinib. Although the U.S. FDA has approved this drug for the treatment of thyroid cancer, some patients do not respond [59]. Starenki et al. [59] reported that increasing the concentration of cabozantinib in TT and MZ-CRC-1 cells can produce resistant cell lines with increased mitochondrial activity. Thus, cabozantinib combined with mitochondrial-targeting drugs could potentially inhibit medullary thyroid carcinoma.
Immune drugs are divided into immunosuppressive drugs and immune enhancing drugs. The latter can specifically kill or inactivate cancer cells by boosting host immunity, as well as some specialized killer cells. Immunotherapy has fewer side effects than chemotherapy, with most being reversible. Therefore, immunotherapy is usually combined with chemotherapy to increase its effectiveness.
When cancer patients start to receive chemotherapy, the tumour cells evade cytotoxicity using a variety of mechanisms. Immunotherapy is quite similar in this regard, although MDR has been less studied with this treatment approach. Some drugs against PD-1/PD-L1, CTLA-4 and VEGF-A are susceptible to develop resistance [60, 61, 62]. When patients develop resistance to immune drugs in the clinic, switching to another drug or drug combinations can be used to overcome the resistance [63]. Therefore, a major challenge with cancer immunotherapy is to reverse the resistance that develops with immune drugs, thereby prolonging patient survival.
The ATP-binding cassette (ABC) transporter is an ATP-dependent transmembrane protein that is widely involved in MDR and has a highly conserved sequence. ABC transporters are overexpressed in tumour cells and act as drug efflux pumps to induce MDR by reducing the intracellular concentration of anti-cancer drugs [64, 65, 66]. ABC transporter proteins have a typical four-domain structure that includes conserved cytoplasmic nucleotide-binding domains (NBDs) and highly heterogeneous transmembrane structural domains (TMDs) [67]. NBDs are mainly responsible for the hydrolysis of ATP, while TMDs are responsible for the recognition and transport of substrates [67]. Due to the high heterogeneity of TMDs, the ABC transporter family has many members and can recognize numerous different substrates, which ultimately leads to MDR in tumours. At present, there are 48 known members of the ABC transporter superfamily in the human genome. These are divided into 7 subfamilies, namely ABCA, ABCB, ABCC, ABCD, ABCE, ABCF and ABCG [68, 69]. Among them, ABCB1/MDR1 and BCRP/ABCG2 play a decisive role in HCC chemotherapy. The 7 subfamilies that mediate drug resistance in different tumour types are summarized in Table 1 [68, 70].
| Gene | Drug-resistant associated protein | Resistant drugs | Inhibitor |
| ABCB1 | P-gp | Erlotinib [88], adriamycin [89], sorafenib [90], cisplatin [91], fluorouracil [91], paclitaxel [92] | Elacridar [93], Verapamil [94, 95], ONT-093 (Ontogen) [96] |
| ABCC | MRP1, MRP2, MRP3, MRP4 | Sorafenib [97], Taxanes [98], adriamycin [99], cisplatin [100], etoposide [101], vincristine [102] | Ceefourin 1 [103], Ceefourin 2 [104], Indican [105], MK-571 [106], MK-571 sodium [106] |
| ABCG | ABCG1, ABCG2 (BCRP) | Regorafenib [107], adriamycin [108], paclitaxel [109], Tivantinib [54], cisplatin [110], sorafenib [111] | Elacridar [93], Verapamil [94, 95], Ko143 [112], Fumitremorgin C [113] |
P-glycoprotein (P-gp) is the most representative protein of ABC transporters and is referred to as multidrug resistance protein 1 due to its role in modulating MDR in tumour cells. P-gp protein is encoded by the MDR1/ABCB1 gene and has a molecular weight of 170 kDa. It is involved in the transport of anti-cancer drugs such as adriamycin, paclitaxel and 5-FU [5, 11, 16, 71, 72]. P-gp expression is upregulated in HCC and is associated with MDR. Zhao et al. [73] reported that P-gp is upregulated by NF-kB activation and the MAPK/ERK pathway-mediated translocation of Y-box binding protein 1 to the nucleus. Moreover, overexpression of SIRT1 contributes to MDR in HepG2 cells through increased expression of MDR1/P-gp in a process that can be reversed by the natural naphthoquinone compound shikonin [74]. Since P-gp plays an important role in HCC MDR, an increasing number of studies have focused on P-gp inhibitors. Xu et al. [75] found that gambogenic acid (GNA) could reduce P-gp expression by inhibiting the NF-kB and MAPK pathways, suggesting it could be used to inhibit P-gp in MDR. Zhao et al. [76] reported that downregulation of Snail Family Transcriptional Repressor 2 (SNAI2) increased the transcription of ABCB1 and ABCG2 genes in HCC cells, which was followed by the development of MDR. This study implies that SNAI2 is a negative regulatory factor for ABCB1/ABCG2 and MDR in HCC cells. It has also been reported that astragaloside IV (ASIV) may be used to reverse P-gp-mediated MDR [5].
Human breast cancer resistant protein (BCRP/ABCG2) is the second member of the G subfamily of the large ABC transporter superfamily. BCRP was initially discovered in MDR breast cancer cell lines, where it confers resistance to chemotherapeutic agents. BCRP/ABCG2 accelerates the efflux of sorafenib from cells, indicating that BCRP/ABCG2 expression may be a predictor of sensitivity to sorafenib in HCC [15]. Similar to ABCB1, BCRP/ABCG2 is also involved in ADM efflux, thereby facilitating the resistance of HCC cells to ADM [77]. Downstream EGFR signalling by Akt may regulate both the protein expression and membrane distribution of BCRP/ABCG2, thereby affecting its efflux ability. Of note, EGFR has been reported to determine the sensitivity of HCC cells to sorafenib treatment [78].
P-gp and ABCG2 are thought to be the two most important determinants of MDR in response to chemotherapy in HCC. For example, focus transferase (FUT) 4-, FUT6- and FUT8-mediated MDR in drug-resistant BEL7402/5-FU cells is associated with activation of the PI3K/Akt pathway and the expression of MRP1, but not of P-gp [79]. SLAMF3 is a tumour suppressor receptor in HCC, and overexpression of SLAMF3 in Huh-7 cells specifically induces MRP1 dysfunction [80]. The deubiquitinating enzyme ubiquitin-specific protease 22 (USP22) is a marker for CSCs, and also causes MDR in HCC by activating the SIRT1/AKT/MRP1 pathway [81]. Antigen-processing associated transporter 1 (TAP1) is another member of the ABC transporter family. TAP1 and P-gp show high homology in their transmembrane domain responsible for substrate specificity. TAP1 mRNA is highly expressed in breast, lung, liver and ovarian cancers, and TAP1-induced MDR has also been found in gastric cancer cells [82]. Further research is likely to reveal more ABC transporters that participate in MDR mechanisms in tumours. Research into the different subtypes of ABC proteins involved in drug resistance and their relevant inhibitors should provide additional options for reversing MDR in the clinical setting of HCC.
Multidrug resistance-associated proteins (MRPs/ABCC) also form a complex membrane protein system. The four main members of the MRP family are highly correlated with tumour resistance, namely MRP1/ABCC1, MRP2/ABCC2, MRP3/ABCC3, and MRP4/ABCC4. These proteins are structurally similar, and their overexpression can confer resistance to multiple drugs [83]. MRP1, MRP2 and MRP3 are all involved in the MDR of HCC. Nies et al. [84] reported that MRP2 and MRP3 were localized on the plasma membrane of these cancer cells, whereas MRP1 was only expressed on some HCC cell membranes. Therefore, MRP2 and MRP3 may be more suitable for the prediction of MDR in HCC [84]. As with the ABC transport family, increased expression of the MRP family also contributes to the development of MDR in HCC. Gu et al. [85] reported that bufalin can significantly block the cell cycle in BEL-7402/5-FU cells, while also reducing the expression of TS by down-regulating MRP1 to inhibit drug effector pump activity. TS is an MDR-related gene, and reducing its expression can reverse the resistance of BEL-7402/5-FU cells. Qu et al. [86] reported that ASIV enhances the anti-tumour effect of cisplatin in liver cancer by inhibiting the expression of MRP2. Furthermore, Tomonari et al. [87] showed that siRNA knockdown of MRP3 in the resistant cell line PLC-PRF5-R2 restored its sensitivity to sorafenib.
Not all ABC transporters are associated with HCC. So we summarized Table 1 (Ref. [54, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113]). The substrates and inhibitors of ABC transporters associated with HCC were summarized in Table 1.
The ultimate goal of anticancer therapy is to induce apoptosis of cancer cells [114]. Dysregulation of cell cycle checkpoints and of apoptotic signals are the main causes of MDR [114]. Experimental studies have discovered a variety of apoptosis signalling molecules that are closely related to MDR in liver cancer, including p53 and the Bcl-2 family of proteins.
The p53 tumour suppressor gene is a well-known factor that regulates apoptosis in a wide variety of cells and tissues. The main function of p53 is to induce apoptosis in response to DNA damage. Zhang et al. [115] reported that p53 suppressed the growth of MDR HepG2 cells by increasing the expression of Bax and decreasing the expression of Bcl-2. p53 also induced G2/M arrest through p21-mediated inhibition of the CDK1/cyclin B complex. In addition, the Nogo-B receptor (NgBR) activates the PI3K/Akt/MDM2 pathway, which then promotes p53 protein degradation via the ubiquitin proteasome pathway leading to chemoresistance in HCC cells [116]. Overexpressed p53 mutants can inhibit apoptosis and reverse the anticancer effects of chemotherapeutic agents, including ADM and cisplatin, resulting in MDR in cancer cells [117].
The Bcl-2 family is also involved in MDR of HCC cells. Bcl-2 family proteins play a critical role in the mitochondria-mediated apoptosis pathway. These proteins share similar domains and can be classified into three main groups: anti-apoptotic proteins (Bcl-2, MCL-1, BCL-XL), pro-apoptotic proteins (Bax, BAK), and regulatory proteins (BH3 proteins) [118]. Bcl-2 overexpression contributes to the resistance to anticancer drugs [61]. Hung et al. [119] reported that hepatitis B virus pre-S2 large mutant surface antigen (HBV pre-S2D) promotes resistance to 5-FU-induced cell death via increased expression of Bcl-2.
DNA damage can be triggered by Ultraviolet (UV) light or chemicals and is the first step in cancer development. Although cells can perform DNA repair, the probability of gene mutation increases with more frequent repair, thereby increasing the risk of cancer.
Many conventional cancer chemotherapeutics induce DNA double-strand breaks (DSBs). The pathways involved in DNA repair can create barriers to the treatment of cancer and lead to drug resistance [120]. Dysregulation of DNA damage repair and cell cycle checkpoints, known as the DNA damage response (DDR), is associated with predisposition to cancer and with chemoresistance to DNA-damaging anticancer therapy. However, the DDR can also protect against genomic instability and may enable cancer cells to become resistant to chemotherapy drugs by enhancing DNA repair [121].
Therefore, the amplification of drug-induced DNA damage may be an effective
approach for overcoming MDR in cancers. For example, bleomycin (BLM) boosts
14-3-3
Many biological and pathological mechanisms have been implicated in chemoresistance. In addition to the dysregulation of anti-apoptotic proteins and aberrations in DDR, accumulating evidence indicates that epigenetic alterations are essential for the development of chemoresistance in cancer cells [125]. DNA methylation in the promoter of tumour suppressor genes leads to transcriptional inactivation, which then accelerates tumour development and malignant progression. Epigenetic changes are usually reversible and are also susceptible to external factors. Thus, the targeting of epigenetic changes may be used as monotherapy or in combination with other anticancer drugs for the treatment of MDR cancers [117]. Depletion of ADAMTSL5 attenuated the tumour-like properties of HCC cells and sensitized them to drugs currently used in the clinic to treat HCC, including crizotinib, lenvatinib, regorafenib, and sorafenib [126]. Moreover, the CDH1 gene promoter is hypomethylated in HepG2 cells, but hypermethylated in the ADM-induced MDR HCC cell line R-HepG2 [127]. Because CpG methylation in the promoter usually correlates with gene silencing, evaluation of the CDH1 gene promoter methylation status may be predictive of MDR in HCC patients. Indeed, overexpression of CDH1 resulted in decreased P-gp expression and increased ADM uptake in R-HepG2 cells, accompanied by sensitization to ADM [127] . Epigenetic changes are especially important for the emergence of plasticity in many tumour-initiating cell subpopulations. This suggests that targeting of epigenetic changes may be a key intervention against HCC drug resistance.
Current studies indicate that autophagy is a double-edged sword in cancer cells. Basal autophagy acts as a tumour suppressor by maintaining genomic stability in normal cells. However, once cancer is established, activated autophagy contributes to the survival of cancer cells under a variety of stresses, thus promoting tumour development. Autophagy is also considered to be an important mechanism of drug resistance by promoting the survival of tumour cells under therapeutic stress [128]. Hence, the suppression of autophagy could sensitize HCC cells to chemotherapeutic drugs [129]. Consistent with this, Yuan et al. [130] found that both MALAT1-specific siRNA and miR-216b mimics were able to reduce the IC50 of 5-FU, ADM and mitomycin C (MMC) in BEL-7402/5-FU cells, as well as the level of intracellular autophagy. This suggests the MALAT1-miR-216b axis affects MDR in HCC cells by modulating autophagy. Current understanding of the role of autophagy in cancer progression and in the response to therapy remain controversial. Therefore, further studies are needed to determine how cellular autophagy can impact chemotherapeutic sensitivity during HCC treatment.
The TME is comprised of diverse types of normal cells in addition to cancer cells, including fibroblasts, blood vessel cells, mesenchymal stem cells, and immune cells of myeloid or lymphoid origin. These cells regulate tumour growth, metastatic spread, and response to treatment [131]. Accumulating evidence indicates a critical role for TME factors in both the response and resistance to various anticancer drugs. The HCC TME is recognized as a key contributor to cancer progression and drug resistance. Due to the tissue specificity of the liver, long-term exposure to a high concentration of 27-hydroxycholesterol (27HC) is a special characteristic of the TME in HCC and has been shown to induce MDR in HepG2 cells [132]. Further study showed that 27HC induced MDR via the activation of GRP75 to modulate redox balance and cause metabolic reprogramming of HepG2 cells [133].
The extracellular matrix (ECM) has long been associated with drug resistance and is a key component of the TME. ECM proteins are secreted by blood endothelial cells, lymphatic endothelial cells, mesenchymal cells and immune cells [134]. Stiffening of the ECM is a crucial indicator of a local change in the microenvironment and is considered a hallmark of many diseases, including HCC. Aggregation of protein-encapsulated hyaluronic acid (HA) gel-like structures can prevent drug uptake and delivery to intra-tumour areas, thereby altering the TME [135]. The stiffness of ECM promotes the formation of CSC ecological niches, a hypoxic environment within the TME, and increased vascular infiltration, all of which cause drug resistance [135, 136]. You et al. [137] showed that increased ECM stiffness activates the Akt/mTOR/SOX2 pathway and subsequently enhances oxaliplatin resistance in Hep3B and Huh7 cells. The fate of cancer cells and immune cells can be regulated by mechanical feedback from the ECM. This could be used in future studies of tumour drug resistance and in vitro drug screening applications.
Similar to the ABC transporter family, the solute carrier (SLC) transporter family are membrane transporters. However, SLC does not rely on ATP hydrolysis to transport small molecules. SLCs include more than 400 different transporters that are grouped into 65 families based on their sequence homology and transport function [138, 139]. The SLC family is classified according to the Hugo Gene Nomenclature Committee of the Human Genome Organization (HGNC) [140]. SLC structures are grouped into four categories based on their protein folds, which include MFS (major facilitator superfamily) fold, LeuT fold, other anti-parallel folds, and others [139]. The SLC family is responsible for the transport of inorganic ions, amino acids, lipids, sugars, neurotransmitters and drugs [139]. When SLCs are absent, normal cells can transform into drug-resistant cells. Most SLC proteins responsible for drug uptake belong to the SLC22A family, including organic anion transporters (OATs), organic cation transporters (OCTs), and organic anion transporting polypeptides (OATPs) from the SLCO family [141, 142]. Moreover, some members of the SLC family can affect the invasion and migration of cancer cells. For example, Fang et al. [143] showed that knockdown of SCL2A1 or SLC22A15 significantly reduced the migration and invasion ability of Hep3B cells. Experiments by Zhao et al. [144] indicated that the IC50 of sorafenib was reduced in resistant cells. This suggests that overexpression of SLC46A3 results in more uptake of sorafenib, thereby improving the clinical prognosis of HCC patients, reducing sorafenib resistance, and enhancing drug response [144]. Herraez et al. [145] reported the MRP2 promoter can overcome chemotherapy resistance in gastrointestinal and liver tumours by increasing the expression of the drug transporter OATP1B1. Moreover, liver cancer can be divided into a high glycolytic type and a low glycolytic type [146]. High glycolytic HCC has poor prognosis and is prone to drug resistance [146]. Patients with low glycolysis showed better clinical outcomes than patients with low oxygen and high glycolysis [146]. Kim et al. [146] showed that SLC13A5 is highly expressed in HCC with low glycolysis, which may be more evidence that overexpressed SLC protein can attenuate MDR in HCC.
CSCs are a small subset of the cancer cell population and are thought to play an important role in cancer cell self-renewal, metastasis, and response to therapy [147]. They are inherently drug-resistant, which is also a hallmark feature [148, 149]. CSCs show resistance to a variety of chemotherapeutic and targeted drugs, including cisplatin and sorafenib [150]. They are inherently resistant to chemotherapy and radiation [151] and often develop drug resistance during dormancy, thus making effective cancer treatment a major challenge [152]. Touil et al. [153] reported that dormant CSCs can express a phenotype similar to that of 5-FU-resistant cancer cells. Such a phenotype includes markers of self-renewal and tumour dissemination potential [153]. These authors also observed that some CSCs become dormant when exposed to chemotherapeutic drugs [153]. Therefore, decreasing cancer cell stemness can to some extent reverse the development of drug resistance in cancer cells. Nanog is an important transcription factor that regulates cell pluripotency and can also induce CSC properties [150]. The stem cell properties induced by Nango can lead to resistance to regorfenib and cisplatin. Alemohammad et al. [150] reported that siRNA knockdown of Nanog in HepG2 cells, in combination with cisplatin treatment, downregulated the expression of stem cell-related genes and upregulated the expression of apoptosis-related genes. Therefore, the property of cancer cell stemness can promote drug resistance in cancer cells.
EMT is a developmentally-related, multistep molecular and cellular reprogramming process that is hijacked by cancer cells to acquire aggressiveness. Activated EMT inhibits apoptosis. An increasing number of studies have also shown that activation of EMT can promote drug resistance in cancer cells [154, 155]. Strong correlations have been reported between EMT-related gene expression profiles and treatment resistance [155]. Liao et al. [156] found that Y-Box Binding Protein-1 can promote EMT in sorafenib-resistant HCC. Moreover, sorafenib can promote YB-1 phosphorylation through the EGFR/PI3K/AKT pathway, leading to a significant increase in the metastasis of HCC cells [156]. Cyclin B1 promotes cell growth and inhibits apoptosis in tumor necrosis factor (TNF)-related, apoptosis-inducing and ligand-resistant HCC cells [157]. Lv et al. [157] also reported that mesenchymal epithelial transition factor can promote TNF-associated apoptosis and induce the growth of drug-resistant HCC cells by regulating cyclin B1. The mechanisms by which EMT cause chemoresistance have been described. RHOJ is a small GTPase that is preferentially expressed in EMT cancer cells. Debaugnies et al. [158] showed that RHOJ modulates EMT-associated chemoresistance by enhancing the response to replication stress and activating the DDR, thereby allowing tumour cells to rapidly repair chemotherapy-induced DNA damage.
Owing to the multifactorial nature of MDR, the blocking of one mechanism is insufficient to overcome resistance. Hence, other drug-resistance mechanisms should be considered to prevent therapeutic failure in HCC patients. The different pathways and mechanisms relating to MDR in HCC are summarized in Fig. 1.
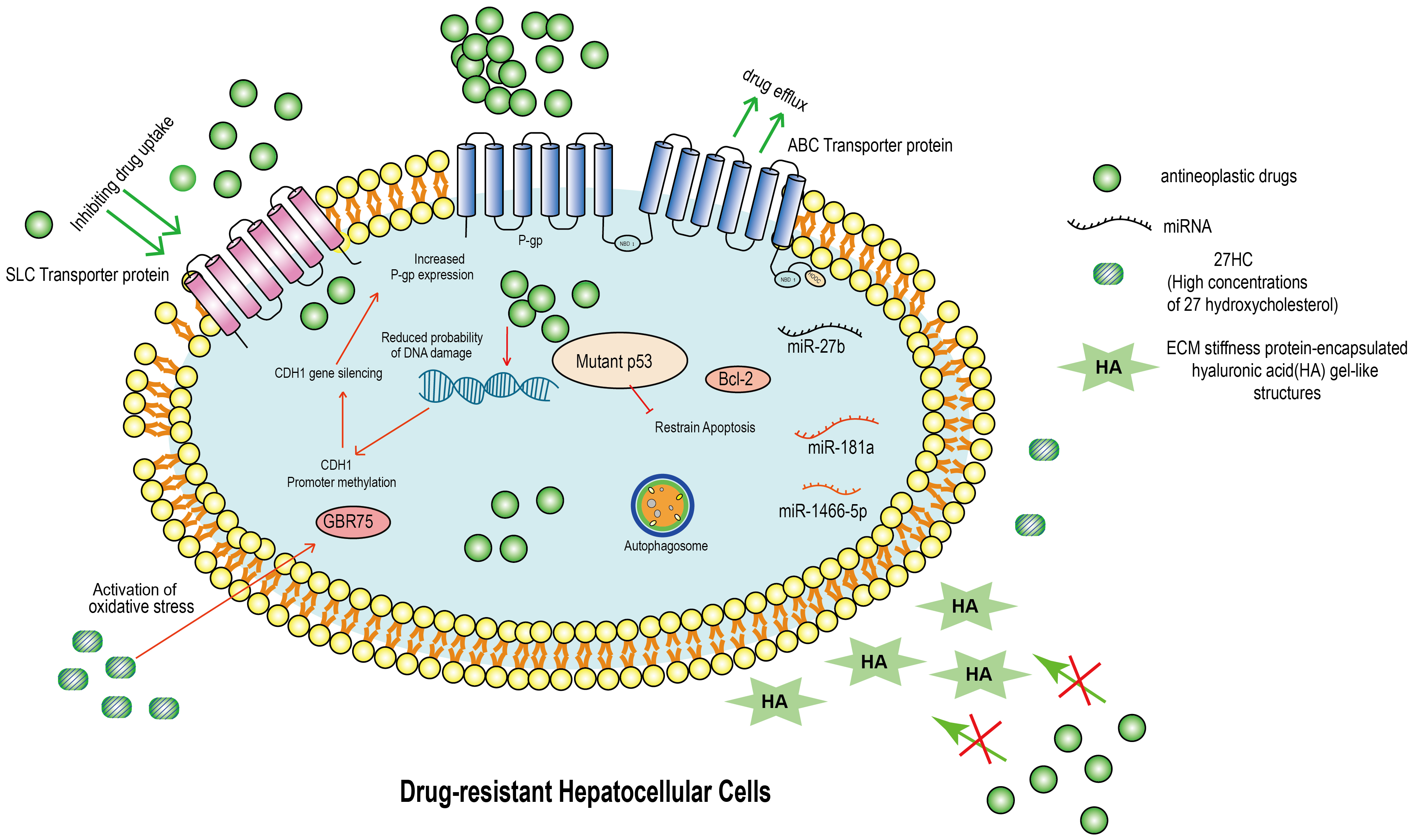 Fig. 1.
Fig. 1.Molecular mechanism of drug resistance in hepatocellular carcinoma (HCC). This figure summarizes the molecular mechanism of drug resistance in HCC. ABC transporter protein acts as a transmembrane protein and inhibits the entry of anticancer drugs into cells, leading to drug resistance. Inhibition of the SLC family delivers drugs to cancer cells and causes HCC cells to develop MDR. Drug-resistant HCC will highly express mutant p53 and bcl-2, which prevent HCC’s apoptosis. In addition, when the drug enters the drug-resistant HCC, it does not cause DNA damage. Not only that, in drug-resistant HCC, the CDH1 promoter is hypermethylated, leading to CDH1 silencing and increased P-gp protein expression. Importantly, in drug-resistant HCC, the autophagic system is activated and is an important mechanism of drug resistance. In addition, miRNA down-regulation in MDR HCC is one of the mechanisms to generate MDR. In TME, hepatocellular carcinoma cells are surrounded by large amounts of 27HC (27-hydroxycholesterol) and HA (ECM stiffness protein-encapsulated hyaluronic acid gel-like structures). 27HC leads to an increase in intracellular GRP75 and HA leads to an increase in ECM stiffness, which result HCC drug resistance. ABC, ATP-binding cassette; SLC, solute carrier; MDR, multidrug resistance; TME, tumor microenvironment; ECM, extracellular matrix; HA, hyaluronic acid; miRNA, microRNA.
Since MDR is prone to occur in HCC, its reversal is considered to be the key to improving the efficacy of chemotherapy. The screening of active ingredients from plant-derived natural products has recently become the focus of research to reverse MDR in HCC. These must have low toxicity and minimal side-effects for normal cells, but high efficiency against tumour cells. For example, a chromatography extract and defatted extract from P. americana effectively increases drug accumulation and reverses MDR in 5-FU-resistant BEL-7402 cells [159]. Astragalus polysaccharide (APS) is an extract from the Chinese medicinal herb Astragalus membranaceus root. This agent exerts a synergistic anti-tumour effect with ADM in H22 cancer cell-bearing mice via the suppression of MDR1 and P-gp expression levels [160]. Yao et al. [161] reported that a new derivative of betulinic acid, B5G1, triggered the apoptosis of MDR HepG2/ADM and MCF-7/ADR cells by activating mitophagy. This suggests that B5G1 may be a potential candidate for treating different types of MDR cancer cells. In addition, the marine-derived steroid methyl spongoate (MESP) exhibits powerful activity against MDR HCC cells, independent of the expression of ABC transporter protein [162]. This observation suggests that MESP may be used to overcome ABC transporter-mediated MDR in HCC.
Transcatheter arterial chemoembolization (TACE) is currently one of the most commonly used non-surgical treatments for HCC. TACE blocks the supply of oxygenated blood from short blood vessels to HCC tissues [163]. Due to its minimally invasive characteristics, TACE can prolong the survival time and improve the quality of life for patients with advanced liver cancer [164]. However, MDR is one of the main causes of treatment failure with conventional targeted arterial embolization interventions for HCC. Therefore, it is important to understand how best to use the TACE approach for HCC in the presence of MDR. Huang et al. [164] reported that verapamil-targeted arterial perfusion could reverse MDR and enhance the effect of chemotherapy in HCC patients. Although TACE has relatively mild adverse effects, future studies should investigate the optimal dose of TACE required to overcome MDR in HCC, while causing minimal side-effects.
Because of the critical role of selectively delivering anticancer agents, investigators are becoming increasingly focused on vascular permeability in tumour biology. Functional nanosized drug delivery systems (NDDSs) have become important in tumour treatment by enhancing permeability and retention (EPR) effects, thus enabling the delivery of high drug doses to tumour tissues while reducing adverse reactions [165]. NDDSs mainly deliver submicron particles in cancer therapy, including lipids, inorganic nanomaterials (INM), and polymer nanosystems [166]. This system has the advantages of high metabolic stability, membrane permeability, bioavailability, and activity prolongation. These properties not only reduce high-dose adverse effects, but also reverse MDR by bypassing drug efflux, controlling drug release, and interfering with metabolism [166, 167, 168]. For example, triblock polymer micelle-mediated co-delivery of ADM and P-gp siRNA into the MDR HCC cell line HepG2/ADM markedly increased the accumulation of ADM-siRNA-micelles in the tumour region in vivo and inhibited tumour growth with synergistic effects [169]. Liang et al. [170] reported a nano-delivery system based on AS1411 aptamer-functionalized micelles for the simultaneous co-delivery of doxorubicin and miR-519c. The AS1411 aptamer specifically recognizes overexpressed nucleolin protein in tumour cells. Subsequently, miR-519c represses ABCG2-dependent drug efflux, leading to an increase in the intracellular doxorubicin concentration and reversal of MDR in HepG2 cells. Consistent with this result, the lipid-coated, hollow, mesoporous silica nanoparticle co-delivery of ADM and miR-375 improves MDR in HCC cells.
Since the underlying mechanism of MDR in HCC is complex and regulated by multiple factors, the application of NDDS alone is unlikely reach a sufficiently high drug concentration at the tumour site. Thus, NDDS can be combined with other anticancer strategies to overcome MDR in HCC.
Currently, the efficacy of single-agent chemotherapy for liver cancer is limited. Therefore, the combination of multiple chemotherapeutic drugs that target different molecules or pathways has become the standard clinical practice for cancer treatment [171, 172, 173]. Plant-derived natural products have been evaluated as potential anticancer drugs due to low side-effects and high anti-tumour efficacy. It is therefore of great importance to study the combination of plant-derived drugs and chemotherapeutic drugs in the treatment of HCC with MDR. For example, curcumin can significantly enhance the sensitivity of Hep3B cells to paclitaxel and improve drug resistance [174]. Jiang et al. [175] showed that resveratrol can effectively reverse paclitaxel resistance in HepG2 cells. Coumarin derivative 50 is a novel MARK4 inhibitor that directly interacts with MARK4 to reduce microtubule kinetics and induce apoptosis of HCC cells in vitro. This drug significantly enhances the response to paclitaxel treatment and overcame resistance in HCC cells, suggesting that MARK4 is an attractive target for the reversal of MDR [176]. In recent years, a growing number of plant-derived natural products have been used in combination therapy with conventional chemotherapeutic agents against liver cancer. Although combination therapy has improved clinical efficacy through the reversal of MDR, it also results in undesirable side-effects [177]. Thus, further studies are need to explore the use of suitable combination therapies.
FOLFOX4 chemotherapy is a common regimen for HCC therapy consisting of 5-FU, leucovorin and oxaliplatin [178, 179]. This regimen is convenient, well tolerated and safe, and is widely used in stomach, colorectal and liver cancer [180]. In recent years, FOLFOX4 therapy has been used to treat drug-resistant liver cancer. He et al. [181] reported that sorafenib combined with hepatic artery infusion chemotherapy (HAIC) of oxaliplatin, 5-FU and leucovorin (FOLFOX) prolonged the median overall survival of HCC patients to 13.3 months, compared with 7.13 months for sorafenib alone. The survival benefit observed in this study may in part be due to the synergistic, anti-tumour effects of sorafenib and FOLFOX. In particular, the synergism between oxaliplatin and 5-FU is attributed to 5-FU-mediated suppression of drug transporters, while sorafenib can further suppress the activity of MDR-associated transporters [181].
Recently, combination therapies based on immune checkpoint inhibitors (ICIs)
have dominated clinical studies of all stages of HCC [182]. In contrast to
targeted drugs, immunotherapy activates the body’s immune system together with
large numbers of immune cells to destroy tumour cells. The most commonly used
immune-combination therapy for HCC in the clinic is T+A therapy, which is a
combination of anti-PD-L1 antibody (atezolizumab) and anti-VEGF antibody
(bevacizumab). Finn et al. [183] reported that atezolizumab in
combination with bevacizumab showed better overall survival than sorafenib alone.
In addition to target-free therapy, dual-free therapy is also a novel type of
immune combination therapy. O+Y therapy (4 doses of nivolumab 1 mg/kg +
ipilimumab 3 mg/kg every 3 weeks, followed by nivolumab 240 mg every 2 weeks) is
a dual-immune therapy that has been approved by the U.S. FDA for the second-line
treatment of HCC [184]. O+Y therapy has a controlled safety profile and
longer-lasting effects. However, as with other anti-tumour agents, immunotherapy
is limited by the development of drug resistance. For example, drug resistance
can easily develop when atezolizumab alone is used to treat HCC. Moreover, the
use of certain anti-PD-1/PD-L1, anti-CTLA-4 and anti-VEGF-A drugs (e.g.,
pablizumab, nabolutumab, atezolizumab, epirimizumab, and bevacizumab) has been
associated with the development of resistance [60, 62, 185, 186, 187]. ICB (Immune
Checkpoint Blockade) is an immunotherapy method. It aims to activate the
patient’s own immune system to attack tumor cells by removing the tumor’s
suppressive effect on the immune system. ICB therapy with PD-1 enhances the
expression of
Over the past decade, cancer treatment has mainly focused on the development of
novel targeted drugs and immunologic agents. Although combination therapies based
on ICIs have dominated all phases of clinical research in HCC, targeted drugs
remain the most commonly used treatment for HCC. Meanwhile, the replacement of
targeted drugs for patients showing chemoresistance is still a feasible approach.
The development of resistance to a certain class of targeted drugs does not mean
the patients are also resistant to other types of drugs. In fact, almost all
targeted drugs have multiple targets containing the same or similar molecules.
For example, sorafenib targets KIT, VEGFR, PDGFR, RAF, MEK, and ERK [188, 189, 190, 191].
Lenvatinib inhibits VEGF receptors 1-3, FGF receptors 1-4, PDGF receptor
HCC is one of the most common malignant tumours worldwide. Currently, surgical resection is the main treatment for early-stage HCC. Due to the masked onset of HCC and the lack of obvious early symptoms, most patients (about 65–70%) are diagnosed with intermediate or advanced stage disease. Such patients are ineligible for radical therapies and are therefore reliant on chemotherapy. However, chemotherapeutic drugs inevitably develop resistance, leading to the failure of HCC treatment. The detailed mechanisms of MDR include enhanced efflux of drugs, increased DNA repair capacity, genetic factors (gene mutations, epigenetic alterations, etc.) and downstream adaptive responses such as apoptosis and autophagy. Amongst these factors, the drug efflux pump transports anti-cancer drugs out of the cell, thereby reducing intracellular drug concentrations and contributing to MDR. In recent years, improved understanding of the tumour/TME relationship has highlighted the key role of the TME in tumour progression, local drug resistance, and immune escape. Due to the large number of immunosuppressive cells in the TME, immunotherapy that targets these cells has become a new strategy for tumour treatment and reversal of drug resistance. The ECM has recently been recognized as a hallmark of cancer. It serves not only as a physical support for tumour cells but also in the regulation of cell‒cell and cell-matrix cross-talk. The pathological ECM enhances tumour cell growth, survival and invasion, while altering the behaviour of fibroblasts and immune cells to favour metastasis formation and chemotherapy resistance. ECM stiffness is closely related to drug resistance because the stiffened matrix forms a physical barrier to drug infiltration, thereby preventing the entry of drugs into the central core of the tumour tissue. ECM stiffness also induces a hypoxic environment that enhances the stem cell-like properties of tumour cells, leading to drug resistance. A rapidly increasing number of studies are focused on designing chemotherapeutics that can evade or reverse MDR. Because MDR is regulated by complex factors, further studies are needed to better understand the mechanism of MDR, especially in relation to the TME and ECM. The emergence of many new therapeutic drugs and strategies targeting various components of the TME suggest that combination therapy is likely to become an increasingly important therapeutic approach in cancer.
YL completed the writing of the original manuscript, and XH completed the analysis of data and the drawing of pictures and tables. YL and XH collected and organized the literatures. JL and CM conceived and revised the manuscript. All authors contributed to editorial changes in the manuscript. All authors revised and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This study was supported by the National Natural Science Foundation of China (No. 82170645), Project of Chengdu Health and Family Planning Commission (No. 2021076 and 2022043), Department of Science and Technology of Sichuan Province (CN) (No. 24NSFSC2357 and 22NSFSC0766), Disciplinary Construction Innovation Team Foundation of Chengdu Medical College (CMC-XK-2103).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.