
- Academic Editor
†These authors contributed equally.
Background: Cancer treatment has recently shifted towards metabolic approaches aimed at enhancing therapeutic efficacy. Somewhat surprisingly, a known regulator of energy metabolism in normal tissues, PPP3CB, is down-regulated in bladder cancer. This suggests that PPP3CB could exert an inhibitory effect on bladder cancer through its role in energy metabolism. Methods: To explore the above hypothesis, we employed non-targeted metabolism screening in bladder cancer cells with knockdown of PPP3CB. Glucose uptake and lactate production were carefully measured using specialized assay kits for glucose/lactic acid content. Western blot analysis was also used to evaluate the expression levels of pyruvate dehydrogenase kinase 1 (PDHK1) and p-PDHA1 in cells with PPP3CB knockdown. To substantiate the findings, co-immunoprecipitation (co-IP) experiments were performed to validate the interaction between PPP3CB and PDHK1. Various in vitro assays were also performed, including clone formation assay and Cell Counting Kit-8 (CCK8) viability assays. The in vivo anti-tumor potential of PPP3CB in bladder cancer was also studied using a nude mouse tumorigenesis model. Results: Significant down-regulation of PPP3CB was observed in bladder tumors, and potent anti-tumor effects of PPP3CB were observed in vitro. Investigation of the underlying mechanism by which PPP3CB hampers glycolysis in bladder cancer cells revealed that it interacted with PDHK1 to inhibit its protein stabilization. PDHK1 thus appears to be a crucial mediator through which PPP3CB exerts its inhibitory effects on bladder cancer cells. Conclusions: In summary, PPP3CB exerts strong inhibitory influences on bladder cancer cell proliferation and glycolysis via its destabilization of PDHK1. These results highlight the potential of PPP3CB as a novel regulator of the Warburg effect. Interestingly, the downregulation of PPP3CB in bladder cancer cells increases the Warburg effect, thereby generating more lactic acid and reshaping the tumor microenvironment so as to promote tumor cell proliferation.
Bladder cancer is one of the most prevalent tumor types, with over 430,000 cases diagnosed annually worldwide [1]. Known risk factors for the development of bladder cancer are male sex, advanced age and cigarette smoking [2]. Most cancers are characterized by high rates of glucose metabolism and glycolysis. During tumor growth, cells at the center of the tumor become hypoxic and oxidative phosphorylation (OXPHO) decreases [3]. This aerobic, glycolysis-dependent metabolism is referred to as the Warburg effect [4]. Metabolomic studies have revealed that metabolites such as lactic acid, glutamate and pyruvic acid are upregulated in bladder cancer, indicating the crucial role of metabolism in bladder cancer progression. Furthermore, upregulation of genes that promote the pentose phosphate and fatty-acid synthesis pathways, together with decreased activities of AMP-activated protein kinase (AMPK) and the Krebs cycle, are characteristics of bladder cancer metabolism [5]. Several alterations in glucose metabolism have also been reported in bladder cancer. These include stimulation of GLUT-1 activity [6], upregulation of glycolysis [7], increased pyruvate metabolism and lactate production [8, 9], increased expression of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) [10] and steroid receptor coactivator-3 (SRC-3) [11], and increased pentose phosphate pathway activity [12].
PPP3CB is a class of serine threonine calcineurin (CN) enzymes that act
by dephosphorylating substrates. Calcineurin (CN) is a heterodimer consisting of
a 58–59 kDa catalytic subunit (CNA) that is bound to calmodulin, and a 19 kDa
regulatory subunit (CNB) that binds Ca
Pyruvate dehydrogenase kinase (PDHK) comprises four types of tissue-specific
isoforms. These are mostly located in the mitochondria, and PDHK activity can be
regulated by hormones, hypoxia, nutrients, etc. [17]. PDHK is activated in
hypoxic tumor microenvironments through phosphorylation of its E1
We reasoned that PPP3CB might impact the proliferation of bladder cancer cells by regulating glycolysis, with the PDC being a possible target. Here we show for the first time that downregulation and inhibition of PPP3CB can suppress cell proliferation glycolysis in bladder cancer. Further investigation revealed the mechanism for this PPP3CB-mediated suppression was via regulation of PDHK1. Co-immunoprecipitation confirmed that PPP3CB interacts directly with PDHK1 to promote its degradation.
The siRNA used in this study was obtained from GenePharma (Shanghai, China).
Rabbit polyclonal antibody targeting PPP3CB was acquired from GeneTex
(Shanghai, China). Mouse monoclonal antibodies against
Human bladder cancer cell lines T24 and human embryonic kidney cells HEK-293T
were obtained from the American Type Culture Collections (ATCC). All cell lines
were grown in Dulbecco’s Modified Eagle’s Medium (Gibco, Thermo Fisher
Scientific, New York, NY, USA) supplemented with 10% fetal bovine serum (FBS)
(Gibco, Thermo Fisher Scientific, USA) and 1% penicillin/streptomycin (PS)
(Hyclone, cytiva, Logan, UT, USA). Cell lines were cultured in a humidified
incubator at 37 °C and with 5% CO
The coding sequence (CDS) for h.PPP3CB was amplified from complementary
DNA (cDNA) obtained from HEK-293T. Forward primer: 5
The coding sequence (CDS) for h.PDHK1 was amplified from cDNA
obtained from HEK-293T cells. Forward primer: 5
Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) was used for siRNA transfection as per the manufacturer’s instructions. HEK-293T cells were transiently transfected using Linear Polyethylenimine following the manufacturer’s instructions. For the immunoprecipitation assay, T24 cells were transfected using Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA) as recommended by the manufacturer. For lentivirus infection, the cells were grown to 50–60% confluence and subsequently treated with medium containing lentivirus. They were then harvested 48 hours after transfection and incubated overnight. Virus particles were collected and used to infect T24 cells in the presence of 8 µg/mL polybrene (Sigma, Saint Louis, MO, USA). After infection, stable clones were selected using puromycin (Invitrogen, Carlsbad, CA, USA).
Cell proliferation assays were conducted 24 hours after transfection. Cells were seeded at a density of 2000 per well in a 96-well plate. The CCK-8 reagent was diluted with DMEM to a 10% concentration and then added to each well. The absorbance at 450 nm was measured at 24-hour intervals.
T24 cells (1000 per well) were seeded into 6-well plates and incubated under normothermia conditions for 2 weeks, with the culture medium refreshed every 3 days. Following this, cell colonies were fixed with 4% paraformaldehyde (Sangon, Shanghai, China) for 15 minutes and subsequently stained with 0.5% crystal violet (Beyotime, Shanghai, China) for 20 minutes at room temperature. The images of colonies were recorded using a microscope (Leica, DMC5400, Wetzlar, Germany).
The isolation of total RNA from cells was carried out using Trizol reagent
(Invitrogen, Carlsbad, CA, USA). cDNA synthesis was performed using an
oligonucleotide primer and transcription kit (Thermo Scientific, Waltham, MA,
USA). Subsequently, quantitative real-time PCR was conducted using Ultra SYBR
Mixture (CWBIO, Beijing, China). All experiments were normalized to internal
control (18 s). The following primer pairs were utilized for quantitative
real-time PCR: human PPP3CB, forward:
5
Protein A agarose beads (Millipore, Billerica, MA, USA) were washed three times with PBS before the addition of anti-GFP antibody. This mixture was incubated for 3 hours at 4 °C. Protein was extracted from HEK-293T cells using the following lysis buffer: 50 mM Tris–HCl pH 7.5, 0.1% SDS, 1% TritonX-100, 150 mM NaCl, 1 mM dithiothreitol, 0.5 mM EDTA, 100 mM PMSF, 100 mM leupeptin, 1 mM aprotinin, 100 mM sodium orthovanadate, 100 mM sodium pyrophosphate, and 1 mM sodium fluoride. Cell extracts were centrifuged at 13,000 g for 10 min and the resulting supernatants were added to the bead and antibody mixture. Following overnight incubation at 4 °C, the immunocomplexes were washed three times with lysis buffer and then subjected to immunoblotting using specific antibodies. Image Lab software (Bio-Rad, version 3.0, Berkeley, CA, USA) was used to quantify the band intensity.
Approximately 3000 cells were seeded into a 24-well plate and grown for 24 hours at 37 °C. The cells were then fixed with 4% paraformaldehyde (Sigma) for 10 min, followed by permeabilization with 0.1% Triton X-100 (Sigma) for 10 minutes and then blocking with 10% bovine serum albumin (BSA) (Sigma) at room temperature for 1 hour. The primary antibodies anti-GFP, anti-PDHK1 and anti-PDHA1 were diluted (1:100) in 2% BSA and 20 µL was added to each sample and incubated overnight. After washing, secondary antibodies diluted with 2% BSA and mixed with DAPI were added to each sample and incubated for 1 h. The cells were then viewed using a Leica TCS SP8 confocal microscope.
Sections of bladder tissue on slides were purchased from Shuangxuan company
(Wuhan, China). The slides were deparaffinized by immersing twice in xylene for 5
minutes each, and then immersed twice in 100% alcohol for 3 minutes each. This
was followed by sequential immersion in 95%, 85%, and 75% alcohol for 3
minutes each. Antigen retrieval was achieved by boiling the slide in citric acid
(pH = 6.0) for 20 minutes, then cooling to room temperature. Endogenous
peroxidase activity was blocked by incubating the sections in a 3%
H
Extracellular lactate and glucose concentrations were quantified under normoxic
conditions using lactate (solarbio, BC2230, Beijing, China) and glucose
(solarbio, BC2505) assay kits, respectively. Briefly, 1
The measurement of glucose content involved three reagents within the kit. Use as described by the manufacturer of each kit.
The animal studies conducted in this research were approved by the Animal
Research Ethics Committee of Chongqing Medical University. Male BALB/c mice aged
6-weeks were housed and maintained in the experimental animal center of Chongqing
Medical University. A total of 4
A total of 5
T24 cells were digested and centrifuged at 1000 rpm, washed three times with PBS, snap frozen with liquid nitrogen and then stored at –80 ℃. Untargeted metabolomics were performed using Ultra High Performance Liquid Chromatography (UHPLC) (1290 Infinity II, Agilent Technologies, Santa Clara, CA, USA) equipped with a quadrupole time-of-flight mass spectrometer (AB Sciex TripleTOF 6600) at Shanghai Applied Protein Technology Co., Ltd. (Shanghai, China). Cell metabolites were separated on an ACQUITY UHPLC BEH Amide column under optimized conditions. The samples were kept at 4 °C throughout the UHPLC-Mass Spectrometry (UHPLC-MS) analysis. Mass spectrum analysis was performed using the electrospray ionization (ESI) source in both positive and negative modes. Metabolite identification was carried out by converting the raw MS data to mzXML files and analyzing with XCMS software (Mass Consortium, version 4.0.0, La Jolla, CA, USA). Metabolites were identified by comparing their m/z values and MS/MS spectra with an in-house database of authentic standards and metabolic reaction network. All metabolomic sample readings were averaged.
Statistical analysis was performed using Graphpad Prism 8 software (GraphPad
Software Inc., La Jolla, CA, USA). * p
PPP3CB was recently reported to be a key regulator of epithelial-mesenchymal transition (EMT) in G401 cells [15]. However, the function of PPP3CB in bladder cancer remains unclear. Bioinformatic analysis of the TCGA cohort revealed that PPP3CB is significantly downregulated in bladder tumor tissue (Fig. 1A,B). To confirm this result, we performed immunohistochemical (IHC) staining of bladder cancer tissue using tissue microarrays. This revealed that most tumor tissues have lower PPP3CB expression levels compared with their paired normal tissue (Fig. 1C,D), indicating that PPP3CB is a potential suppressor in bladder cancer.
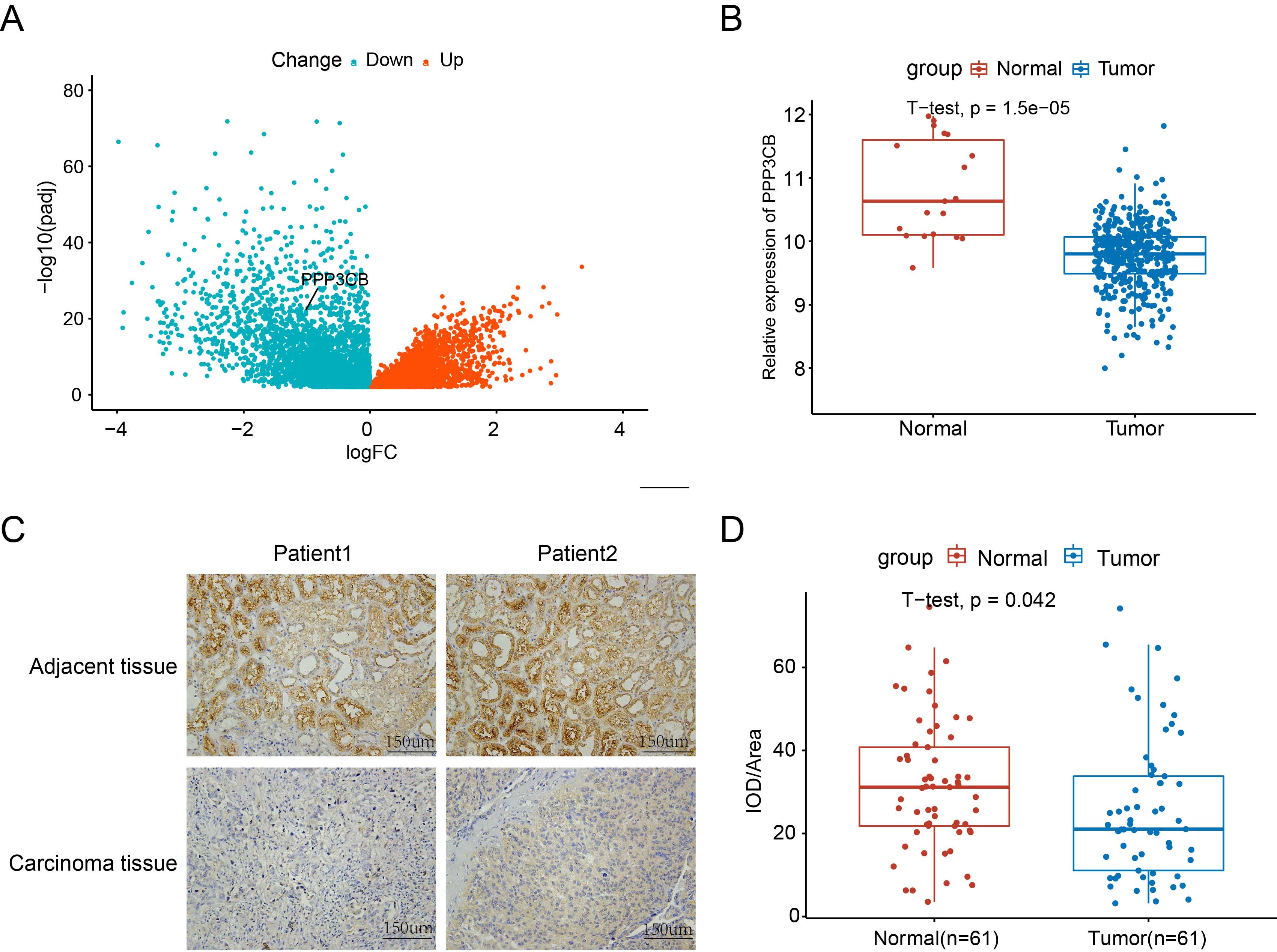 Fig. 1.
Fig. 1.PPP3CB expression is downregulated in bladder cancer.
(A) PPP3CB expression in bladder cancer is low in the The Cancer Genome Atlas (TCGA) cohort. (B)
The PPP3CB expression level in bladder cancer tissue was significantly
lower than in para-cancerous tissue. (C) Immunohistochemical (IHC) staining
showing lower PPP3CB expression in the tumor tissue of bladder cancer
patients compared to adjacent normal tissue. (D) Average integrated optical
density (IOD) in bladder cancer tissue was lower than in adjacent tissue. Error
bars represent the mean
In order to validate the anti-tumor effect of PPP3CB in vitro, we performed PPP3CB overexpression and knockout experiments in T24 cells. Flow cytometry showed that overexpression of PPP3CB promotes the apoptosis of T24 cells (Fig. 2A–C). In contrast, knockout of PPP3CB inhibited the apoptosis of T24 cells. We also investigated the effect of PPP3CB on the proliferation of T24 cells (Fig. 2D–F). Cell viability assays revealed that PPP3CB overexpression inhibited the growth of T24 cells (Fig. 2G), whereas knockout of PPP3CB promoted their proliferation (Fig. 2J). Consistent with these findings, the colony formation assay revealed that PPP3CB overexpression suppressed the colony formation ability of T24 cells (Fig. 2H,I), whereas knockout of PPP3CB promoted colony formation (Fig. 2K,L). Collectively, these results suggest that PPP3CB can inhibit the proliferation of bladder cancer cells in vitro and promote their apoptosis.
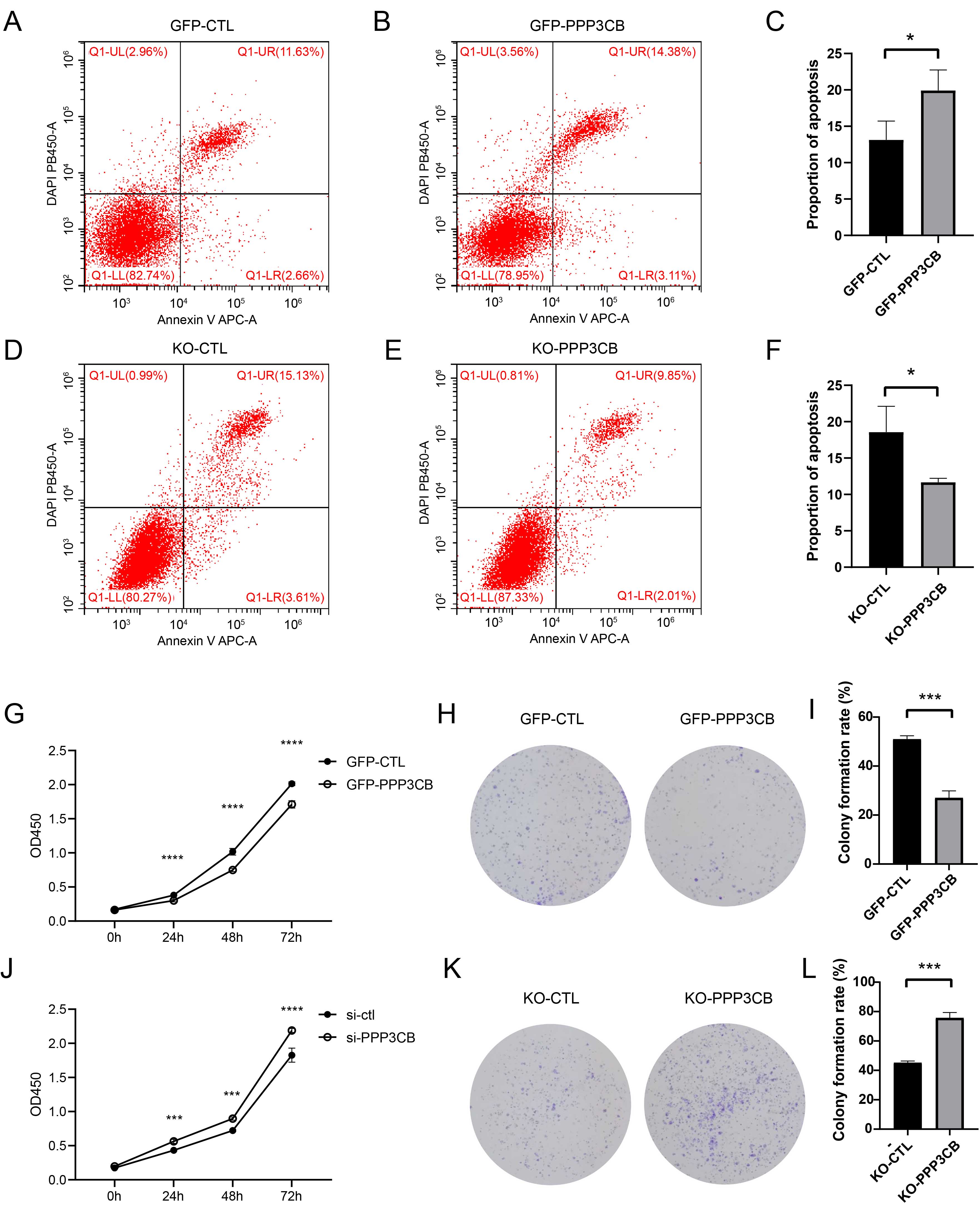 Fig. 2.
Fig. 2.PPP3CB inhibits the proliferation of bladder cancer
cells and promotes their apoptosis. (A–C) PPP3CB overexpression
promotes the apoptosis of T24 cells. (D–F) Knockout of PPP3CB inhibits
the apoptosis of T24 cells. (G) PPP3CB overexpression inhibits the
proliferation of T24 cells. (H,I) Overexpression of PPP3CB inhibits
colony formation of T24 cells. (J) Knockout of PPP3CB promotes the
proliferation of T24 cells. (K,L) Knockout of PPP3CB promotes colony
formation by T24 cells. Error bars represent the mean
To investigate whether PPP3CB deficiency promotes the proliferation of T24 cells through glucose metabolism, we first examined glucose metabolism after knockdown of PPP3CB. Specifically, metabolomics was performed to investigate differences in intracellular metabolites between the negative control and PPP3CB knockdown cells (Fig. 3A). The increase in glycolysis metabolites after PPP3CB knockdown (Fig. 3B) indicates a pivotal role for PPP3CB in T24 cell glucose metabolism. To confirm these results, we also measured glucose uptake and lactate production. Consistent with the above findings, overexpression of PPP3CB reduced glucose uptake and lactose production (Fig. 3C,D), whereas knockdown of PPP3CB increased glucose uptake and lactate production (Fig. 3E,F). Collectively, these results suggest that PPP3CB is a negative regulator of glucose metabolism in T24 cells.
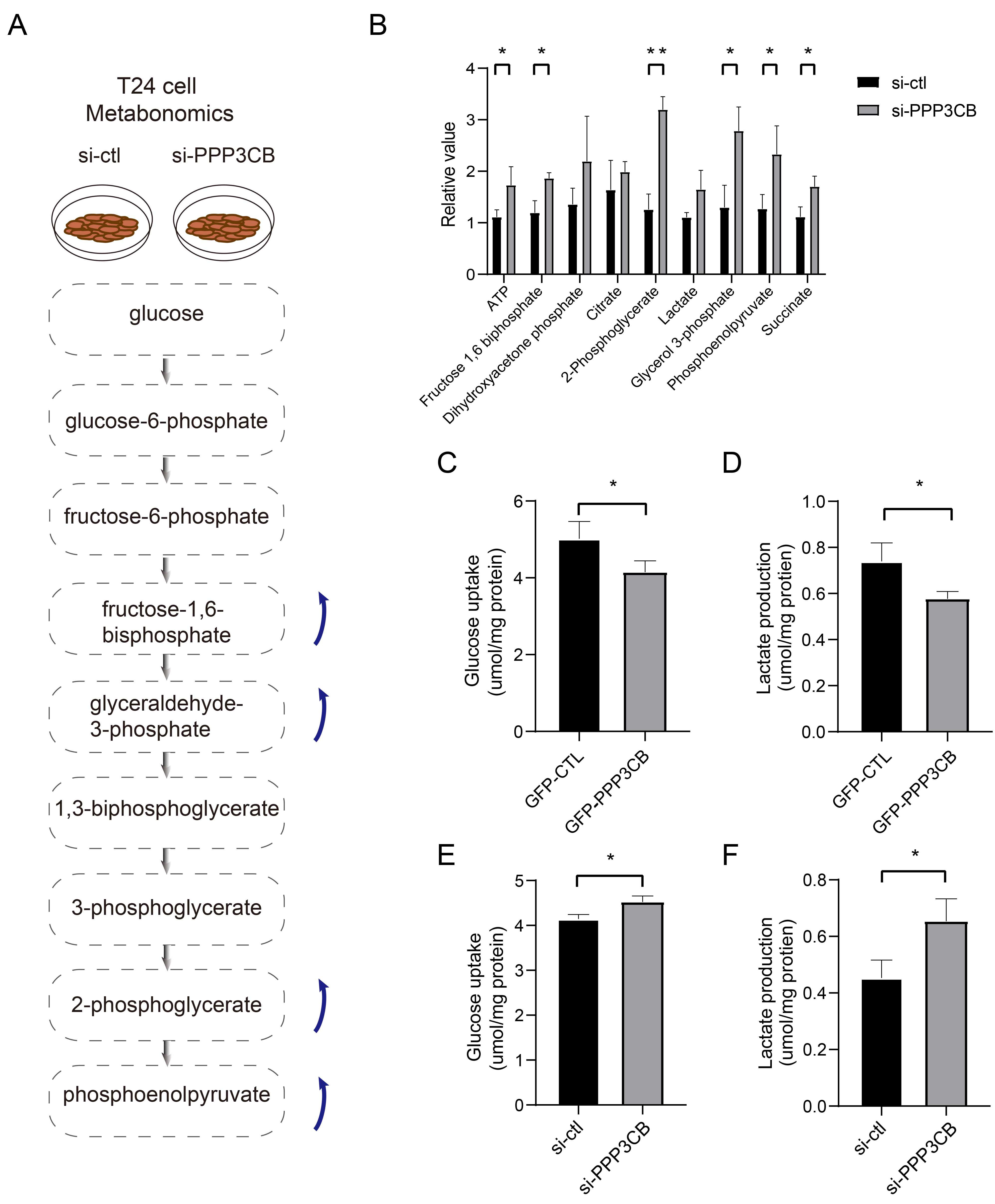 Fig. 3.
Fig. 3.PPP3CB inhibits glycolysis in bladder cancer cells.
(A) Schematic representation of the metabolic pathways and specific metabolites
affected by PPP3CB knockout in T24 cells. (B) Knockdown of
PPP3CB upregulates glycolysis intermediates. (C,D) Overexpression of
PPP3CB decreases glucose uptake and lactate production. (E,F) Knockdown
of PPP3CB enhances glucose uptake and lactate production. Error bars
represent the mean
Due to the upregulation of lactate, we deduced that PPP3CB may affect the expression of LDHA. However, the results showed no difference in LDHA level between the two groups (Supplementary Fig. 1). Therefore, we next examined whether the change in glucose metabolism could be attributed to changes in pyruvate dehydrogenase (PDH). For this, we evaluated the expression levels of pyruvate dehydrogenase E1 subunit alpha1 (PDHA1), phospho-PDHA1, pyruvate dehydrogenase E1 subunit beta (PDHB), and pyruvate dehydrogenase kinase 1 (PDHK1). Interestingly, knockdown of PPP3CB in T24 cells resulted in increased expression levels of PDHK1 and p-PDHA1, but not PDHB or PDHA1 (Fig. 4A, Supplementary Fig. 3). In line with this, knockout of PPP3CB also increased the expression of PDHK1 and p-PDHA1 (Fig. 4B,C). In contrast, the overexpression of PPP3CB in T24 cells led to a reduction in the expression of PDHK1 and p-PDHA1 (Fig. 4D,E). We next investigated whether increased expression of PDHK1 was due to changes in mRNA or protein stability. Quantitative real-time PCR showed the mRNA level increased after knockdown of PPP3CB (Supplementary Fig. 2). Knockdown of PPP3CB also attenuated the degradation of PDHK1 after cycloheximide (CHX) treatment (Fig. 4F,G). These results indicate that knockdown of PPP3CB enhances PDHK1 protein expression by increasing its transcription level and stabilizing protein expression.
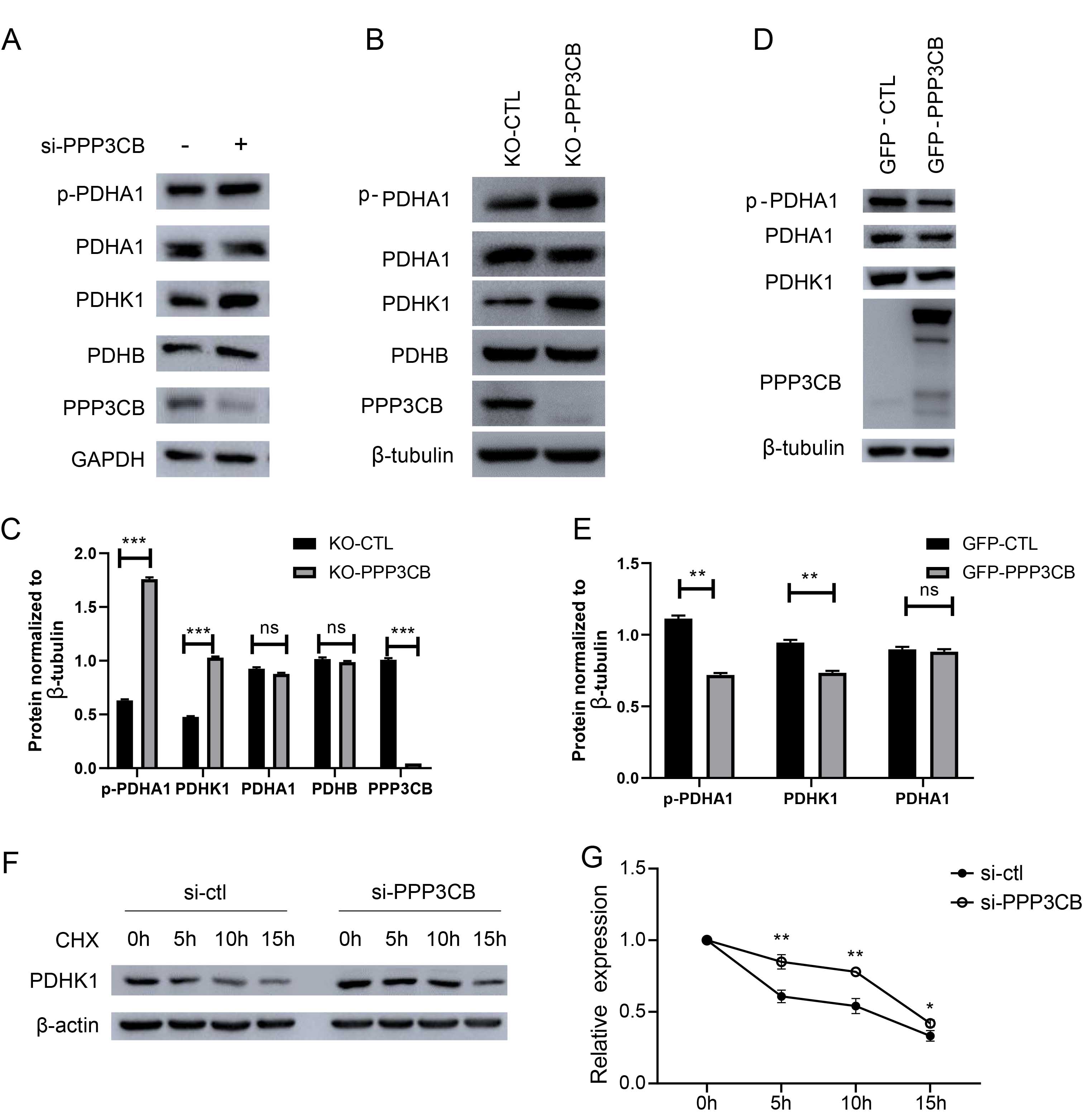 Fig. 4.
Fig. 4.PPP3CB decreases the expression pyruvate dehydrogenase
kinase 1 (PDHK1). (A) Knockdown of PPP3CB increased the
expression of PDHK1 in normoxia and in CoCl
To further study the mechanism by which PDHK1 expression increased, immunofluorescence (IF) was performed to evaluate the co-localization of PPP3CB and PDHK1, while co-immunoprecipitation (co-IP) was used to examine the interaction between these proteins. IF showed that PPP3CB co-localized with PDHK1 in T24 cells (Fig. 5A). Exogenous PDHK1 was also found to interact with endogenous PPP3CB in T24 cells (Fig. 5B). Moreover, exogenous PDHK1 interacted with both exogenous PPP3CB (Fig. 5C) and endogenous PPP3CB (Fig. 5D) in HEK-293T. In line with this, co-IP revealed that exogenous PPP3CB interacted with exogenous PDHK1 in HEK-293T (Fig. 5E). To identify the precise interaction domain of PPP3CB, we constructed plasmids with different deletion mutations of PPP3CB (Fig. 5F). co-IP experiments with HEK-293T cells showed that PPP3CB interacted with the PDHK1 catalytic domain and with the calcineurin B binding domain (Fig. 5G).
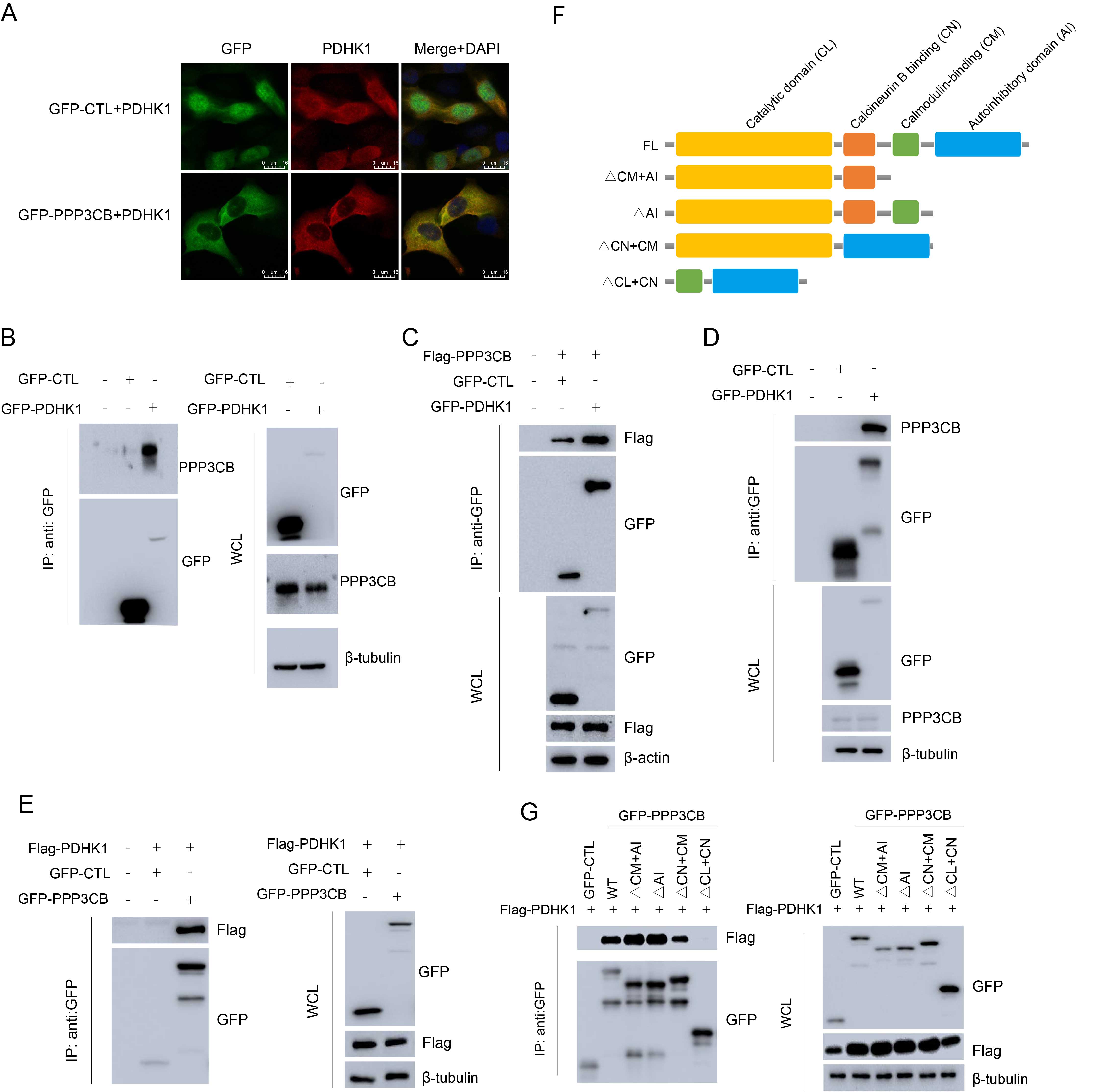 Fig. 5.
Fig. 5.PPP3CB interacts with PDHK1. (A) Immunofluorescence showing the co-localization of PPP3CB with PDHK1 in T24 cells. (B) GFP-PDHK1 interacting with endogenous PPP3CB in T24 cells. (C) GFP-PDHK1 interacting with Flag-PPP3CB in HEK-293T cells. (D) GFP-PDHK1 interacting with endogenous PPP3CB in HEK-293T cells. (E) GFP-PPP3CB interacting with Flag-PDHK1 in HEK-293T cells. (F) The structural domain of PPP3CB. (G) PPP3CB mainly interacts with the catalytic domain of PDHK1 in HEK-293T cells.
Taken together, these results indicate that PPP3CB interacts with PDHK1, resulting in the destabilization and degradation of PDHK1. Hence, the low expression of PPP3CB in bladder cancer cells leads to reduced degradation of PDHK1, thereby promoting the Warburg effect.
Since PPP3CB interacts with PDHK1, we inferred that PPP3CB inhibits glycolysis and the proliferation of bladder cancer cells through PDHK1. We therefore treated T24 cells with DCA, which is an inhibitor of PDHK1 [24]. Both PDHK1 and p-PDHA1 expression were significantly downregulated by DCA (Fig. 6A), with flow cytometry also showing increased apoptosis (Fig. 6B–D). In line with this, the cell viability and colony formation assays showed that proliferation of T24 cells was suppressed by DCA (Fig. 6E–G). Furthermore, treatment with DCA inhibited glucose uptake and lactate production in T24 cells (Fig. 6H,I). As stated previously, overactivated PDHK1 can inhibit PDC, thereby switching tumor cells from oxidative phosphorylation to glycolysis and promoting bladder cancer cell proliferation. The present results confirm that inhibition of PDHK1 with DCA blocks glycolysis and the proliferation of bladder cancer cells.
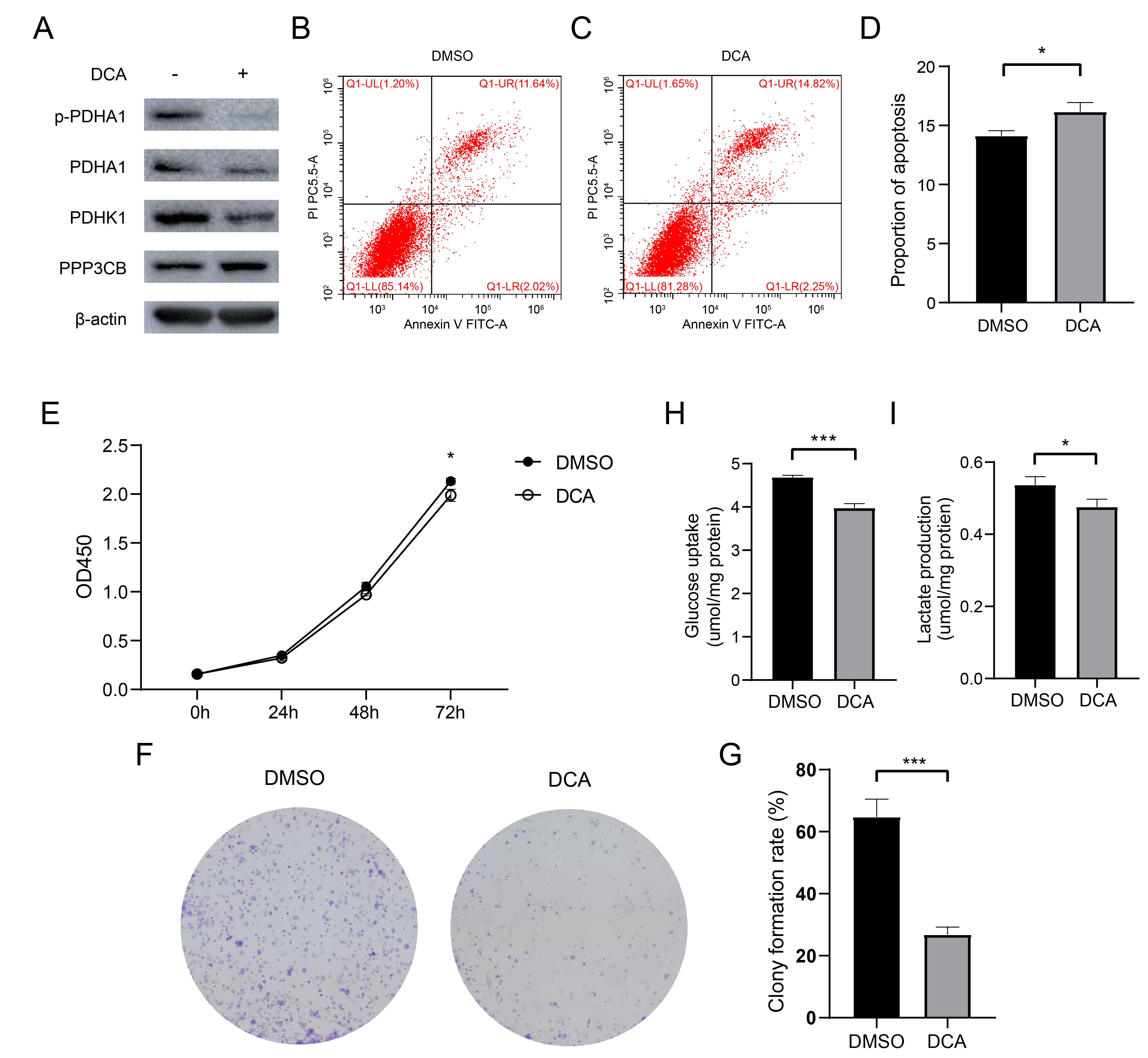 Fig. 6.
Fig. 6.Dichloroacetate (DCA) promotes apoptosis and inhibits proliferation and glycolysis
in T24 cells. (A) Suppression of PDHA1 phosphorylation by DCA treatment of T24
cells for 2 hours. (B–D) DCA treatment increases apoptosis. (E) DCA treatment
inhibits the proliferation of T24 cells. (F,G) Decreased colony number after DCA
treatment. For the Cell Counting Kit-8 (CCK-8) and clonal formation assays, DCA
was added 24 hours after cell seeding. (H,I) DCA treatment (0.5 mmol/L) decreased
glucose uptake and lactate production. *p
In order to confirm that increased glycolysis and cell proliferation induced by PPP3CB knockdown were due to PDHK1, we conducted immunoblot analysis to determine the expression level of p-PDHA1 in T24 cells. DCA was used to inhibit PDHK1 due to its relative specificity for this enzyme. The upregulation of p-PDHA1 after PPP3CB knockdown was found to be blocked by DCA treatment (Fig. 7A, Supplementary Fig. 4). This indicates that PPP3CB upregulates the expression of both PDHK1 and p-PDHA1, whereas DCA treatment reversed this effect. Next, flow cytometry revealed that DCA could block apoptosis resulting from PPP3CB knockdown (Fig. 7B–E). In line with this, the proliferation of T24 cells after PPP3CB knockout was attenuated by DCA treatment (Fig. 7F–H). As expected, glucose uptake and lactate production were significantly upregulated after PPP3CB knockdown, but DCA treatment blocked this effect (Fig. 7I,J). Nevertheless, our findings indicate that PPP3CB deficiency increases glycolysis and the proliferation of T24 cells by upregulating the expression of PDHK1.
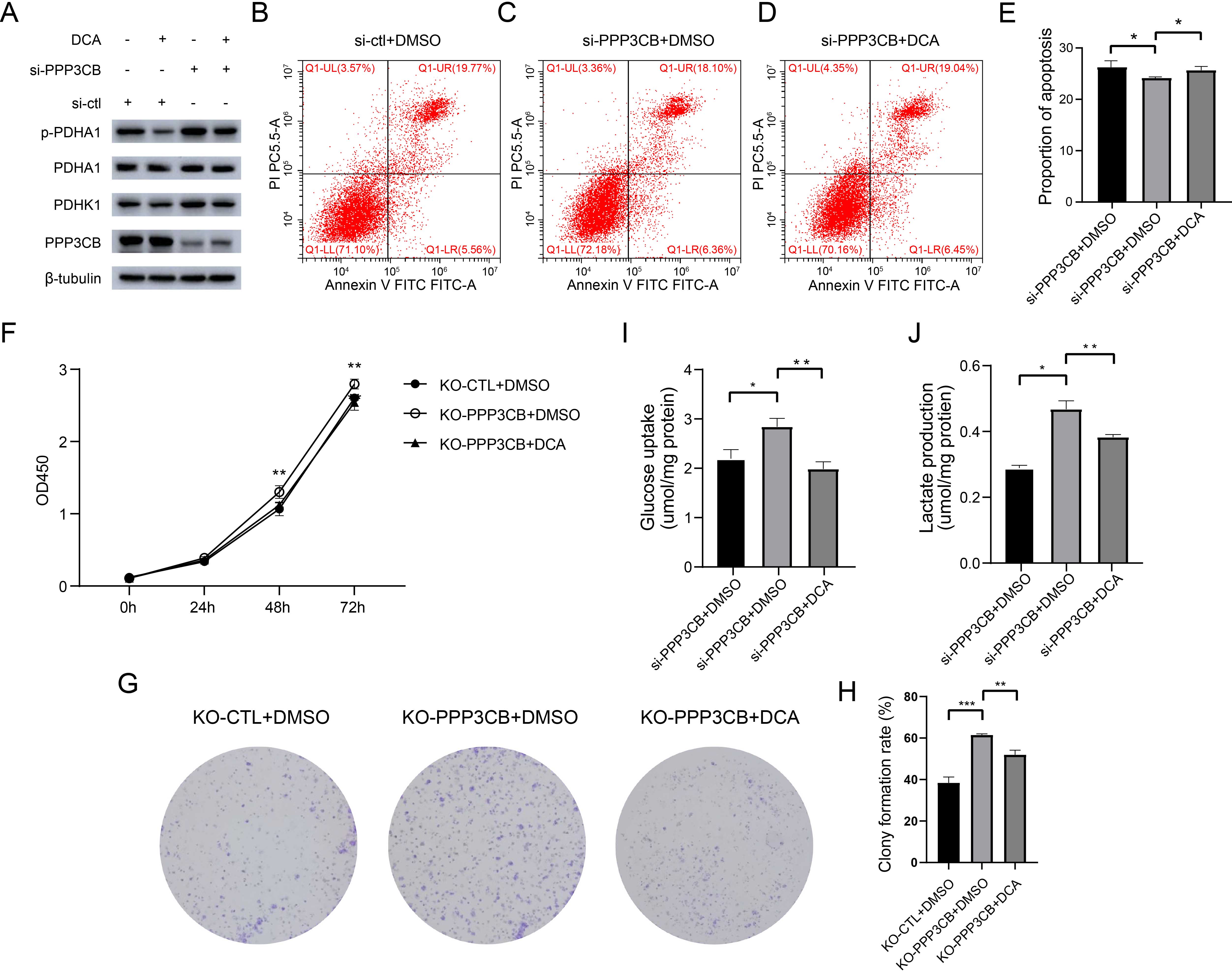 Fig. 7.
Fig. 7.PPP3CB knockdown promotes cell proliferation and
inhibits apoptosis by upregulating PDHK1. (A) DCA blocks the
upregulation of PDHK1 after PPP3CB knockdown in T24 cells. (B–E) DCA
abolishes the effect of PPP3CB knockdown on apoptosis. (F) DCA blocks
the increased proliferation of T24 cells after PPP3CB knockout.
Statistical analysis was performed using a two-way ANOVA with Bonferroni’s post
hoc test. (G,H) DCA abolishes the increased number of T24 cell colonies induced
by PPP3CB knockout. (I,J) DCA blocks the increase in glucose uptake and
lactate production induced by PPP3CB knockdown. *p
To investigate the tumor-promoting effect of PPP3CB deficiency, we established a xenograft mouse model by injecting T24 cells with or without PPP3CB knockout (Fig. 8A, Supplementary Fig. 5). The results showed that injection of T24 cells with PPP3CB knockout resulted in significantly higher tumor weight and volume in nude mice (Fig. 8B,C), thus demonstrating the anti-tumor effect of PPP3CB in vivo.
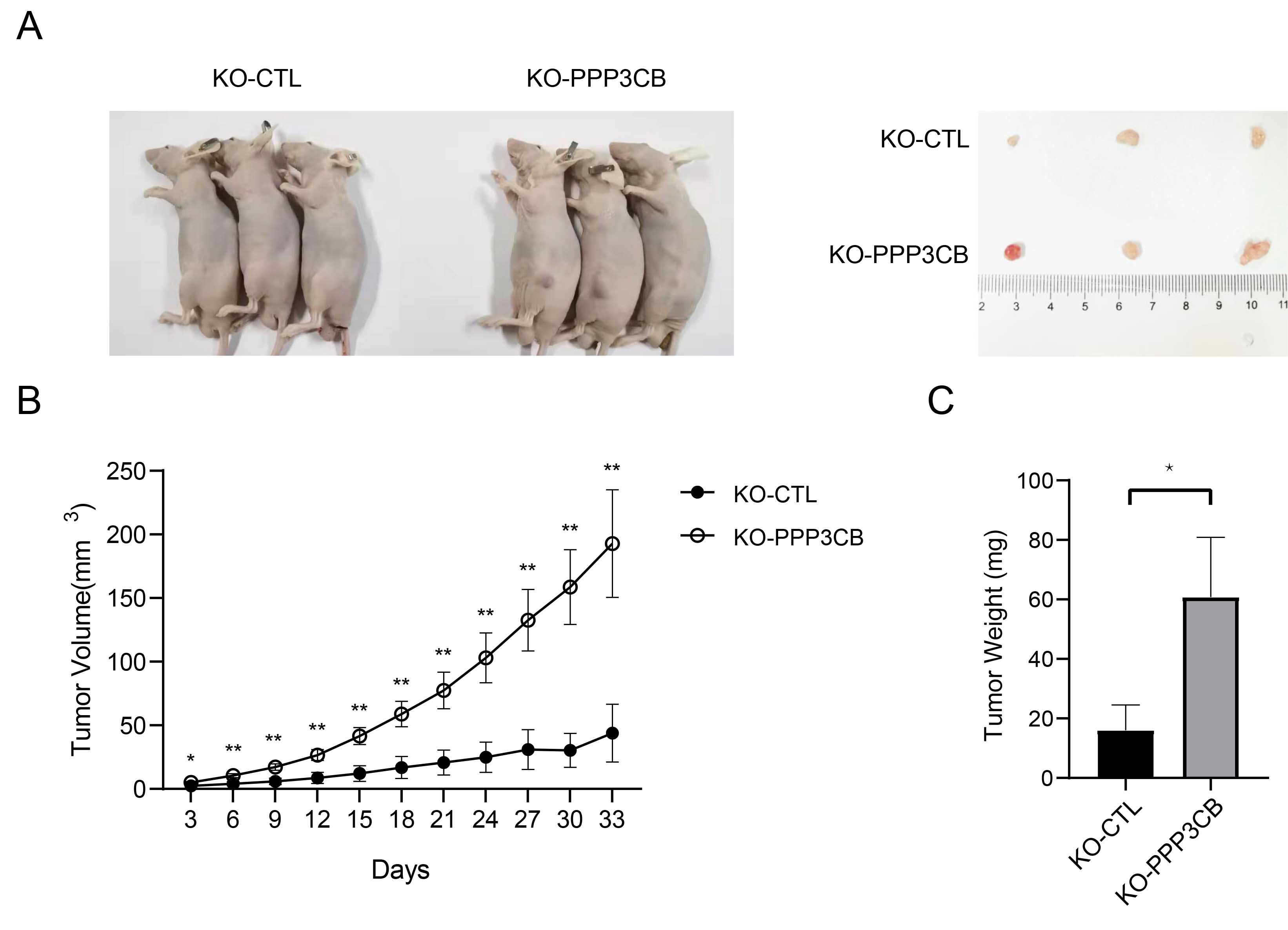 Fig. 8.
Fig. 8.Knockout of PPP3CB promotes the growth of bladder
tumors formed by injection of T24 cells. (A) Sacrificed mice and the
corresponding tumors. (B) Greater tumor volume in PPP3CB knockout
tumors. (C) Increased tumor weight of PPP3CB knockout tumors. Error bars
represent the mean
As shown in Fig. 9, deficiency of PPP3CB stabilizes PDHK1, thereby increasing phospho-PDHA1. This deactivates the PDC, leading to an enhanced Warburg effect and faster growth of bladder tumors.
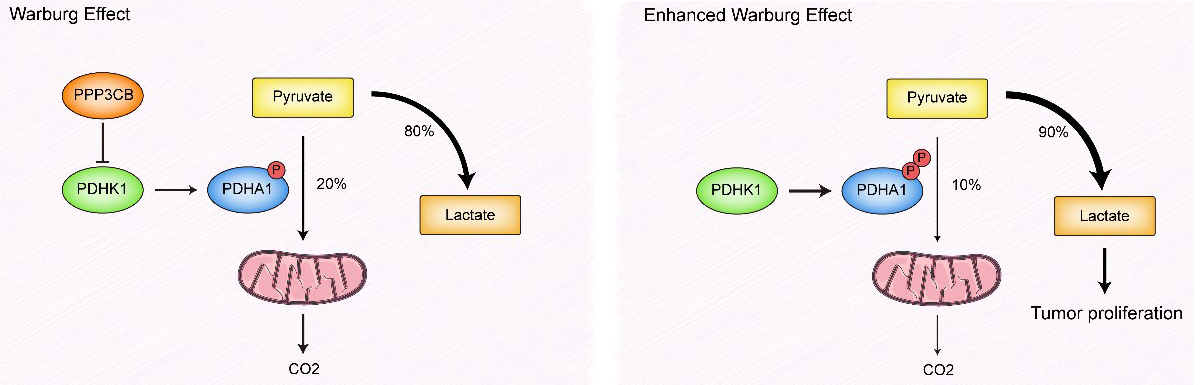 Fig. 9.
Fig. 9.Working model derived from the study results. P represents phosphorylation of PDHA1.
The reprogramming of glucose metabolism in cancer cells towards “aerobic
glycolysis” has received considerable attention in the scientific community,
with the “Warburg effect” recognized as one of the established hallmarks of
cancer [25]. Unlike most normal cells that metabolize glucose to carbon dioxide
instead of lactate, tumor cells produce large amounts of lactate independently of
oxygen supply [26]. This metabolic switch is regulated by oncogenes and tumor
suppressor genes and enables the high rate of proliferation and division of tumor
cells [27]. Bladder cancers also exhibit high lactate production and
downregulation of pyruvic acid [8]. Many biomarkers have been associated with
glucose metabolism in bladder cancer. Cao et al. [28] reported that ROCR
regulates cell proliferation, glucose metabolism and chemoresistance in bladder
cancer, specifically through its involvement in the PD-L1/ITGB6/STAT3 signaling
axis. In addition, Wang et al. [29] highlighted the pivotal role of the
HIF-1
Protein phosphatase 2B (PP2B) can act on a large cross-section of protein
substrates in response to intracellular calcium [30]. Recent research has
revealed that PP2B plays a vital role in cancer. Chen et al. [31]
reported that PP2B dephosphorylates c-Jun in A431 cells to regulate c-Jun/Sp1
interactions. As one of the catalytic subunits of PP2B, PPP3CB has been
reported to inhibit the migration of G401 cells [15]. Furthermore, suppression of
PPP3CB transcription leads to Herceptin resistance in HER2-positive
breast cancer [32]. These studies provide novel insights into the role of
PPP3CB in cancer. PPP3CB is also involved with metabolism.
Pfluger et al. [14] reported that PPP3CB deficiency increases
mitochondrial elongation and respiration, and protects mice from diet-induced
obesity, thus indicating its critical function in lipid metabolism. Although Vasu
et al. [33] reported that hyperglycemia could elevate the mRNA
expression level of PPP3CB in a human pancreatic
Here, we studied the potential involvement of PPP3CB in the glucose
metabolism and cell proliferation of bladder cancer. Based on the TCGA database
and the analysis of human tissues, our findings indicate that PPP3CB
expression is significantly lower in bladder cancer than in paracancerous
tissues. This result was verified by in vitro experiments
showing that PPP3CB inhibited the proliferation of bladder cancer cells
and induced their apoptosis. Subsequently, non-targeted metabolomic analysis of
T24 cells revealed that PPP3CB knockdown can upregulate sugar
metabolites. In a similar manner, knockdown of PPP3CB in bladder cancer
cells resulted in increased lactate production and glucose uptake, whereas
overexpression of PPP3CB had the opposite effect. These findings
indicate that PPP3CB is involved in the regulation of glycolysis in T24
cells. Since the level of LDHA did not change following PPP3CB
knockdown, we assessed the protein expression levels within the PDC, with a
particular focus on PDHK1. PDHK1 has been reported to impair CD8
We therefore propose that increased glycolysis and enhanced proliferation in cells with PPP3CB knockdown are due to elevated PDHK1 expression. DCA, an inhibitor of PDHK1, maintains PDC in an unphosphorylated and active state, thereby reversing the Warburg effect in tumor cells [38]. The present study found that DCA treatment significantly downregulated p-PDHA1 expression. In addition, apoptosis was promoted and proliferation was inhibited in bladder cancer cells treated with DCA, consistent with the results of a previous study [39]. As expected, DCA treatment inhibited glucose uptake and lactate production. Furthermore, we found that DCA treatment reversed the effects of PPP3CB knockdown, i.e., increased cell proliferation, reduced apoptosis and increased glycolysis. These results demonstrate that PPP3CB regulates proliferation and glycolysis in bladder cancer cells through PDHK1. Finally, our in vivo experiments confirmed the pro-tumor effect of PPP3CB deficiency. A limitation of this study is that the underlying mechanism for the increased PDHK1 stability after PPP3CB knockdown is still unclear and requires further in-depth investigation. Whether other molecular mechanisms are involved in the degradation of PDHK1 also requires additional study.
The summary of our findings and conclusions are shown in Fig. 9. A range of
tumorigenic factors have been shown to induce PDC dysfunction in bladder cancer
cells. This results in the majority of pyruvate being directed towards anaerobic
metabolism to produce lactate, rather than CO
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work. QL and TS designed the project, XQ and ZJ performed the experiments, XQ wrote the manuscript, YL interpret part of data for the work, DT analyzed the data. All authors contributed to the revision of this manuscript and approved the final manuscript.
The Ethics Committee of Chongqing Medical University has reviewed the proposed use of human subjects in the projects. It is recognized that the rights and the welfare of the subjects are adequately protected; the potential risks are outweighed by potential benefits. The species, strains, grade, specification and number of the animals to be used are justified. Appropriate animal care throughout the experiment, including anesthetics, sedatives should be used. Disposition of animals at the end of study euthanasia criteria and method is in accordance with the code of practice for the care and use of animals for scientific purposes.
Not applicable.
This work was supported by (1) Chongqing Natural Science Foundation project (NO. cstc2021jcyj-msmX0019). (2) Project Supported by Scientific and Technological Research Program of Chongqing Municipal Education Commission (NO. KJQN202000438). (3) Chongqing Municipal Key Project for Technological Innovation and Application Development (NO. cstc2019jscx-dxwtBX0032).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.