




 , Jian Zhuang 1,2,*
, Jian Zhuang 1,2,*1 Department of Cardiovascular Surgery, Guangdong Cardiovascular Institute, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, 510080 Guangzhou, Guangdong, China
2 Guangdong Provincial Key Laboratory of South China Structural Heart Disease, 510080 Guangzhou, Guangdong, China
3 School of Medicine South China University of Technology, 510006 Guangzhou, Guangdong, China
4 School of Medical Imaging, Tianjin Medical University, 300203 Tianjin, China
5 Guangdong Beating Origin Regenerative Medicine Co., Ltd., 528231 Foshan, Guangdong, China
6 Pen-Tung Sah Institute of Micro-Nano Science and Technology, Xiamen University, 361102 Xiamen, Fujian, China
7 Guangdong Provincial First Disabled Veterans Hospital, 510260 Guangzhou, Guangdong, China
†These authors contributed equally.
Abstract
Background: Myogenin is well known as a crucial transcription factor in
skeletal muscle development, yet its other biological functions remain
unexplored. Previous research showed that myogenin suppresses apoptosis induced
by angiotensin II in human induced pluripotent stem cell-derived cardiomyocytes,
and offered a new perspective on myogenin’s role in cardioprotection. However,
the detailed mechanism of this cardioprotection, especially under oxidative
stress, is still unclear. Methods: In this study, hydrogen peroxide
(H
Keywords
- myogenin
- DUSP13
- reactive oxygen species
- p38 MAPK pathway
- apoptosis
- cardiomyocyte
Oxidative stress plays a critical role in the development and progression of
cardiovascular disease [1]. In myocytes under oxidative stress, increased
intracellular production of reactive oxygen species (ROS) causes cardiac
pathogenic effects including apoptosis [2, 3], autophagy [4], aging [5],
hypertrophy [6], fibrosis [7], and myocardial remodeling [8]. Accumulating
evidence suggests that ROS are involved in the pathophysiology of heart failure
[8]; elevated ROS levels were detected in patients with heart failure [9, 10].
Indeed, ROS mediate many heart failure-related pathways [11]. For example, the
angiotensin II (Ang II) signaling pathway promotes NAD(P)H oxidase-dependent ROS
generation through binding to the type 1 Ang II receptor. The increased ROS
activates the Ca
Myogenin, a basic helix-loop-helix myogenic transcription factor, is known to play an essential role in skeletal muscle development, and most studies have focused on its function in myoblast or satellite cells during myogenesis and muscle regeneration [15]. Myogenin is indispensable: MyoG-null mice suffer severe muscle disruption and perinatal lethality [16]. On the other hand, myogenin-overexpressed mice also show high rates of neonatal mortality, although mice that survive into adulthood show no obvious muscle abnormalities [17]. Our previous studies demonstrated that myogenin inhibited apoptosis induced by Ang II in human induced pluripotent stem cell-derived cardiomyocytes [18]. Although RNA-seq data revealed that the expression levels of inflammatory response-related genes were downregulated by a 6-day induction with myogenin [18], the detailed mechanism of its anti-apoptotic effect is not well understood. It is known that excess intracellular ROS causes DNA strand breaks and mitochondrial damage followed by activation of protein kinases, such as MAPK, to initiate apoptosis-related signaling pathways [8, 19]. Therefore we hypothesize that myogenin might exert its cardioprotective effects via ROS-related pathways.
To investigate this hypothesis, cardiomyocytes overexpressing myogenin were
treated with hydrogen peroxide (H
Human cardiomyocytes (hCMs) (BFN608007121, Bluefbio Biotechnology Development
Co., Ltd., Shanghai, China) were plated on gelatin-coated plates (V900863, Sigma,
Darmstadt, Germany), and then cultured in D10 medium: DMEM medium (C22400500BT,
Gibco, Shanghai, China) supplemented with 10% fetal bovine serum (fsp500, Excell
BIO, Shanghai, China), 1
The plasmid pCW-Cas9-Blast (83481, Addgene, Watertown, MA, USA) was modified by removing Cas9 cDNA and replacing the blasticidin S resistance gene with a puromycin resistance gene. Then human MYOG cDNA and an enhanced green fluorescent protein (EGFP) gene, which was linked by a T2A sequence, were subcloned into the plasmid. Lentivirus was prepared using a third-generation lentivirus packaging system as described elsewhere [18]. After 24 hours of transduction, the medium was replaced with fresh D10 medium supplemented with Dox (to induce myogenin expression) or DMSO (controls). After 2 days of induction, EGFP expression was observed by fluorescence microscopy, and the EGFP-positive cells were sorted by flow cytometry (FACSAria III, BD Biosciences, San Jose, CA, USA) and cultured further in D10 medium. After 5–7 days of incubation, single clones were picked at random for further expansion, to be used in the following experiments.
Cells were fixed with 4% paraformaldehyde at room temperature for 20 minutes
and washed three times with 1
Total RNA was isolated from cells using the EZ-10 Total RNA Mini-Preps Kit
(B618583-0100, Sangon Biotech, Shanghai, China) followed by treatment with
RNase-Free DNase (B618253, Sangon Biotech). cDNA was synthesized using the
Hifair®Ⅲ 1st Strand cDNA Synthesis Kit (11139ES60, YEASEN,
Shanghai, China). Quantitative real-time PCR was performed using a Light
Cycler® 480 (Roche) with TB Green® Fast qPCR Mix
(RR430B, Takara, Beijing, China). Data were analyzed by the relative standard
curve method and normalized to GAPDH. Quantitative PCR primers and their
sequences, designed by Primer3 [20], are listed below (from 5
MYOG-RT-F: GCCCAAGGTGGAGATCCT;
MYOG-RT-R: GGTCAGCCGTGAGCAGAT;
FOXA3-RT-F: TGGGCTCAGTGAAGATGGAG;
FOXA3-RT-R: GGGGATAGGGAGAGCTTAGAG;
ATOH8-RT-F: GAGATCAAAGCCCTGCAGC;
ATOH8-RT-R: TCGGCACTGTAGTCAAGGTC;
DUSP13-RT-F: CTTCCCCAACCGAGGCTT;
DUSP13-RT-R: CAGGGAGTAAGGACCAGCTC;
GAPDH-RT-F: CGCTCTCTGCTCCTCCTGTT;
GAPDH-RT-R: CCATGGTGTCTGAGCGATGT.
Apoptosis was detected using APC annexin V (640920, Biolegend, San Diego, CA,
USA). Cells were seeded into 6-well plates with 5
Cells were washed twice with ice-cold 1
Potential binding sites of myogenin in the DUSP13 promoter region
(–2500 to 0 bp) were predicted by the JASPAR database [21]. Fragments with
different lengths of DUSP13 promoter were amplified from the genomic DNA
of hCMs via PCR. The mutant (Mut) sequence of the DUSP13 promoter was
synthesized by Sangon Biotech. These DNA fragments were then subcloned into the
pGL6-TA luciferase reporter vector (D2105, Beyotime). MYOG cDNA was
subcloned into the expression vector pCMV-N-Myc-Puro (D2791, Beyotime). HEK293T
cells were transfected with luciferase reporter vectors, Renilla pRLTK control
reporter vector (D2760, Beyotime), and MYOG expression vector using the
EZ-Trans transfection reagent (AC04L071, Life-iLab, Shanghai, China). After 48
hours of transfection, Dual-Lumi™ Luciferase Reporter Gene Assay
Kit (RG089S, Beyotime) was used to detect luciferase activity. Each experiment
was performed in triplicate. Results are shown as firefly luciferase activity
normalized to Renilla luciferase activity. The PCR primers are listed below (from
5
DUSP13-HindIII-R: GTAAGCTTGTCATTTCTCCTTCCCAG;
DUSP13-500-XhoI-F: TACTCGAGCCTGACCTCCGTAATTCAGAG;
DUSP13-1000-XhoI-F: AACTCGAGCGTCTCCCCATTTGACAT;
DUSP13-1350XhoI-F: AACTCGAGCGACTTCAATGGTTCTGA;
DUSP13-1500-XhoI-F: AACTCGAGCTGTATTTTCCTCACTCC;
DUSP13-2500-XhoI-F: AACTCGAGAGTTCGACAACCTCCATTTG.
ChIP assay was performed using the Pierce™ Magnetic ChIP Kit
(26157, Thermo Scientific, Eugene, OR, USA). Cells were first treated with 2
µg/mL Dox for 72 hours to induce myogenin expression, then cross-linked
with 1% formaldehyde for 10 minutes at room temperature and incubated with a
final concentration of 0.2 M glycine for 5 minutes to terminate cross-linking.
After washing with PBS, cells were collected and resuspended in Membrane
Extraction Buffer on ice for 10 minutes. The cell lysates were resuspended in
MNase Digestion Buffer Working Solution supplemented with MNase (10
U/µL) for chromatin digestion (37 °C, 15 minutes), and the
digestion was terminated by adding MNase Stop Solution for 5 minutes on ice.
Lysates were sonicated on ice using 3 W power for three 20 s-pulses (JY92-IIN,
SCIENTZ, Ningbo, China). One-tenth of supernatant was used as Input. The
remaining solution was divided into 3 fractions and incubated with antibodies
overnight at 4 °C (Negative control IP: 1–2 µL of Rabbit IgG
(included in the kit); Target-specific IP: 10 µg Myogenin Rabbit pAb
(A17427, Abclonal, Wuhan, China). Magnetic beads (20 µL) were added to each
IP sample and incubated at 4 °C for 2 hours. The beads were collected by
a magnetic scaffold, washed three times with IP Wash Buffer I, once with IP Wash
Buffer II, then mixed with IP Elution Buffer. Sodium chloride (5 M) and
Proteinase K (20 mg/mL) were added to the supernatant to remove staining and
protein cross-linking. DNA was purified using a DNA Clean-Up column. ChIP DNA was
detected by qPCR using TB Green® Fast qPCR Mix (RR430A, TaKaRa).
The primers specific for PBS3, PBS4, and PBS5 are as followed (from 5
PBS3-F: TGTCAGACCCAGCATGTCCT;
PBS3-R: TGCAAGTCAGCTACCACCCT;
PBS4-F: CCTCTGCACCCTGGATCTCT;
PBS4-R: AAGGACATGCTGGGTCTGAC;
PBS5-F: GGTCTTGTCCACTCCAGAGGC;
PBS5-R: CCTCTGTGGTCACTCCTCTCT.
DUSP13A and DUSP13B cDNA were subcloned into the expression
vector pcDNA3.1 (HG-VPI0001, HonorGene, Changsha, China). HCMs were seeded into
6-well plates with 2.5
DUSP13A-NheI-F: TATAGCTAGCACCATGGCTGAGACCTCTCTCC;
DUSP13A-BamhI-R: TATAGGATCCTCAGCTCTGGCCGGCAC;
DUSP13B-NheI-F: TAGCTAGCACCATGGACTCACTGCAGAAGCAG;
DUSP13B-BamhI-R: TATAGGATCCTCAGAACCGCCCCGTCTCC.
RNA-seq was performed by Novogene Co., Ltd., in three biological replicates for
each group. 150-bp paired-end RNA-seq reads were obtained from the Illumina
sequencing platform. First, adaptors and low-quality bases were trimmed by Trim
Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Then,
gene expression was quantified by Kallisto [22] and the GENCODE database (v32).
DESeq2 [23] was used to identify DEGs. The list of significant DEGs was defined
at a false discovery rate (FDR)
According to the mRNA sequence of human DUSP13B gene (Accession number: NM_016364.3), we obtained three small interfering RNA (siRNA). The sequences are shown in Supplementary Table 1. All sequences were synthesized by Genomeditech Company (Shanghai, China) Co., Ltd. Knockdown efficiency was evaluated by western blotting and siRNA-1 was selected to perform the knockdown assay.
HCM
Values are expressed as mean
To obtain stable cell lines expressing myogenin, an hCM cell line in which the
expression of myogenin could be modulated by a Tet-On system (hCM
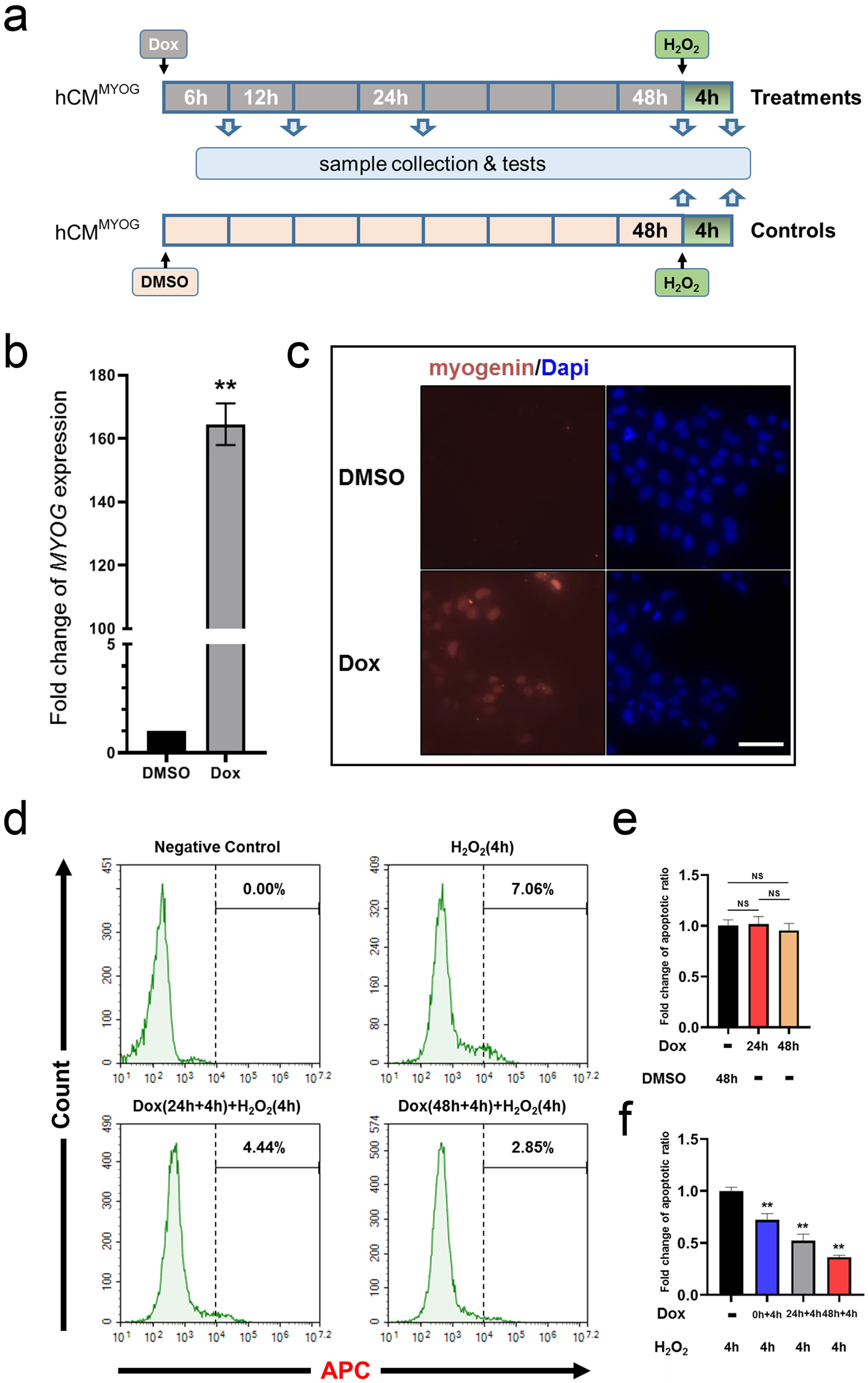 Fig. 1.
Fig. 1.Overexpression of myogenin decreased the rate of
apoptosis after hydrogen peroxide (H
As a transcription factor, myogenin regulates the expression of a series of
genes, including several key components of transcriptional machinery [26]. Some
of the DEGs observed after long-term expression of myogenin may be due to its
regulation of other transcription factors. Therefore it is necessary to monitor
changes in gene expression profile during the early period of myogenin
expression. To this end, hCM
Only protein-coding genes with FDR
 Fig. 2.
Fig. 2.DUSP13 expression was elevated in
myogenin-overexpressing hCM
Next, the DEGs that increased in expression from 6- to 48-hour induction were
analyzed to identify candidate genes directly regulated by myogenin (Fig. 2c).
Among these genes, ATOH8, ASCL2, and ID3 encoded
transcription factors or transcriptional regulators, and the 6-hour induction
only slightly increased their expression (1.42-fold, 1.13-fold, and 1.29-fold,
respectively). By contrast, PHGR1, DUSP13, and VWA5B2,
the top three up-regulated genes after 48-hour induction, exhibited about 2-fold
increased expression after the 6-hour induction. There are few studies about the
molecular functions of PHGR1 and VWA5B, while it has been
reported that DUSP13 dephosphorylates JNK1 and p38
HEK 293T cells were used to examine whether myogenin bound to the promoter
region of DUSP13. A 2500-bp sequence upstream of the 5
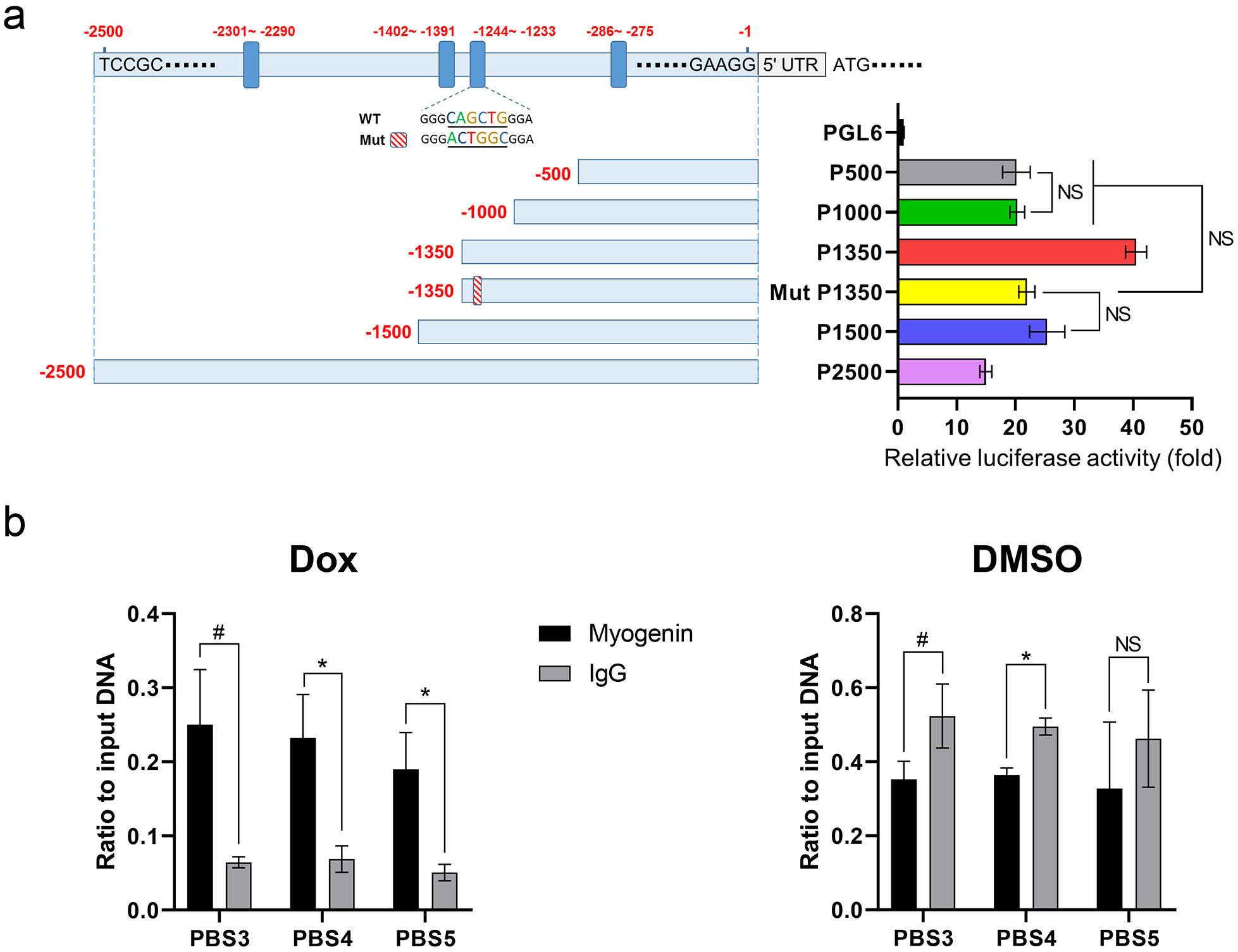 Fig. 3.
Fig. 3.Myogenin bound the DUSP13 promoter. (a) Different
DUSP13 promoter regions (500, 1000, 1350, 1500, and 2500 bp) and a
mutated promoter sequence (Mut p1350) were cloned into luciferase reporter
vectors. Cells transfected with the PGL6 vector were used as the control group.
Luciferase activity was measured 48 hours after transfection. p-values
for the comparisons between each experimental group can be found in
Supplementary Table 3. NS: no significant difference. (b) ChIP analysis
of Dox-treated hCM
To further demonstrate that myogenin inhibited H
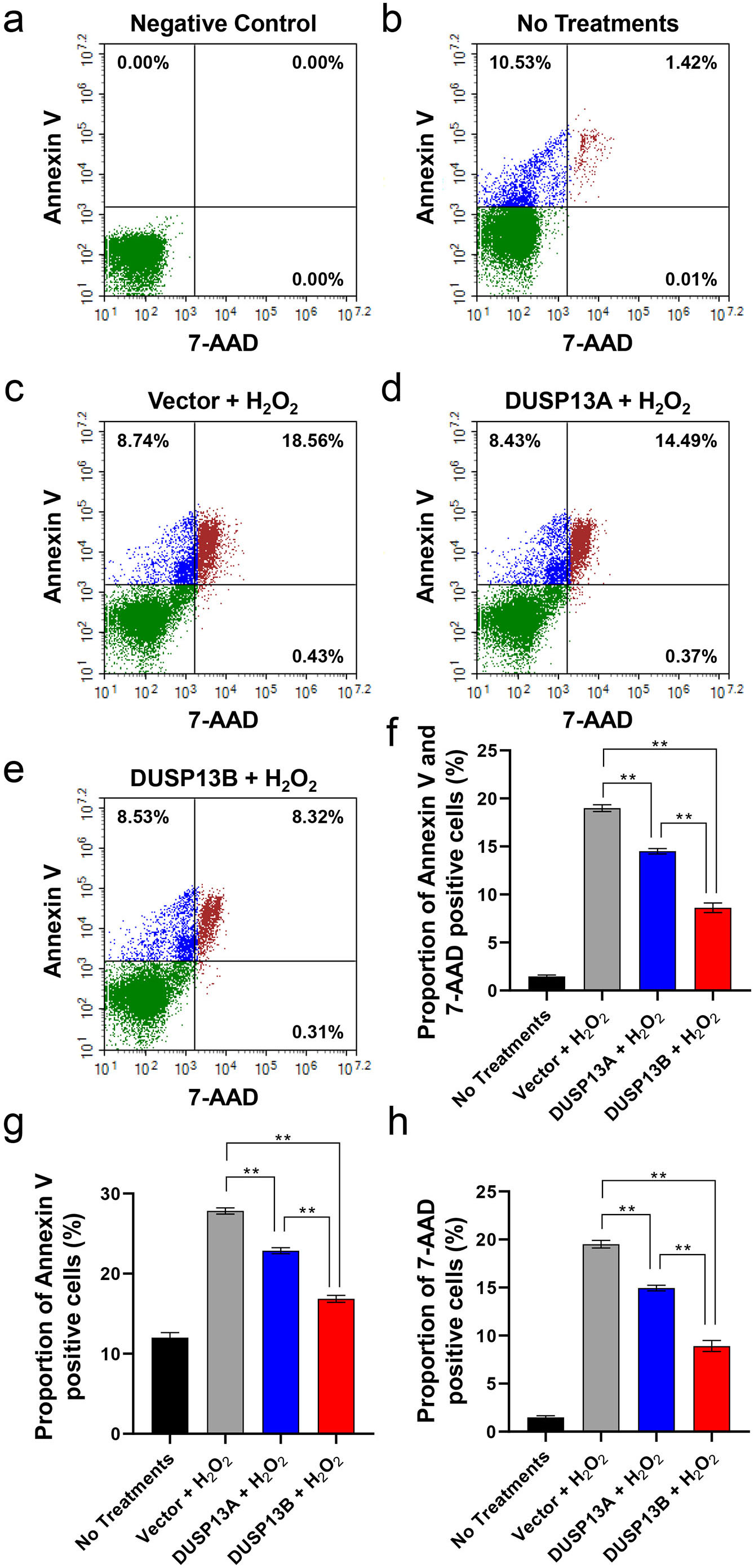 Fig. 4.
Fig. 4.DUSP13 decreased the apoptotic and necrotic ratios of
H
We also knocked down DUSP13B by using siRNA in myogenin-expressing
cardiomyocytes to confirm this conclusion. Three pairs of siRNA targeting
DUSP13B gene were designed and synthesized. According to the knockdown
efficiency evaluated by western blotting, siRNA-1 was selected to perform the
knockdown assay. As shown in Supplementary Fig. 1, in the
myogenin-expressing cardiomyocytes treated by H
At the cellular level, the genes regulated by myogenin overexpression have not been studied systematically. It has been variously reported that overexpression of myogenin downregulates the expression of Pax7 [28], and induces the expression of Mef2c [29], MuRF1 [30], Atrogin-1 [30], MEGF10 [31], and Ncam1 [32]. Most of these genes were not identified in our RNA-seq data, possibly because (i) Many studies were performed in a mouse myoblast cell line (C2C12) that exhibits epigenetic characteristics distinct from human cardiomyocytes; (ii) The present study only considered the first 48 hours of myogenin expression and may have missed genes that require long-term induction. The expression profiles of myogenin-overexpressed human induced pluripotent stem cell-derived cardiomyocytes were described in our previous work [18], in which myogenin-overexpressed cardiomyocytes were treated with Ang II for 6 days. The RNA-seq data from these two studies did not completely agree. For example, DUSP13 did not appear in the DEGs of the original work [18], probably because the gene regulatory networks influenced by complex external factors (such as Ang II treatment and incubation time) eliminated any changes in its expression. These results demonstrate the necessity of time-course analysis of gene expression at the early stages of myogenin expression.
Based on the results from the dual-luciferase reporter assay, P1350 cells exhibited higher relative luciferase activity than P1000 cells, suggesting the existence of a certain motif in the –1350 bp to –1000 bp region that might enhance the transcriptional activity of myogenin. We speculated that this motif was the CAGCTG site in the –1244 to –1233 region. A mutated motif (CAGCTG to ACTGGC) was therefore generated, and its relative luciferase activity was found to be 46% lower when compared to unmutated P1350 cells, and was almost equal with P1000 cells (Fig. 3a). These data confirmed that the –1244 to –1233 region was a key motif that could increase the expression of a downstream gene (DUSP13). We further speculate that the reason that P1500 and P2500 cells had less activity than P1350 cells was due to the presence of repressor elements in the –1500 to –1350 bp or –2500 to –1500 bp regions. To identify these repressor elements, more delicate design experiments will be necessary to carry out in future studies. The lack of knowledge and the complexity of its promoter region are serious impediments to exploring the regulatory mechanisms of DUSP13 gene. For example, an online tool, Find Individual Motif Occurences (Version 5.5.4, FIMO, Seattle, WA, USA), was used to search the potential E-box motifs in the 2500-bp region, and in total 48 E-boxes were identified (Supplementary Table 5). What roles these motifs actually play is still undiscovered.
The p38 MAPK signaling pathway is involved in skeletal muscle development [33].
P38 directly phosphorylates MEF2A and MEF2C [34], and may induce MyoD
through an indirect effect [33]. Although there is no evidence that p38 directly
regulates myogenin, the p38 inhibitor SB203580 decreased the expression of
myogenin and prevented myogenic differentiation [35]. The present study
demonstrated that myogenin might reduce the phosphorylation of p38 by increasing
expression of DUSP13, suggesting that there might be a feedback loop to
regulate p38 MAPK signaling by myogenic regulatory factors. In addition,
overexpression and knockdown assays illustrated the vital role of DUSP13
in the inhibition of apoptosis induced by H
In conclusion, DUSP13 was identified as a direct target of myogenin, and presented a new possible mechanism by which myogenin suppresses ROS-induced apoptosis in cardiomyocytes. Myogenin increased the expression of DUSP13, which in turn dephosphorylated p38 to inhibit ROS-activated MAPK, thereby obstructing apoptosis.
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
JZ and JC contributed to the conception and design of the study. JL, QG, JZ, and JC prepared the study materials. JL, QG, HQ, SZ, WZ, PW, LZ, XL and BL performed the experiments. JL, QG, PW, LZ, BL, LL, FX, RZ, and DS analyzed and interpreted the data. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by the grants from the Guangdong Basic and Applied Basic Research Foundation (2021A1515111152), the National Natural Science Foundation of China (No. U2005214) and the National Key R&D Program of China (2022YFB4600600).
The authors declare no conflict of interest. However, it should be noted that Lishi Zhou and Bin Lin are affiliated with Guangdong Beating Origin Regenerative Medicine Co., Ltd. Steps have been taken to ensure that their involvement in this research did not influence the objectivity and integrity of the study findings.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.