


 , Sinee Disthabanchong 1,*
, Sinee Disthabanchong 1,*
1 Division of Nephrology, Department of Medicine, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Ratchathewi, 10400 Bangkok, Thailand
Abstract
Parathyroid hormone (PTH) is an endocrine peptide found exclusively in the parathyroid glands, whereas parathyroid hormone-related protein (PTHrP) is expressed in a wide range of tissues and organs and exerts endocrine, paracrine, and autocrine actions. PTH and PTHrP have a similar homology, sharing the initial 13 amino acid residues at the N-terminus and binding to the same type 1 PTH receptor (PTH1R), which regulates calcium homeostasis. An abnormal increase in PTH production can occur in primary and secondary hyperparathyroidism, whereas PTHrP can be produced in large quantities by malignant cancer cells from solid organs. In addition to increased bone resorption and hypercalcemia, recent evidence suggests that excess PTH and PTHrP can result in protein-energy wasting, malnutrition, and cachexia. Through binding to PTH1R and activation of cyclic adenosine monophosphate (cAMP)-dependent protein kinase A in white adipose tissue, PTH and PTHrP can stimulate the expression of thermogenic genes causing adipose tissue browning. This change results in an increase in resting energy expenditure, loss of muscle and fat mass, and weight loss. These findings provide a mechanistic link for the long-established relationship between hyperparathyroidism and myopathy, as well as cancer and cachexia. The purpose of this review is to provide a summary of the emerging evidence from both experimental and clinical studies on the role of PTH and PTHrP in protein-energy malnutrition.
Keywords
- protein-energy wasting
- malignancy
- solid tumor
- muscle wasting
- catabolism
- parathyroid
- sarcopenia
- hypoalbuminemia
The parathyroid glands produce parathyroid hormone (PTH), whereas parathyroid hormone-related protein (PTHrP) is produced by a variety of tissue types. They share similar amino-terminal fragments, and the first 13 amino acids bind to the same transmembrane domain of the type 1 PTH receptor (PTH1R). When produced in excess, they both result in increased osteoclast activity, bone resorption, and increased serum calcium [1]. Under physiological conditions, PTH is primarily secreted in response to low-ionized calcium. PTH may be produced in excess in primary and secondary hyperparathyroidism, whereas increased levels of PTHrP is the result of production by various types of solid tumor. Recent findings suggest the role of excess production of PTH and PTHrP in disease-related malnutrition and cachexia. Through binding to PTH1R in adipocytes, PTH and PTHrP activate the thermogenic gene program causing browning of adipose tissue, increased energy expenditure, skeletal muscle wasting, and weight loss [2]. These findings provide an important mechanistic link between hyperparathyroidism and cancer with malnutrition.
This narrative review summarizes the emerging evidence, particularly from the past ten years, including both experimental and clinical studies that link excess production of PTH and PTHrP to malnutrition and cachexia. A literature search was conducted using the PubMed database. The search terms included “PTHrP” with “malnutrition” or “cachexia” and “PTH” with “malnutrition” or “protein-energy wasting”. The background information on the structure and physiological actions of PTH and PTHrP and the physiology and function of adipose tissue in the context of malnutrition, cachexia, and protein-energy wasting are also briefly discussed.
PTH is a peptide hormone produced exclusively by the chief cells in the parathyroid glands. The pre-hormone or pre-pro-PTH (115 amino acids) undergoes processing into a bioactive form, intact PTH (1–84), which is stored in vesicles. The hormone is primarily secreted in response to low ionized calcium, with a half-life of 2–4 minutes which ensures minute-to-minute calcium regulation [3, 4]. A fall in serum calcium triggers the activation of the calcium-sensing receptor, resulting in increased PTH synthesis and secretion [5, 6]. The primary targets of PTH are bone and kidneys, where PTH binds to a G protein-coupled receptor called type 1 PTH receptor (PTH1R). Under normal conditions, the binding of PTH to PTH1R on osteoblasts stimulates osteoblast differentiation and bone formation. However, when PTH is chronically elevated, such as in hyperparathyroidism, the receptor-activated production of nuclear factor kappa-B ligand (RANKL) production by osteoblasts is upregulated, and RANKL stimulates osteoclast precursor cells through binding to the receptor activator of nuclear factor kappa-B (RANK). This binding results in an increase in osteoclast differentiation and bone resorption [7].
In the kidneys, PTH increases serum calcium by promoting active transcellular calcium reabsorption at the distal tubule and increases 1,25-dihydroxyvitamin D synthesis at the proximal tubule. The 1,25-dihydroxyvitamin D then acts on the intestine to enhance calcium absorption [8]. High levels of serum calcium and 1,25-dihydroxyvitamin D, through binding to their respective calcium-sensing and vitamin D receptors in the parathyroid glands, initiate a negative feedback loop to inhibit PTH synthesis [9].
PTH also regulates phosphate metabolism. An increase in PTH leads to the internalization and deactivation of sodium-phosphate co-transporters at the proximal tubule resulting in a decrease in phosphate reabsorption [10]. As a result, hypophosphatemia is commonly observed in patients with primary hyperparathyroidism and normal kidney function [11]. In contrast, patients with secondary hyperparathyroidism from end-stage kidney disease usually have hypercalcemia with hyperphosphatemia due to the limited ability of the kidneys to excrete dietary phosphate load adequately. Increased extracellular phosphate has also been shown to directly stimulate PTH synthesis and secretion via unknown mechanisms [12].
PTHrP was discovered in the 1980s through research attempting to explain the PTH-like bioactivity responsible for hypercalcemia associated with malignancy. This PTH-like protein was identified in the blood and tumors of patients with hypercalcemia. The gene was finally cloned and named parathyroid hormone-related protein or PTHrP [13]. Under normal conditions, PTHrP is undetectable in the blood. However, it is expressed in a variety of tissues, including the cardiovascular system, kidneys, lungs, bladder, uterus, placenta, mammary glands, stomach, pancreas, bone, cartilage, and teeth. Further studies have revealed the diverse endocrine, autocrine, and paracrine functions of PTHrP, particularly during normal development. The important autocrine and paracrine roles of PTHrP include mammary gland development, tooth eruption, keratinocyte differentiation for hair follicle development, chondrocyte maturation, and endochondral bone formation.
Alternative RNA splicing of PTHrP generates three isoforms that encode 1–139, 1–141, and 1–173 amino acid proteins. Post-translational processing creates multiple fragments that contain three functional domains: N-terminal (residues 1–34), mid-range (residues 66–94, 88–106), and C-terminal (residues 107–139, 107–111, 122–139) [14]. Although several have been characterized, little is known about the cell-specific processing of PTHrP or the biological importance of the various PTHrP peptides [15]. N-terminal fragments of PTHrP (1–36) share some amino acid sequences with PTH (1–34). However, beyond this point, the sequences are dissimilar (Fig. 1A). The first 13 amino acids of PTHrP bind to the transmembrane domain of PTH1R and activate receptors enabling PTHrP to have the same effect as PTH. The remaining 14–36 residues are used to dock ligands to the extracellular domain of the receptors [16]. The mid-region PTHrP (38–106) contains a nuclear localization signal that mediates nucleus and nucleolar translocation for both the full-length and the mid-region PTHrP from the cytoplasm. These nuclear proteins can activate the cell cycle, induce vascular smooth muscle cell proliferation, and prolong chondrocyte survival.
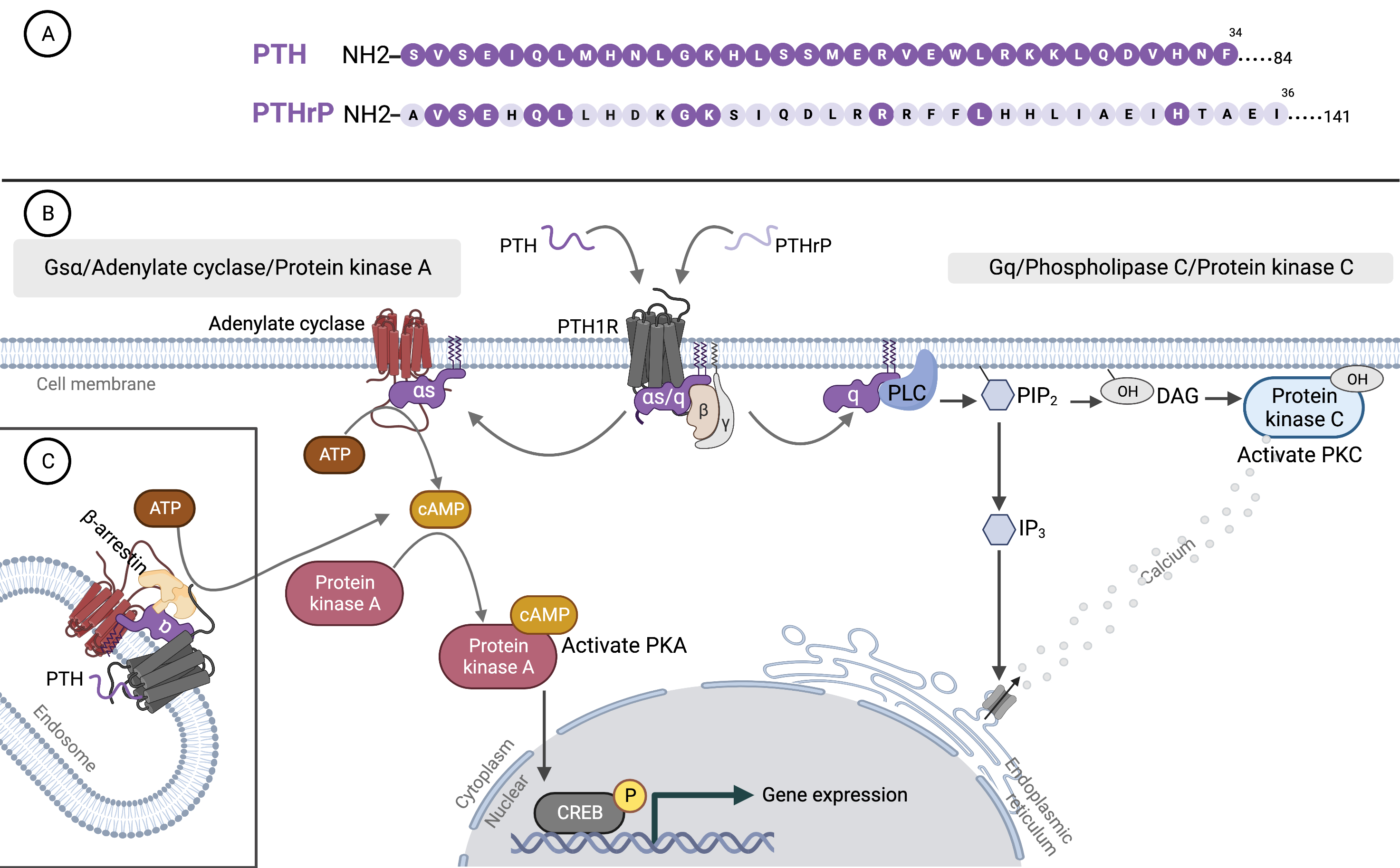 Fig. 1.
Fig. 1.Parathyroid hormone, parathyroid hormone-related protein, and binding to type 1 PTH receptor. (A) Amino acid sequences of the N-terminal region of PTH and PTHrP. The shared sequences with PTH are indicated by dark purple circles on PTHrP. (B) Binding of PTH and PTHrP to G protein-coupled receptor (PTH1R). (C) The two principal signaling pathways of PTH1R: the cyclic adenosine monophosphate (cAMP) and phosphatidylinositol pathways. PTH, parathyroid hormone; PTHrP, parathyroid hormone-related protein; PTH1R, type 1 PTH receptor; ATP, adenosine triphosphate; CREB, cAMP response element binding protein; PLC, phospholipase C; PKC, protein kinase C; PIP2, phosphatidylinositol 1,4,5 trisphosphate; DAG, diacylglycerol; IP3, inositol 1,4,5 trisphosphate; PKA, protein kinase A. The figure was created by BioRender.com.
An additional role of PTHrP in regulating RNA metabolism has been suggested. PTHrP has been reported to bind directly to RNA through a nuclear localizing signal in the nuclei [17]. The C-terminal fragments have been shown to stimulate osteoblast function and regulate bone resorption [18, 19, 20]. One of the most well-studied paracrine functions of PTHrP is the regulation of normal bone remodeling. PTHrP is produced locally by osteoblast progenitors to promote the differentiation of mature osteoblasts and bone formation. Osteoblast-derived PTHrP, in turn, stimulates osteoclast differentiation to increase bone resorption. In cancer, excess production of PTHrP by cells from solid tumors such as lung, breast, and kidney cancers can cause bone resorption and hypercalcemia similar to the actions of PTH. PTHrP-expressed tumor cells that metastasize to the bone also secrete PTHrP locally, which drives osteoclast differentiation via the RANKL-RANK system, causing further bone destruction [21]. The increase in PTHrP production in cancer causes bone resorption and hypercalcemia and promotes cancer progression and metastasis. One important mechanism is the ability of PTHrP to induce the proliferation and survival of cancer cells via autocrine, intracrine, and paracrine actions with and without binding and activation of PTH1R [17].
PTH1R is a transmembrane protein that belongs to family B of the G
protein-coupled receptors [22]. The binding of PTH/PTHrP to PTH1R induces a
conformational change in the receptor and activates G-proteins. The cyclic
adenosine monophosphate (cAMP) and the phosphatidylinositol pathways are the
principal signal transduction pathways. G
In addition to bone and kidneys, PTH1R is expressed at low levels in other tissues at various periods of development. In adults, the action mediated by PTH1R binding in bone and kidneys results in the homeostatic maintenance of blood calcium levels. This is distinct from that mediated by PTH1R in developing tissues. Fetal expression of PTH1R enables the paracrine function of PTHrP during normal development, as mentioned previously [25]. The existence of PTH1R in adipose tissue and the ability of PTH to induce adipocyte lipolysis through the activation of the cAMP pathway has been documented previously [26]. The emerging role of excess production of PTH and PTHrP in the browning of white adipose tissue will be discussed in the following section.
Currently, adipose tissue is considered an endocrine organ as it secretes
several bioactive adipokines and lipokines into blood circulation. There are two
types of adipose tissue, white and brown. White adipose tissue is found below the
skin (subcutaneous) or in the trunk (visceral) and plays a key homeostatic role
in ensuring efficient energy storage in the form of triglycerides which can be
mobilized quickly to meet metabolic demands. Brown adipose tissue functions to
dissipate energy in the form of heat (thermogenesis). Adipose tissue is dynamic
and can adapt to different environmental, nutritional, and energy requirements.
During periods of energy demand, it mobilizes lipids and activates lipolysis and
fatty acid
Malnutrition is a state resulting from the lack of intake or uptake of nutrients that leads to weight loss and altered body composition. Malnutrition can be classified according to etiology into three types: disease-related malnutrition with inflammation, disease-related malnutrition without inflammation, and malnutrition without disease [28]. Cachexia is no longer considered the end-stage of malnutrition but rather a multifactorial syndrome associated with underlying illnesses. More recently, cachexia has been conceptualized as a type of disease-related malnutrition associated with chronic inflammation [29]. Cachexia is a common manifestation of several serious health conditions, including cancers, end-stage heart, lung, and kidney disease, severe brain injuries, chronic infections, and autoimmune diseases. Cachexia is one of the main contributors to human morbidity and mortality [30]. Cancer cachexia occurs due to complex interactions between systemic inflammation, imbalance of catabolic and anabolic pathways, host immune responses, and genetic factors. The manifestations of cancer cachexia include anorexia, loss of appetite, fatigue, and weight loss due to muscle and fat mass loss. Cancer cachexia may be progressive and cannot be fully reversed by nutritional support alone but may be ameliorated by eliminating underlying causes [31].
The concept of protein-energy wasting was proposed in 2007 by the International Society of Renal Nutrition and Metabolism as a state of nutritional and metabolic derangements in patients with chronic kidney disease. Protein-energy wasting is characterized by the simultaneous loss of systemic body protein and energy stores, leading to the loss of muscle and fat mass [32]. Protein-energy wasting should be differentiated from malnutrition because, in addition to inadequate nutrient intake due to anorexia and/or dietary restrictions, factors related to chronic kidney disease, including hypercatabolism, inflammation, uremic toxins, dialysis, and hyperparathyroidism also contribute to this condition [33]. The presence of protein-energy wasting is a significant risk factor for weakness, low quality of life, hospitalization, and mortality [34, 35].
In patients with primary hyperparathyroidism, the production of PTH is unregulated primarily due to the presence of a parathyroid adenoma or hyperplastic parathyroid glands. In chronic kidney disease, the increase in PTH production is secondary to the decline in renal function. An increase in fibroblast growth factor-23 production, suppression of 1,25-dihydroxy vitamin D synthesis, and phosphate retention contribute to secondary hyperparathyroidism. Chronic stimulation of parathyroid glands results in glandular enlargement from hyperplasia and nodular transformation [36]. Myopathy has been described in primary hyperparathyroidism since 1974 [37]. Previous animal studies have reported impaired energy production, transfer, and utilization along with enhanced proteolysis of skeletal muscle by PTH [38, 39]. In patients with kidney failure and severe secondary hyperparathyroidism, resting energy expenditure increases compared to mild to moderate hyperparathyroidism [40]. Elevated resting energy expenditure leads to skeletal muscle wasting through enhanced fat and protein catabolism. Parathyroidectomy has been shown to result in a decline in resting energy expenditure within six months, followed by an increase in body weight and an improvement in muscle weakness [40, 41, 42]. The influence of primary and secondary hyperparathyroidism on glucose, fat, and protein catabolism has been explored since the 1980s. Patients with kidney failure and secondary hyperparathyroidism had increased catabolism with increased rates of glucose and fat turnover, whereas patients with primary hyperparathyroidism showed limited suppression of endogenous glucose turnover and higher rates of net protein loss [43].
Recently, an elegant study performed in both animals and patients with primary
hyperparathyroidism has revealed the mechanisms underlying these findings.
Adipose tissue browning, a phenotypic change from white adipose tissue to a brown
adipose-like phenotype, appears to play a major role. The browning of white
adipose tissue is associated with an increase in the expression of mitochondrial
uncoupling protein 1 (UCP-1), which favors thermogenesis instead of ATP
synthesis, leading to lipid mobilization and increased energy expenditure. In a
mouse model of primary hyperparathyroidism, overexpression of the PTH
gene resulted in significant weight loss not due to reduced food intake or
increased activity but rather the increase in resting energy expenditure,
especially at night. White adipose tissue in these mice showed morphological
transition with increased expression of Ucp 1 as well as other
thermogenic genes, including Pgc1a, Cidea, and Dio2
(Fig. 2). In the same study, the direct effects of the active PTH fragments
(1–34) and full-length PTH (1–84) on the transition to brown adipocytes were
confirmed in vitro. Further investigation into the correlation between
serum PTH levels and body mass index (BMI) in a cohort of 496 patients with
primary hyperparathyroidism revealed lower BMI among patients in the highest
tertile of PTH compared with those in the lowest tertile. The prevalence of low
BMI (
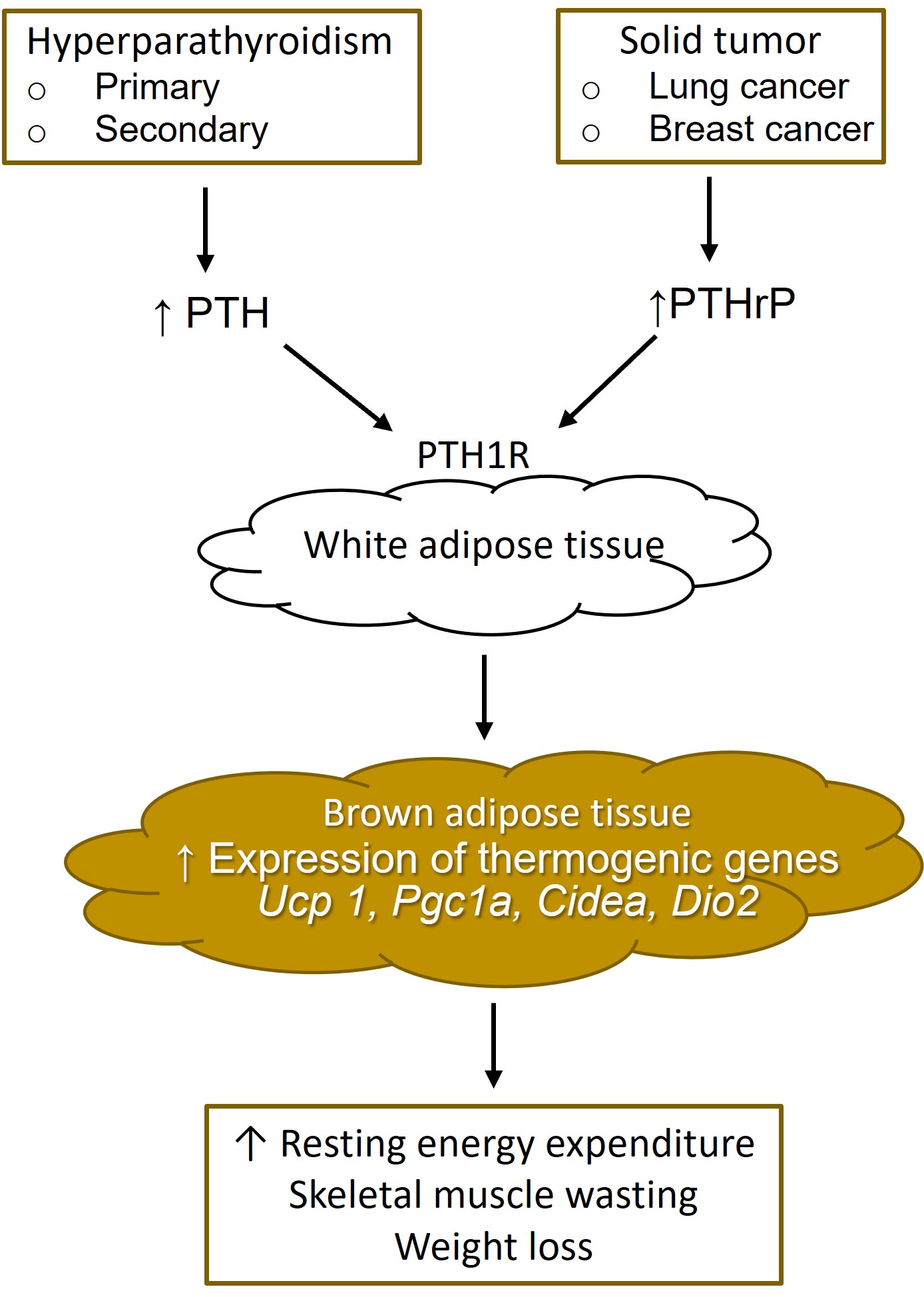 Fig. 2.
Fig. 2.White adipose tissue browning from increased parathyroid hormone and parathyroid hormone-related protein. PTH, parathyroid hormone; PTHrP, parathyroid hormone-related protein; PTH1R, type 1 PTH receptor.
In secondary hyperparathyroidism, a 5/6 nephrectomy mouse model of kidney failure, which involved the removal of one kidney and two-thirds of the other, was used to investigate the role of PTH in adipose tissue browning and protein-energy wasting. Similar to the mouse model of primary hyperparathyroidism, nephrectomized mice with elevated PTH levels suffered from weight loss accompanied by an increase in energy expenditure that was not due to reduced food intake or increased activity. The weight of white adipose tissue and muscle mass also decreased significantly. An increase in the expression of thermogenic genes, including Ucp1, Dio2, Cidea, and Pgc1, was observed. PTH (1–34) and full-length PTH (1–84) directly stimulated Ucp1, Dio2, and Pgc1a mRNA levels in primary inguinal fat cells. The stimulation of UCP1 protein expression and cellular respiration occurred through a mechanism involving the downstream PKA signaling cascade of PTH1R. Furthermore, PTH administration led to an induction of thermogenic genes in various fat tissues. To further explore the role of PTH signaling through PTH1R in adipocytes, fat cell-specific PTH1R-knockout mice were created. PTH failed to upregulate Ucp 1 in isolated fat cells from these mice. These findings indicated that PTH depended on PTH1R to drive the thermogenic gene program. The PTH1R-knockout, 5/6 nephrectomy mice were also resistant to adipose tissue browning and muscle wasting [2].
Recent clinical studies have confirmed the correlation between an increased PTH
level and protein-energy wasting, especially among patients with an advanced
stage of chronic kidney disease. The proportion of patients with low skeletal
muscle mass index examined by bioelectrical impedance analysis was substantially
higher in patients with chronic kidney disease stage 5 compared with stages 1–4
and a high PTH level was an independent risk factor for low skeletal muscle mass
[47]. Epidemiological data from 42,319 patients with end-stage kidney disease
receiving hemodialysis revealed an inverse association between baseline PTH level
and 12-month weight change. Higher degrees of weight loss were observed among
patients with higher PTH levels, and the relationship was more pronounced among
those with preserved appetite [48]. The relationship between increased PTH level
and appetite remains unclear. Another study that included older hemodialysis
patients (average age 60.6
Cachexia is common in patients with cancer. The mechanisms of cancer-associated cachexia are complex and remain poorly understood. Cancer cells produce a proteolysis-inducing factor that inhibits protein synthesis and accelerates proteolysis in skeletal muscle. Proinflammatory cytokines also accelerate proteolysis via the ubiquitin-proteasome pathway and enhance glycogen synthesis in the liver via insulin resistance. Lipid-mobilizing factor produced by cancer cells promotes lipolysis converting triglycerides into fatty acids and browning of white adipose tissue, which is more thermogenic. Furthermore, the consumption of glucose by cancer cells depletes glycogen in the liver, further increasing glycogenesis and promoting the degradation of fat and skeletal muscle. Adipose tissue wasting is characteristic of cancer-associated cachexia, and the loss of adipose tissue is typically faster than the loss of skeletal muscle [57].
High levels of PTHrP can be produced by solid tumor cells such as lung, breast, and kidney cancers. An earlier experimental study had linked PTHrP to reduced food intake through an effect in the hypothalamus. Peripherally administered PTHrP in rats activated hypothalamic urocortins 2 and 3 through vagal afferent pathways causing suppression of gastroduodenal motor activity and delayed gastric emptying. This resulted in negative energy balance, weight loss, and decreased fat and skeletal muscle mass [58].
A landmark study that used a Lewis lung carcinoma (LLC) cell model that readily forms tumors in mice has unraveled an additional role of PTHrP in cancer cachexia. These mice suffered from increased resting energy expenditure, elevated heat production, wasting of adipose tissue and skeletal muscle, weight loss, and cachexia. Both white and brown fat exhibited elevated expression of thermogenic genes. The skeletal muscle showed increased expression of atrophy-related genes, including myostatin (Mstn), atrogin-1 (Fbxo32), and MuRF-1 (Trim63). Further cell culture experiments confirmed that LLC tumor-derived factors induced thermogenic gene expression in white adipocytes. Using a combination of cell cloning and gene expression profiling of LLC cells, among several members of the epidermal growth factor (EGF) family, PTHrP emerged as the inducer of thermogenic gene expression comparable to norepinephrine, the classic thermogenic catecholamine produced by the sympathetic nervous system (Fig. 2). Injecting the tumor-bearing mice with a neutralizing antibody against PTHrP prevented adipose tissue and skeletal muscle wasting, weight loss, and cachexia implicating the causal role of PTHrP in thermogenesis and fat wasting. The treatment with anti-PTHrP antibody in cachectic mice also lowered oxygen consumption, improved physical activity, and reduced heat production. In a cohort of patients with metastatic non-small cell lung cancer or colorectal cancer, lower lean body mass and higher energy expenditure were observed in patients whose PTHrP was detected in the blood compared with those lacking detectable PTHrP [59]. Further study by the same group also confirmed the signaling of PTHrP through PTH1R. Deletion of PTH1R in fat tissue prevented the upregulation of thermogenic genes and atrophy-related genes, resulting in the preservation of fat, muscle mass, and muscle strength. The upregulation of thermogenic genes, adipose tissue and skeletal muscle wasting, weight loss, and cachexia were prevented in the LLC model of PTH1R knockout mice [2].
In another study using LLC cell culture, LLC cell-derived extracellular vesicles (EVs) could induce lipolysis in 3T3-L1 adipocytes. EVs fused directly with 3T3-L1 adipocytes and transferred PTHrP, activating the PKA/cAMP signaling pathway. Blocking PTHrP activity using neutralizing antibodies and knocking down PTH1R expression prevented lipolysis. Inhibition of the PKA signaling pathway also prevented the lipolytic effects of EVs. In vivo, suppression of LLC-EVs release by knocking down Rab27A halted white adipose tissue browning and lipolysis [60]. In lung cancer, EGF receptor (EGFR) activation leads to lung cancer progression. EGFR tyrosine kinase inhibitors, which block the activation of downstream signaling induced by EGFR, are currently the first-line treatment of non-small-cell lung cancer. LLC mice treated with EGFR tyrosine kinase inhibitor showed decreased PTHrP mRNA expression and improved muscle mass and strength [61]. Two clinical studies in patients with different types of cancer confirmed an association between positive PTHrP level and weight loss [62, 63]. A recent study comparing men with gastrointestinal or genitourinary cancer with weight loss, without weight loss, and non-cancer patients revealed cancer patients with weight loss had less adipose tissue and higher levels of adipose triglyceride lipase and browning markers when compared with the other two groups. In contrast to the previous studies, there was no difference in resting energy expenditure, circulating free fatty acids, PTHrP, thermogenic genes or protein expression. Moreover, circulating PTHrP was not correlated with the expression of thermogenic genes in white adipose tissue [64]. These contradicting results have raised doubt about the generalizability of the findings from the lung cancer model to other types of cancer.
In conclusion, in situations where PTH and PTHrP are produced in excess, the binding to PTH1R in adipocytes upregulates thermogenic genes causing adipose tissue browning and stimulates lipolysis resulting in an increase in resting energy expenditure, loss of fat, and muscle mass. Lowering the PTH level or treating cancer with medications and/or surgery may prevent adipose tissue and skeletal muscle wasting. Neutralizing PTHrP may provide a novel therapeutic approach to mitigate cachexia in patients with incurable cancer.
PS and SD conceptualized, reviewed the intellectual content critically, and drafted the manuscript. PS and SD revised the manuscript. PS and SD have read and agreed to the published version of the manuscript.
Not applicable.
We would like to thank Dr Paul S Edwards for advice and assistance in writing and editing of the manuscript.
This research received no external funding.
SD received honoraria and travel grants from Kyowa Kirin.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.