
 , Mohan P. Singh 4,*
, Mohan P. Singh 4,*1 Centre of Bioinformatics, University of Allahabad, 211002 Prayagraj, Uttar Pradesh, India
2 Centre of Experimental Medicine and Surgery, Institute of Medical Sciences, Banaras Hindu University, 221005 Varanasi, Uttar Pradesh, India
3 Faculty of Biotechnology, University of Agricultural Sciences and Veterinary Medicine, 011464 Bucharest, Romania
4 Centre of Biotechnology, University of Allahabad, 211002 Prayagraj, Uttar Pradesh, India
†These authors contributed equally.
Abstract
Background: Breast cancer is one of the most common types of cancer among women worldwide, and its metastasis is a significant cause of mortality. Therefore, identifying potential inhibitors of proteins involved in breast cancer metastasis is crucial for developing effective therapies. BUB1 mitotic checkpoint serine/threonine kinase B (BUB1B) is a key regulator of mitotic checkpoint control, which ensures the proper segregation of chromosomes during cell division. Dysregulation of BUB1B has been linked to a variety of human diseases, including breast cancer. Overexpression of BUB1B has been observed in various cancer types, and its inhibition has been shown to induce cancer cell death. Additionally, BUB1B inhibition has been suggested as a potential strategy for overcoming resistance to chemotherapy and radiation therapy. Given the importance of BUB1B in regulating cell division and its potential as a therapeutic target, the development of BUB1B inhibitors has been the focus of intense research efforts. Despite these efforts, few small molecule inhibitors of BUB1B have been identified, highlighting the need for further research in this area. In this study, the authors aimed to identify potential inhibitors of BUB1B from mushroom bioactive compounds using computational methods, which could ultimately lead to the development of new treatments for breast cancer metastasis. Methods: This study has incorporated 70 bioactive compounds (handpicked through literature mining) of distinct mushrooms that were considered and explored to identify a suitable drug candidate. Their absorption, distribution, metabolism and excretion (ADME) properties were obtained to predict the drug-likeness of these 70 mushroom compounds based on Lipinski’s rule of 5 (RO5). Screening these bioactive compounds and subsequent molecular docking against BUB1B provided compounds with the best conformation-based binding affinity. The best two complexes, i.e., BUB1B-lepitaprocerin D and BUB1B-peptidoglycan, were subjected to molecular dynamic simulations. Both complexes were assessed for their affinity, stability, and flexibility in protein-ligand complex systems. Results: The molecular dynamic (MD) simulation studies revealed that lepitaprocerin D has an energetically favorable binding affinity with BUB1B. Results showed that the formation of a hydrogen bond between residues ASN123 and SER157, and lepitaprocerin D had strengthened the affinity of lepitaprocerin D with BUB1B. Conclusions: This study identified lepitaprocerin D as a potential and novel inhibitor for BUB1B that could be a plausible drug candidate for identifying and controlling the spread of breast cancer metastasis.
Keywords
- lepitaprocerin D
- molecular docking
- molecular dynamics simulation
- BUB1B
- ADMET
- aneuploidy
- tumorigenesis
The spindle assembly checkpoint (SAC) is deployed by cells during mitosis to prevent segregation errors resulting from unattached or improperly attached chromosomes [1]. The SAC remains active at the kinetochore until it becomes stably attached to the spindle apparatus. Ultimately, SAC satisfaction leads to the release of cell division cycle protein 20 (CDC20) from the inhibitory mitotic checkpoint complex allowing activation of the anaphase-promoting complex, or cyclosome (APC/C) [2]. Once activated by CDC20, the APC/C E3 ubiquitin ligase targets several proteins for degradation, including securin, ultimately leading to sister chromatid separation and triggering anaphase onset. SAC activity at the kinetochore is orchestrated by a network of protein interactions and the activity of several protein kinases, including Mucopolysaccharidosis Type-1 (MPS1), Aurora B, and BUB1. MPS1 phosphorylation of Mellitin (MELT) recruits the knl1 kinetochore protein and enables SAC activation by recruiting other SAC proteins, such as Bub1 [2]. Aurora B is localized to centromeres via a combination of Haspin-mediated phosphorylation of histone H3 (H3pT3) and Bub1-mediated phosphorylation of histone H2A at Thr120 (H2ApT120), where it is required to promote correct kinetochore attachment and regulate the SAC.
Similar to Aurora B, Bub1 has been described as having a dual role in the SAC and chromosome alignment. While its contribution to chromosome alignment has been consistently demonstrated, studies have yielded conflicting results regarding the requirement of Bub1 for the SAC [3]. Generation of conditional knockout mouse embryonic fibroblasts (MEFs) and RNA interference (RNAi) knockdown from Henrietta Lacks (HeLa) and Human retinal pigment epithelial-1 (RPE1) cells found Bub1 to be essential for the SAC. Conversely, initial clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9 (CRISPR-CAS9) genome editing approaches in RPE1 and near-haploid human cell line (HAP1) cells suggested only a minor role for Bub1 in the SAC when cells were sensitized via Mps1 inhibition. These conflicting results were initially reconciled by the discovery that nonsense-associated alternative splicing allows for Bub1 expression following CRISPR-CAS9. The short interfering RNA (siRNA) knockdown of residual BUB1 significantly impaired SAC response in BUB1-disrupted cells. However, more recently, HAP1 cells with several BUB1 exons absent from genomic DNA were created following the use of two guide RNAs for CRISPR-CAS9 [4]. Surprisingly, the SAC remained functional in these cells, even when the more extensive approach was combined with Bub1 siRNA knockdown [4]. However, the generation of a complete BUB1 deletion was only possible in haploid HAP1 cells but not in several other cell lines.
Despite the controversy, these experimental systems have allowed functional evaluation of BUB1. BUB1 has a Bub3-binding domain through which BUB1 is localized to kinetochores [5]. The BUB1 central region acts as a scaffold for BUB1 -mediated localization of the Rod-Zw10-Zwilch (RZZ) complex, mitotic arrest deficiency 1/2 (Mad1/2), and CDC20 localization to kinetochores, and is required for the SAC function of BUB1. Numerous reports have found BUB1 kinase activity dispensable for SAC activation; however, others suggest that Bub1-mediated phosphorylation of CDC20 may directly contribute to APC/C inhibition [6]. Similarly, there are conflicting reports on whether Bub1 kinase activity is required for chromosome alignment. Bub1 H2ApT120 phosphorylation localizes Shugoshin 1/2 (Sgo1/2) and Aurora B, and other proteins of the chromosome passenger complex (CPC), to centromeres and may integrate correction of attachment errors with SAC signaling [7, 8, 9]. Histone H2A threonine 120 phosphorylation (H2ApT120) is required to maintain centromeric Aurora B and SAC activity in the absence of histone H3 at threonine 3 (H3pT3). Finally, Bub1 is also autophosphorylated, both within the kinase domain activation segment and outside of this segment, which may have a role in regulating Bub1 localization [8].
Functional characterization of Bub1 would benefit from small-molecule kinase inhibitors. Drugs targeting Mps1 and Aurora B have been powerful tools for deciphering kinase function and dissection of mitosis. A potent, specific Bub1 kinase inhibitor is of particular value since complete penetrance of genetic deletions or siRNA has been difficult in human cells, and only 4% of residual Bub1 is needed for SAC activity [10]. However, considering multiple conflicting reports regarding the function of Bub1, it is important that the inhibitors used to evaluate its function are properly validated [11]. Availability of potential drugs against BUB1 mitotic checkpoint serine/threonine kinase B (BUB1B) can help to suppress the possibilities of cancer and its metastasis. Thus, it is necessary to screen for the identification of potential molecules which can bind BUB1B and inhibit its activities [12, 13, 14].
Using natural compounds as the drug is always beneficial and preferable as they are comparatively safer in terms of their pharmacokinetics and dynamics. Studies reported that various edible mushrooms had considerable anticancer properties [15]. Over the last two decades, various reports have shown the antitumor properties of these edible mushrooms, which are most remarkable and seek the scientific community’s attention worldwide [1, 16]. These edible mushroom extracts’ cost-effectiveness, natural occurrence, and negligible side effects make them favorable for medicinal use. Pleurotus ostreatus is one of the most cultivated and widely used edible mushrooms [17, 18, 19] which has various medicinal properties that include antitumor [8], antiatherogenic, antioxidative, and hypocholesterolemic activities [2, 3]. Earlier in 2008, Jedinak et al. [14] reported that P. ostreatus possesses an inhibitory effect on human breast and colon cancer [10]; later, P. ostreatus extract was also explored for its anticancer effects on other cancers and found to be potent against erythroleukemia [6, 20], and human gastric cancer [5, 7, 18]. Over the last decade, various structure-based in silico approaches, i.e., homology modeling, molecular docking, and MD simulation, are already in practice as a potential tool to screen compounds against a receptor protein with substantial accuracy [11, 12]. As the 3-D structure of BUB1B (Protein Data Bank (PDB): 2WVI) is available (Fig. 1), it creates opportunities to perform structure-based studies for its molecular interaction. 2WVI has a theoretical weight of 20.06 kDa and is monomeric in nature, having Thr85, Tyr81, Gln58, Asp79, Tyr89, Ser96, Ser157, Tyr139, and Glu161 amino acid residues with a N-terminal domain [21]. The current study aims to screen bioactive compounds from a distinct variety of mushrooms to find potential lead compounds against the BUB1B target. For this purpose, 70 already reported bioactive compounds of mushrooms with anticancer properties were utilized. All 70 compounds were screened for their pharmacokinetics absorption, distribution, metabolism, and excretion (ADMET) properties. Suitable compounds were screened against BUB1B through a structure-based molecular interaction approach, i.e., molecular docking, and further, their stability and affinity were determined via molecular dynamic (MD) simulation.
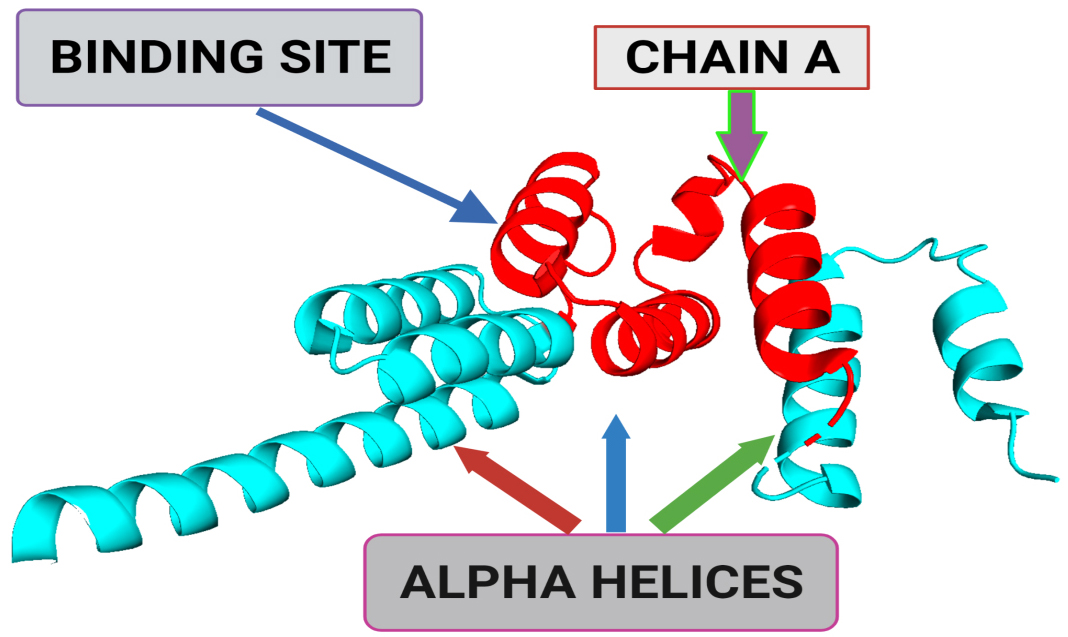
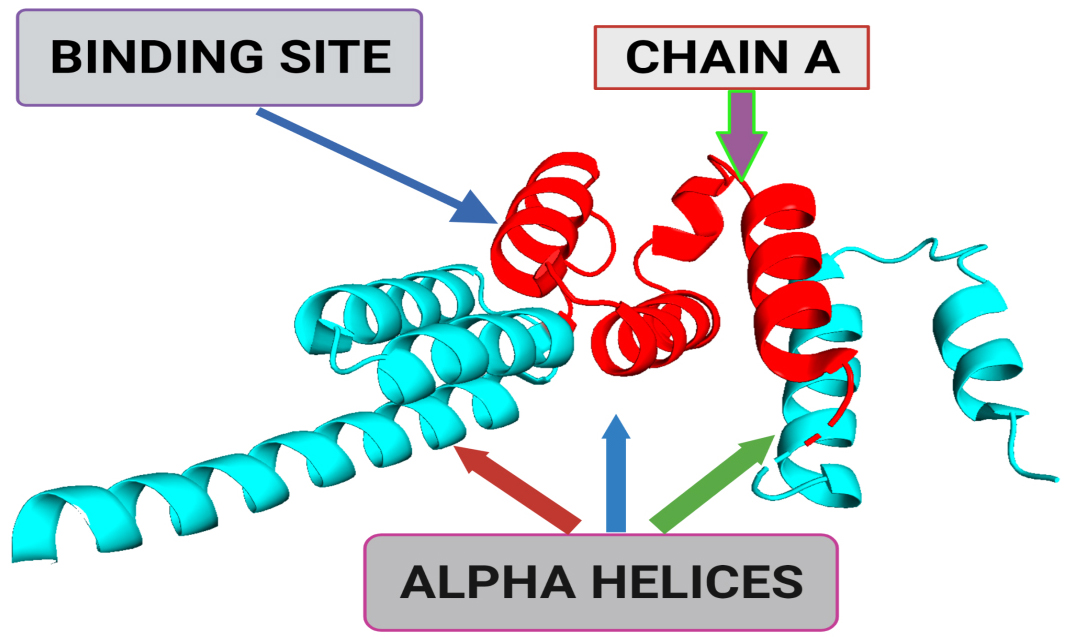 Fig. 1.
Fig. 1.A 3-dimensional structural representation of BUB1 mitotic checkpoint serine/threonine kinase B (BUB1B) (Protein Data Bank (PDB) ID: 2WVI; Resolution = 1.80 Å). The binding site represents the small pocket of 2WVI where ligands bind with the help of weak forces, i.e., non-covalent bonding. Chain A of 2WVI has a sequence length of 164. Alpha-helices are the spiral section of the secondary structure of the 2WVI protein, indicating the spatial arrangement of the main chain of 2WVI.
The 70 bioactive compounds from edible mushrooms were obtained from literature mining. The essential predicted physicochemical descriptors of the selected compounds were obtained by calculating ADMET properties from SwissADME and helped screen compounds with favorable pharmacokinetic properties. A comprehensive analysis of the log P (octanol/water) and QP % (human oral absorption) was obtained from this SwissADME program. Another important element essential for rational drug design is Lipinski’s rule of 5 (RO5) for bioactive compounds, which was also obtained from SwissADME and evaluated the acceptability of the known compounds.
For screening of selected bioactive compounds against BUB1B (PDB: 2WVI), AutoDock Vina [22] was employed. Amongst these bioactive compounds with favorable pharmacokinetic properties, the top five compounds with optimal binding affinity scores were further filtered out, and these were then docked against the BUB1B receptor using AutoDock Vina. PyMol (v 2.5) was used to obtain the active site coordinates and residues of the receptor. This process was based on the pdb file of the receptor-ligand complex. PyMol provided the coordinates (x_center = 0.4537, y_center = –24.6765, z_center = –12.2435, x_size = 60, y_size = 50, z_size = 70) to obtain the grid parameter file, and these coordinates belong to chain-A of the protein; 72–165 residues. During the next step, the grid map files were obtained by running AutoGrid. These grid map files were then used for docking using AutoDock, v4.2.6 suite (The Scripps Research Institute, Prayagraj, Uttar Pradesh, India) [23]. There were several standard parameters set for the docking purpose. These included hybrid Genetic Algorithm with Local Search (GA-LS) runs = 50, number of individuals in the population = 300, rate of gene mutation = 0.02, the maximum number of generations = 27,000, maximum number of energy evaluations = 2,500,000, and rate of crossover = 0.8. The algorithm employed for the AutoDock run was Lamarckian Genetic Algorithm (LGA), which provided RMSD (root-mean-square deviation) values and binding energies. Out of the five compounds, the top two with the best docking score were chosen for further validation using molecular dynamic simulation.
Desmond v3.6 Package [22] was employed for validating the findings of molecular
docking using molecular dynamic simulation. This was performed to elucidate the
effectiveness of these mushroom bioactive compounds for the activation of BUB1B.
For this purpose, the preparation of the lead compounds viz. lepitaprocerin D and
peptidoglycan (muramic acid) with BUB1B protein was completed using the optimized
potentials for liquid simulations 2005 (OPLS2005) force field. To build the
system, the pre-defined transferable intermolecular potential 3P (TIP3P) water
model was used, and these acted as water molecules. The orthorhombic periodic
conditions set at 10 Å units were used for constructing these. Before MD
simulations, the energy minimization of the system was performed using the
steepest-descent method, an optimization algorithm used to find the minimum
energy state of a system by iteratively stepping in the direction of the steepest
decrease in energy. Also, the balancing of Na
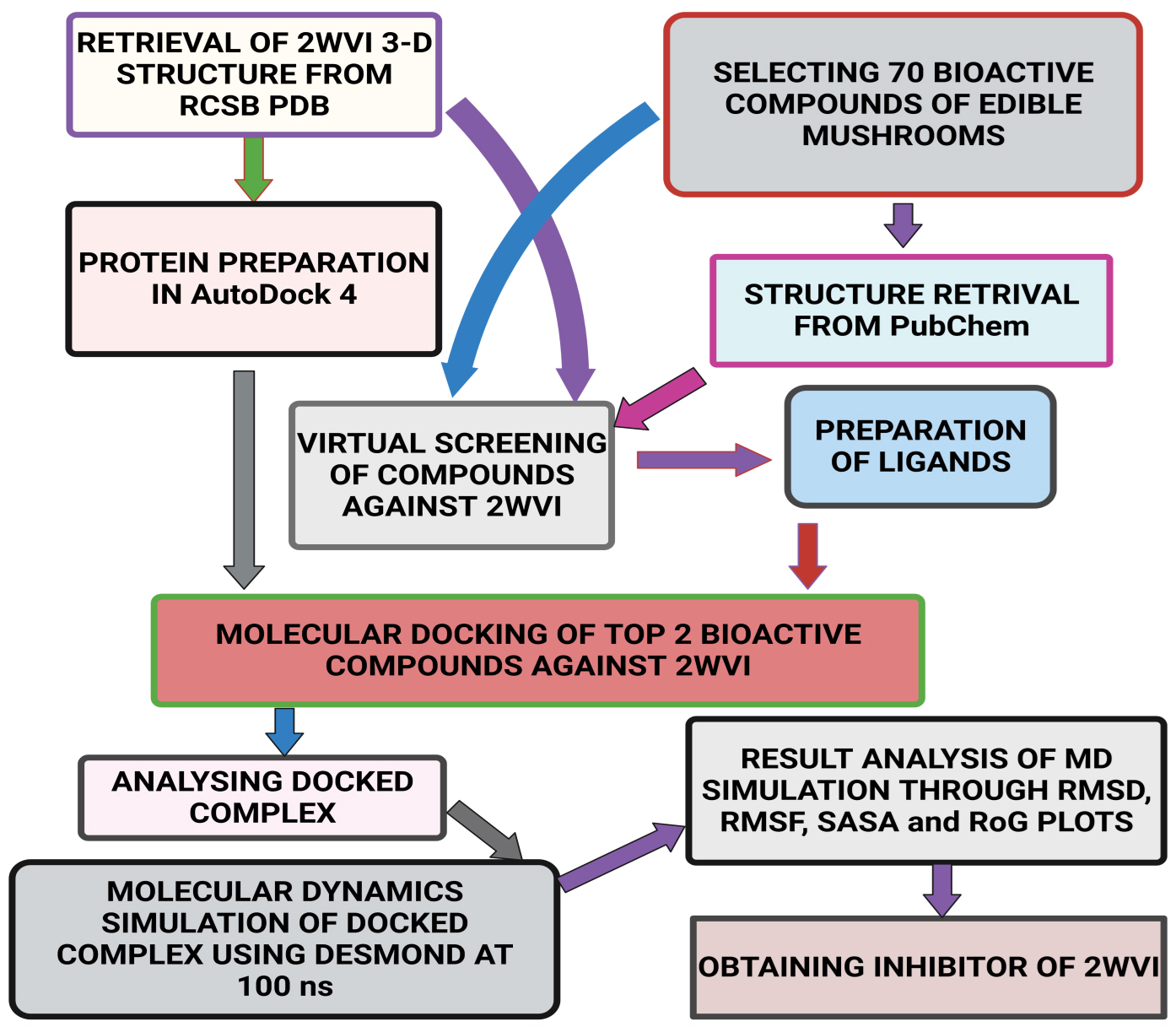
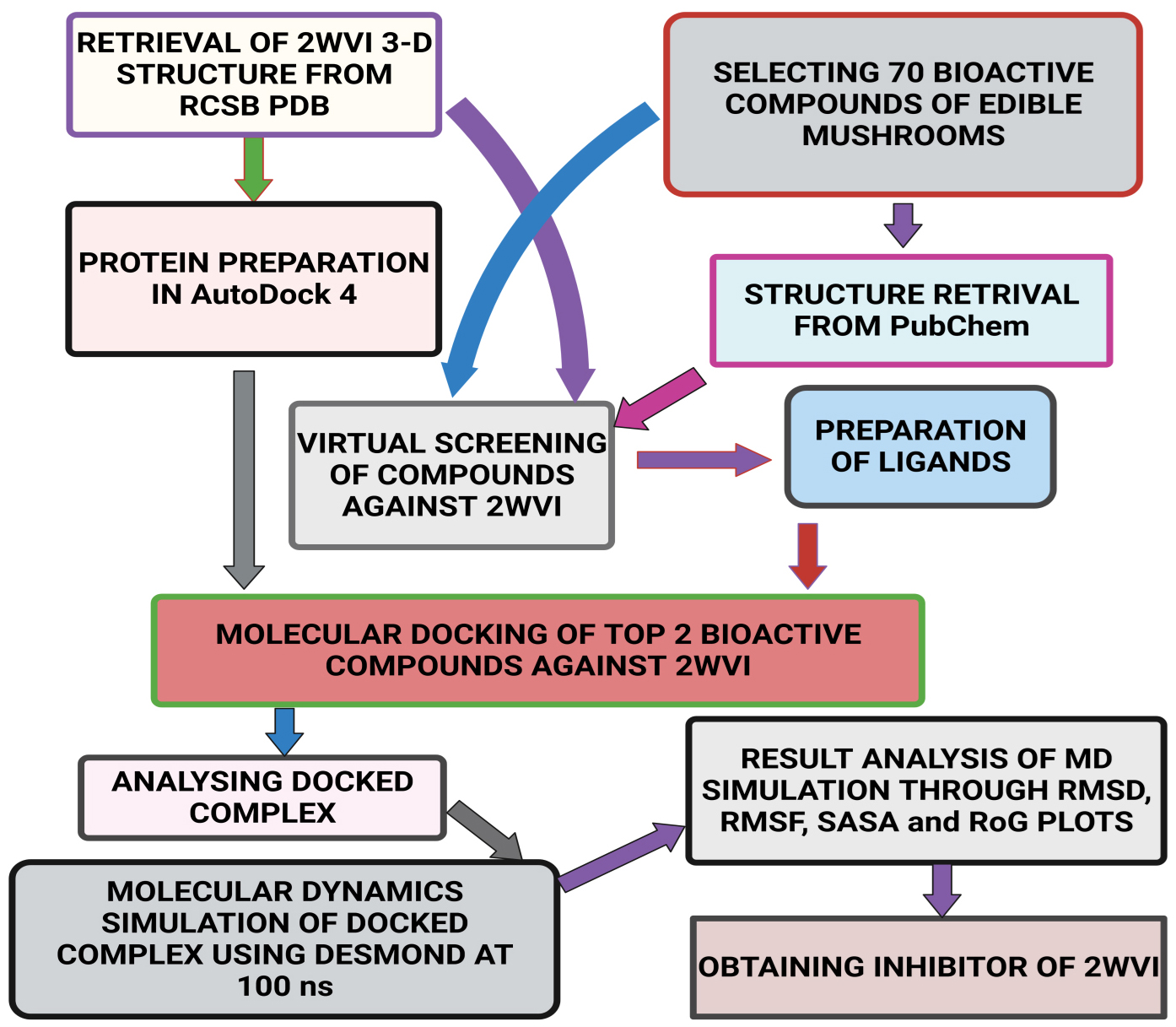 Fig. 2.
Fig. 2.Schematic representation of the methodology followed in the present study.
The 70 bioactive compounds obtained from different mushroom sources have been tabulated in Table 1 (Ref. [23, 24, 25, 26]).
| S.No. | Compound | Source | Structure | Reference |
| 1 | LY-294002 | Tricholoma matsutake |  |
[23] |
| 2 | Hispidine | Phellinus linteus | 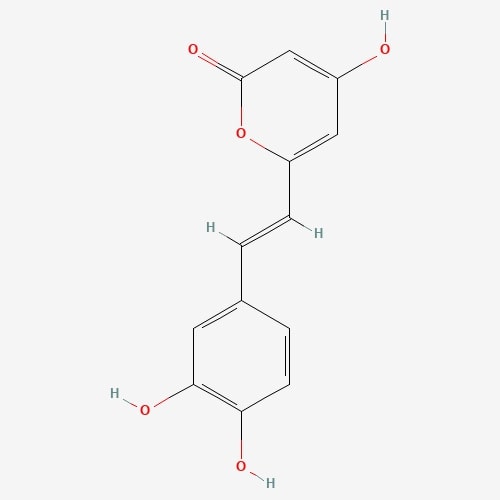 |
[23] |
| 3 | Cordycepin | Cordyceps melitaris | 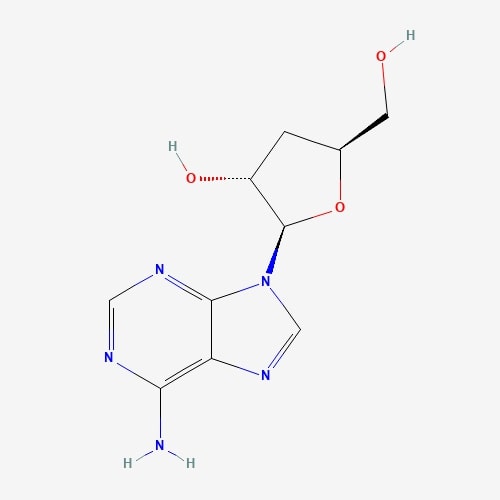 |
[23] |
| 4 | Lucidenic acid | Ganoderma lucidium | 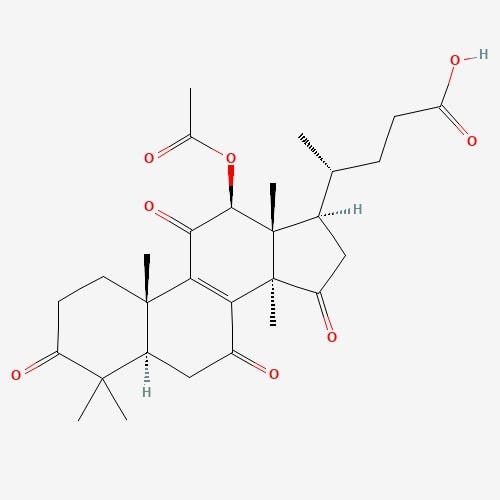 |
[23] |
| 5 | JNJ0966 | Hericium erinaceus | 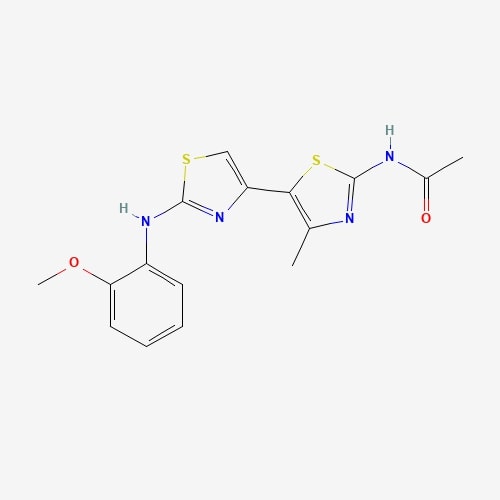 |
[23] |
| 6 | 2-Heptanone | Hericium erinaceus |  |
[23] |
| 7 | Cyclobutanone | Hericium erinaceus |  |
[23] |
| 8 | Cyclopropane | Hericium erinaceus |  |
[23] |
| 9 | 2(5H)-Furanone | Hericium erinaceus |  |
[23] |
| 10 | Formamide | Hericium erinaceus | 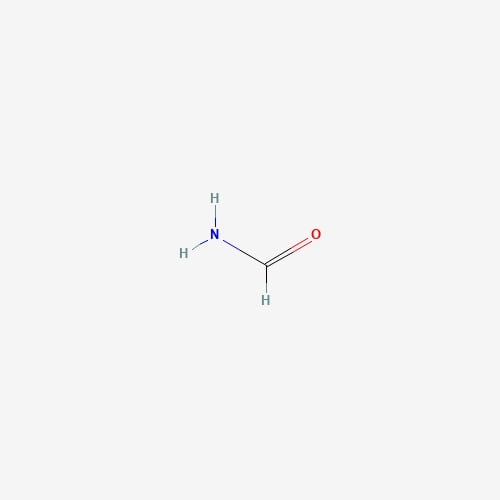 |
[23] |
| 11 | Glycerin | Pleurotus ostreatus | 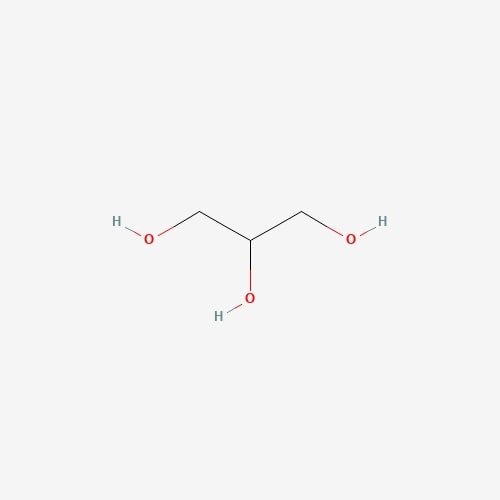 |
[23] |
| 12 | 4-Heptanone | Pleurotus ostreatus | 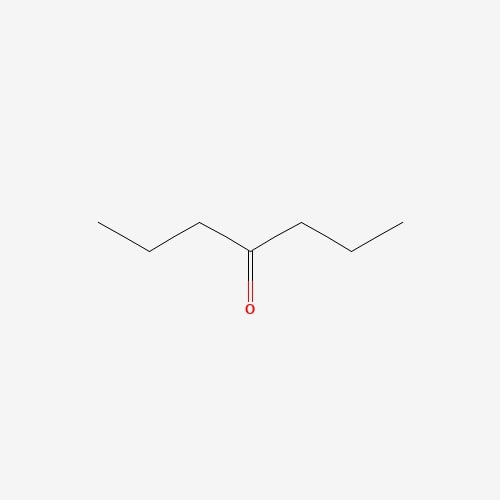 |
[23] |
| 13 | Propanedioic acid | Pleurotus ostreatus | 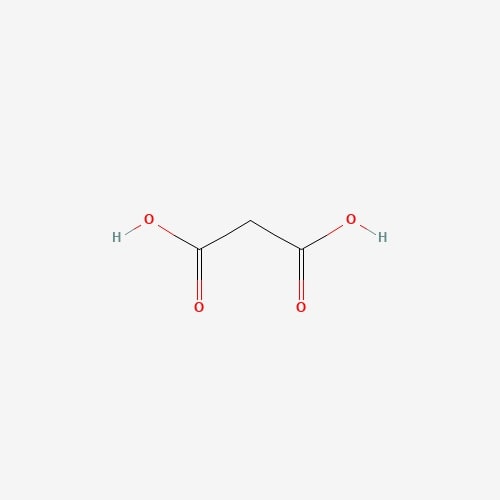 |
[23] |
| 14 | Niacin | Pleurotus ostreatus |  |
[23] |
| 15 | 1,4-Pentanediol | Pleurotus ostreatus |  |
[23] |
| 16 | 3-Methyl-2-Pyrrolidinone | Pleurotus ostreatus |  |
[23] |
| 17 | 5-Methoxypyrrolidin-2-one | Pleurotus ostreatus |  |
[23] |
| 18 | 2-Undecene | Pleurotus ostreatus |  |
[23] |
| 19 | Lovastatin | Pleurotus ostreatus |  |
[23] |
| 20 | Eritadenine | Lentinus edodes |  |
[23] |
| 21 | 1,2-dihydroxymint Lactone | Lentinus sqquarrosulus |  |
[23] |
| 22 | Hydroquinone | Piptoporus betulinus | 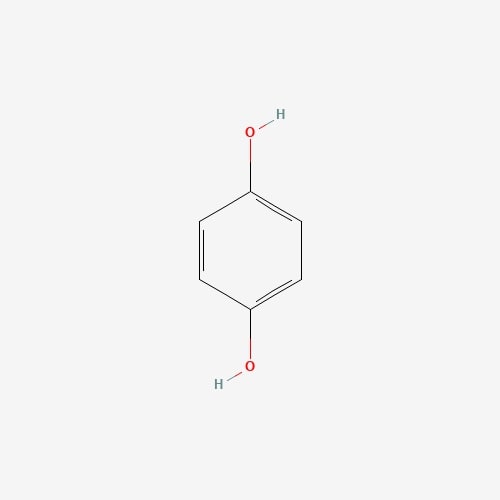 |
[23] |
| 23 | Calvacin | Calvatia gigantean | 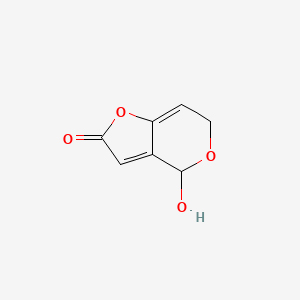 |
[23] |
| 24 | Panepoxydone | Lentinus crinitus | 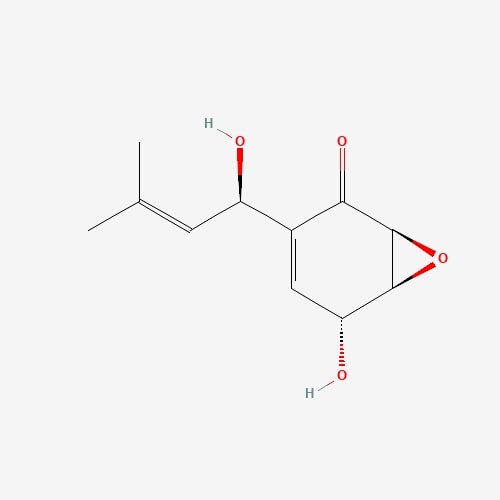 |
[23] |
| 25 | Gliotoxin | Gliocladium fibriatum | 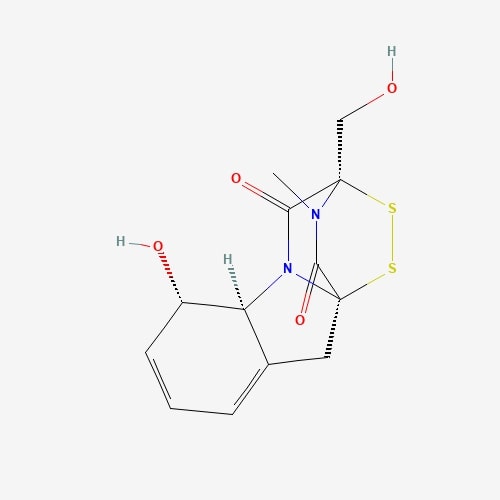 |
[23] |
| 26 | Ganoderic Acid | Ganoderma lucidium |  |
[23] |
| 27 | Gerronemin D | Gerronema sp. |  |
[23] |
| 28 | Ergosterol | Grifola frondosa |  |
[23] |
| 29 | 2-Amino-3H-Phenoxazin-3-one | Lepiota Americana |  |
[23] |
| 30 | 2_Heptanone | Volvariella volvacea |  |
[23] |
| 31 | Antroquinonol | Antrodia Camphorate | 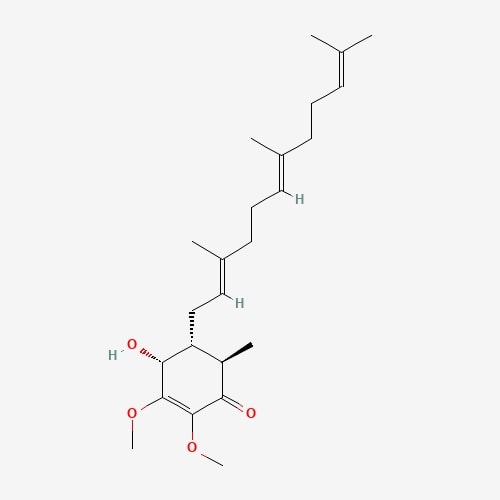 |
[24] |
| 32 | (-)-Catechin | Russula luteotacta |  |
[24] |
| 33 | Erinacin A | Hericium erinaceus |  |
[24] |
| 34 | Ganoderiol | Ganoderma lingzhi |  |
[24] |
| 35 | Ganodermanontriol | Ganoderma leucocontextum |  |
[24] |
| 36 | Grifolin | Albatrellus confluens |  |
[24] |
| 37 | Hispolon | Phellinus linteus |  |
[24] |
| 38 | 4-Hydroxybenzoic Acid | Rusulla emetic | 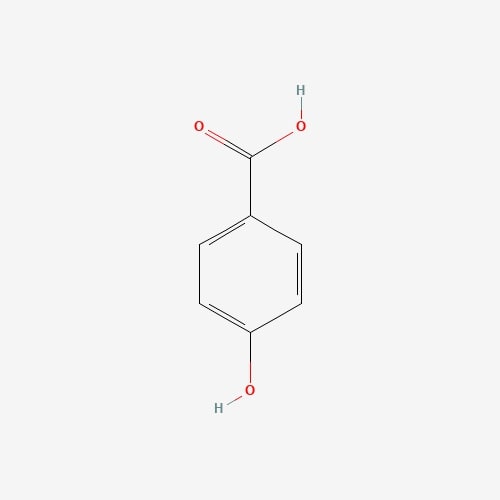 |
[24] |
| 39 | Illudin-S | Omphalotus illudens | 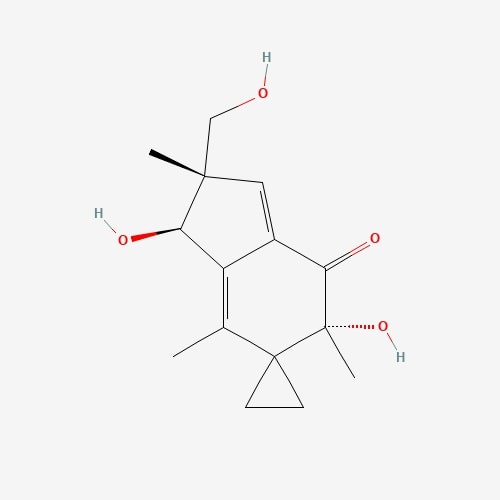 |
[24] |
| 40 | Lepiotaprocerin C | Macrolepiota procera | 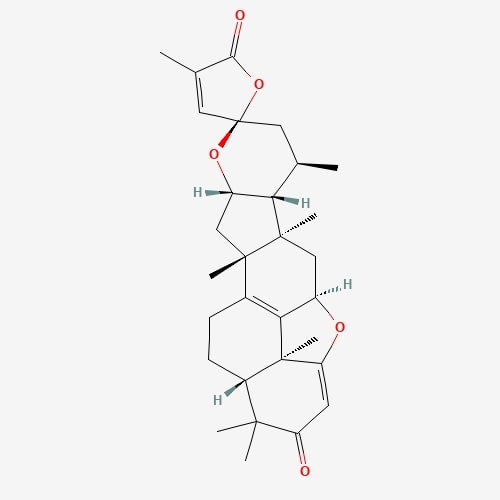 |
[24] |
| 41 | Lepiotaprocerin D | Macrolepiota procera | 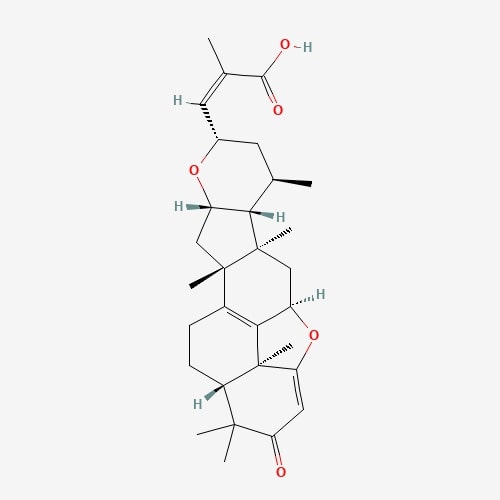 |
[24] |
| 42 | Lepiotaprocerin G | Macrolepiota procera |  |
[24] |
| 43 | Lepiotaprocerin H | Macrolepiota procera |  |
[24] |
| 44 | Lepiotaprocerin I | Macrolepiota procera |  |
[24] |
| 45 | Lepiotaprocerin K | Macrolepiota procera |  |
[24] |
| 46 | Lepiotaprocerin L | Macrolepiota procera |  |
[24] |
| 47 | L-Ergothioneine | Pleurotus ostreatus |  |
[24] |
| 48 | Peptidoglycan | Pleurotus ostreatus |  |
[24] |
| 49 | psilocybine | Psilocybe semilanceata |  |
[24] |
| 50 | Queretin | Pleurotus ostreatus |  |
[25] |
| 51 | Lanostane | Wolfiporia cocos |  |
[25] |
| 52 | Dehydrotrametenolic acid | Wolfiporia cocos |  |
[25] |
| 53 | Termitomycesphins | Termitomyces albuminosus |  |
[25] |
| 54 | Lectin | Xylaria hypoxylon | 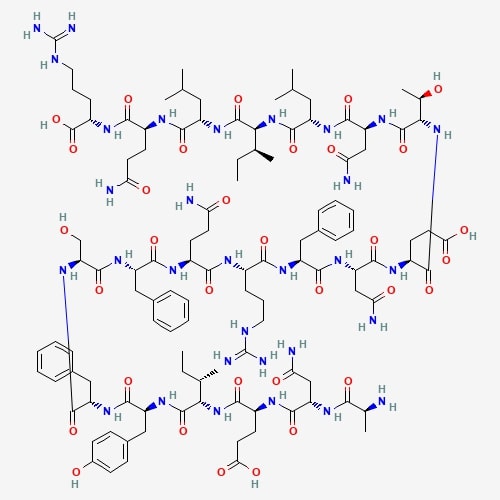 |
[25] |
| 55 | Krestin | Trametes versicolor | 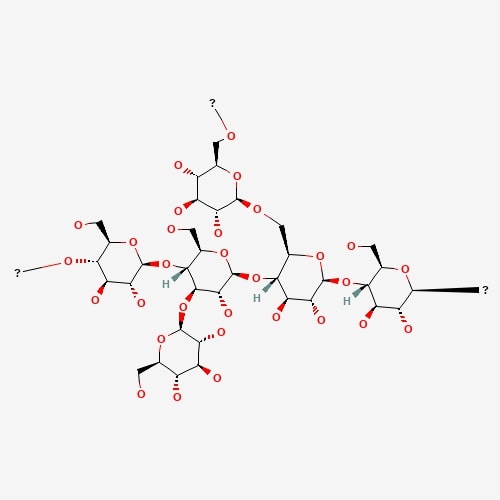 |
[25] |
| 56 | Pleurotine | Pleurotus griseus | 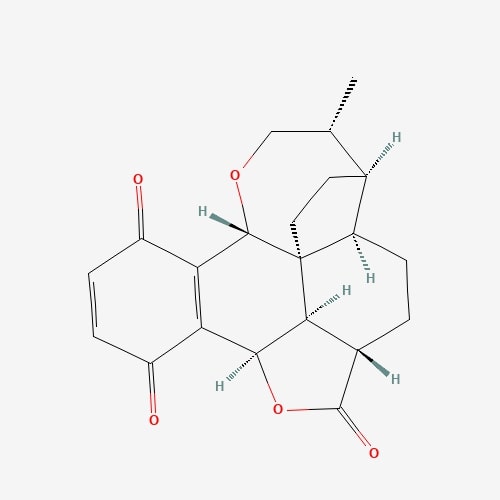 |
[25] |
| 57 | Ling Zhi-8 | Ganoderma lucidum | 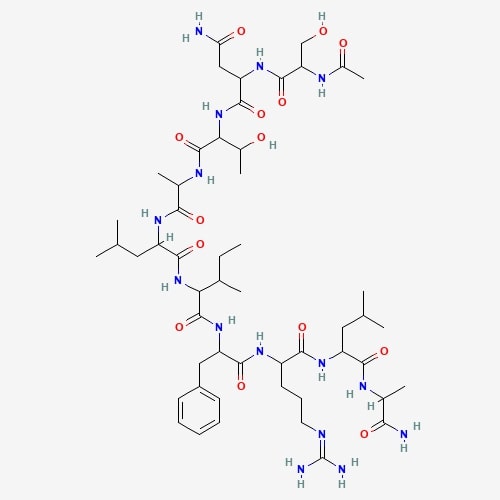 |
[25] |
| 58 | Galactomannan | Morchell esculenta |  |
[25] |
| 59 | Syringic acid | Elaphomyces granulates |  |
[25] |
| 60 | Laccase | Clitocybe maxima |  |
[25] |
| 61 | Ciclosporin | Cordyceps sinensis |  |
[26] |
| 62 | Syringaldehyde | Elaphomyces granulatus | 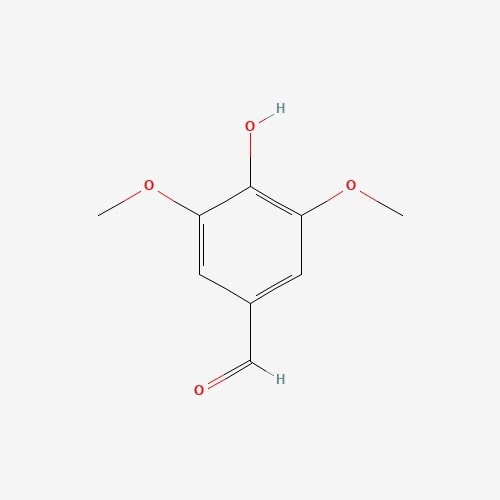 |
[26] |
| 63 | Trehalose | Hypsizygus marmoreus | 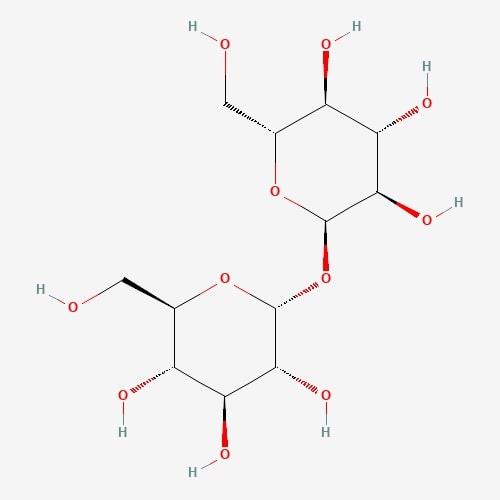 |
[26] |
| 64 | Pyrogallol | Cantharellus cibarius | 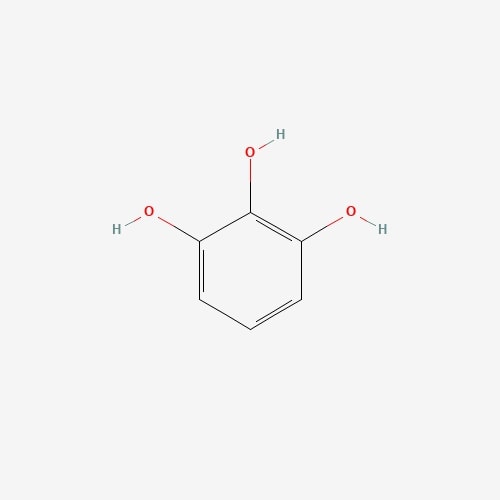 |
[26] |
| 65 | Coprinus comatus |  |
[26] | |
| 66 | Myricetin | Craterellus cornucopiodes | 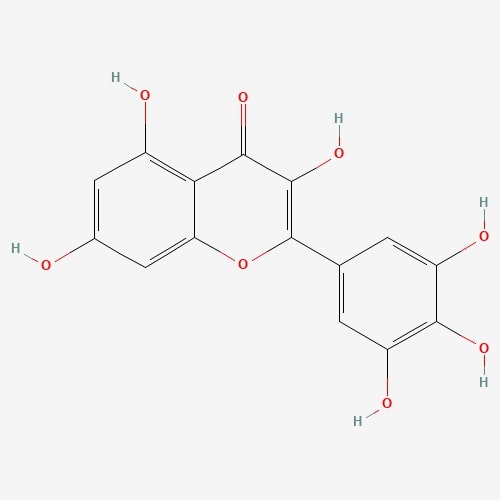 |
[26] |
| 67 | Lanostane | Wolfiporia cocos | 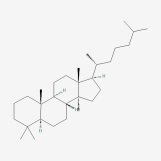 |
[26] |
| 68 | Betulinan A | Lenzites betulina | 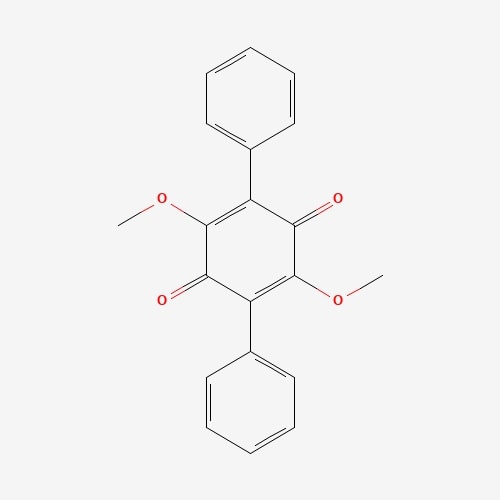 |
[26] |
| 69 | Infractin | Cortinarius infractus | 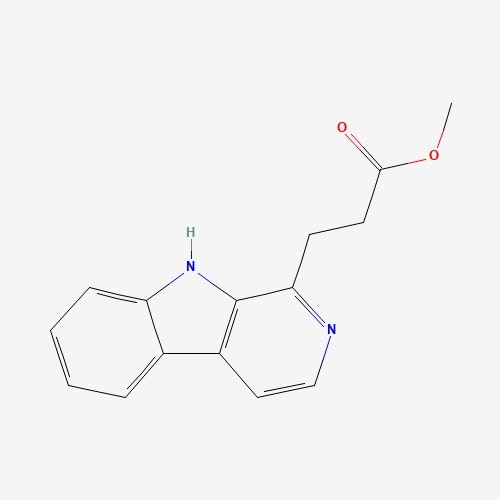 |
[26] |
| 70 | Dilinoleoylphosphatidylethanolamine | Hericium erinace | 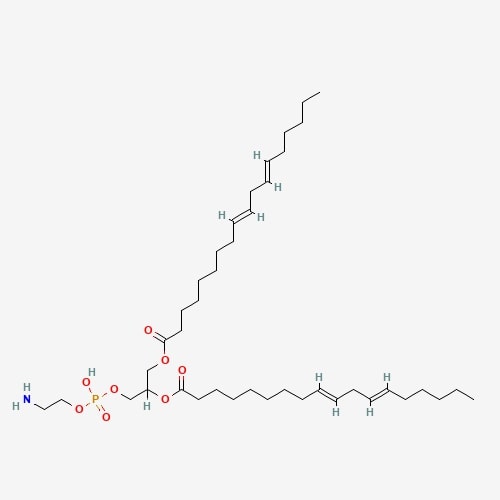 |
[26] |
All compounds administrated orally processed via the absorption, distribution, metabolism, and excretion process. Before considering any bioactive compound as a lead, it is imperative to check its ADME features so that any compound with inappropriate pharmacokinetics properties is filtered out [27, 28]. The drug-like activity of the selected high-molecular-weight (HMW) and low-molecular-weight (LMW) edible mushroom compounds were categorized using the ADME properties shown in Table 2.
| S.No. | Compound Name & Structure | Mol. Volume (m |
Mol. Weight (g/mol) | Rotatable Bonds | VdW PSA (Å |
RO5 Violation | HB Donors | HB Acceptors |
| 1. | Grifolin 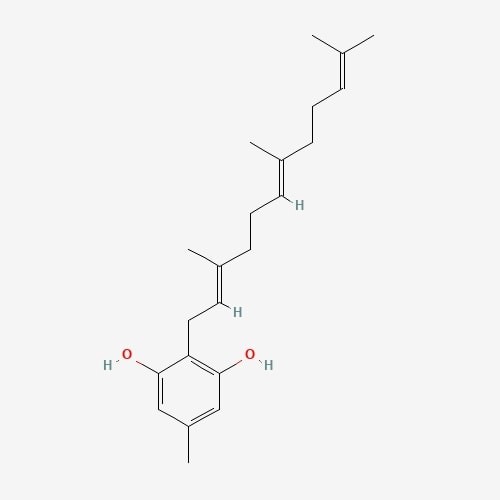 |
195.05 | 328.5 | 8 | 40.46 | 0 | 2 | 2 |
| 2. | Ganoderial 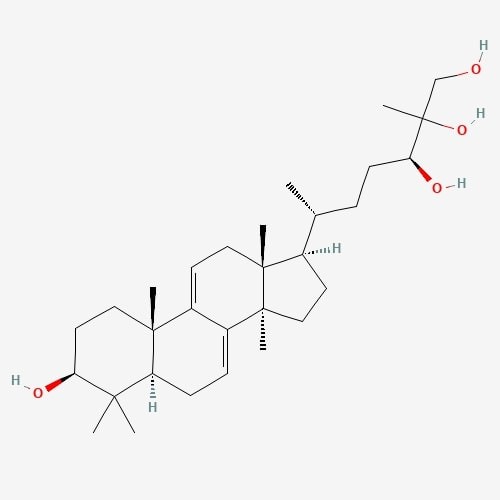 |
217.32 | 474.7 | 6 | 80.92 | 0 | 4 | 4 |
| 3. | Illudin S 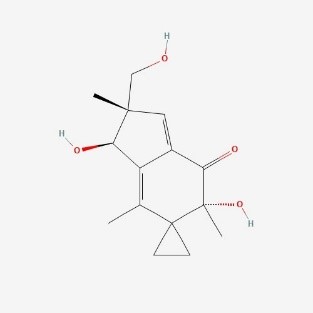 |
121.00 | 264.3 | 1 | 77.76 | 0 | 3 | 4 |
| 4. | Lepiotaprocerin D  |
210.37 | 480.6 | 2 | 72.83 | 0 | 1 | 5 |
| 5. | Peptidoglycan  |
112.38 | 251.2 | 4 | 142.47 | 0 | 5 | 8 |
The physicochemical descriptors include molecular weight, molecular volume,
H-bond donors, H-bond acceptors, and their position according to Lipinski’s rule
of five. Lipinski’s rule of 5 is a rule of thumb to evaluate drug-likeness; if a
chemical compound contains specific pharmacological and biological properties,
then properties would make it an active drug. The rule describes molecular
properties important for a drug’s pharmacokinetics in a living system, including
ADME properties. All these pharmacokinetic parameters are within the acceptable
range defined for human use, thereby indicating their potential as drug-like
molecules. Lepiotaprocerin D had two rotatable bonds with a polar surface area
(PSA) of 72.83 Å
The receptor BUB1B (PDB: 2WVI) was docked with the 70 compounds obtained from the literature survey. The active site of the BUB1B (2WVI) receptor consisted of Thr85, Tyr81, Gln58, Asp79, Tyr89, Ser96, Ser157, Tyr139, and Glu161 amino acid residues. The screening with AutoDock Vina found five compounds, i.e., grifolin, ganoderiol, illudin S, lepiotaprocerin D, and peptidoglycan as the most appropriate leads. These five compounds further docked against the BUB1B to obtain the best binding conformation and affinity. The docking showed well-formed binding of the identified lead compounds with one or more residues (amino acids) in the active site pocket of the 2WVI receptor shown in Fig. 3. Lepiotaprocerin D forms a hydrogen bond with 2WVI along with binding site residues of Ser157 and Glu161, and water bridges along Tyr139, Ser157, and Glu161. Peptidoglycan formed ionic interactions along Tyr81, hydrogen bond along Gln58, Tyr81, Thr85, and Tyr89, and water bridges along Gln58, Tyr81, and Tyr89 (Fig. 4).
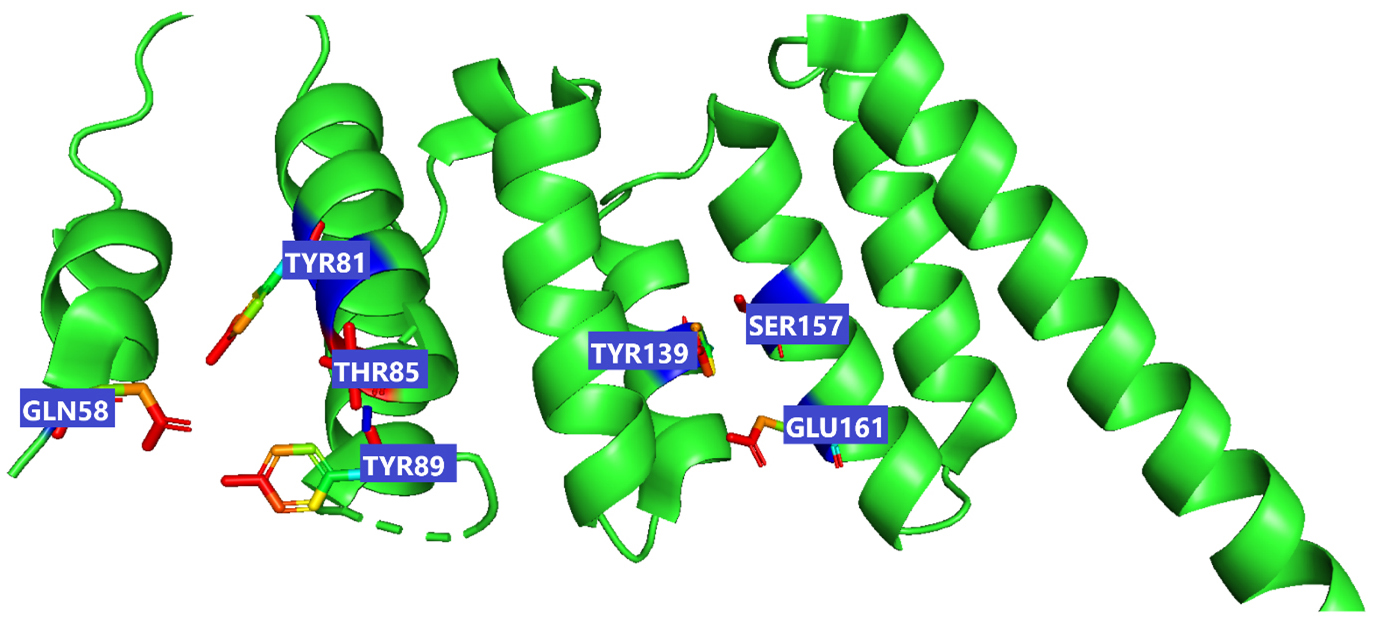
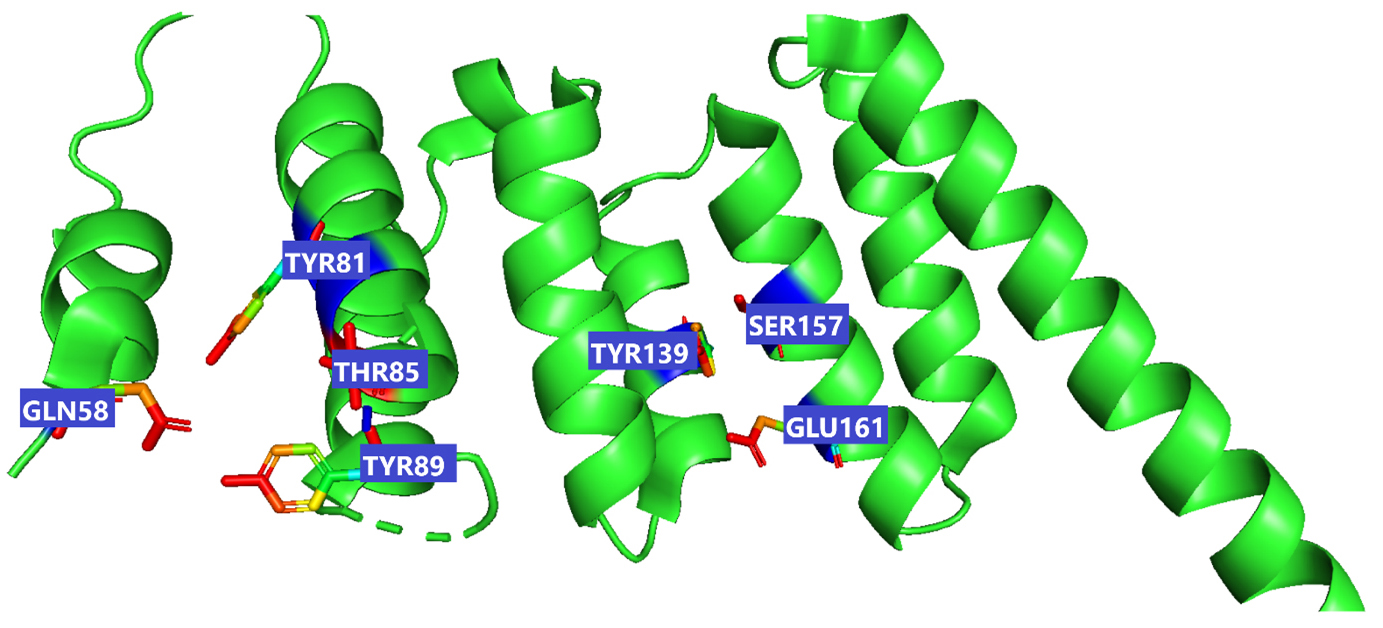 Fig. 3.
Fig. 3.This figure illustrate the active sites of 2WVI obtained from PyMol. The provided coordinates (x_center = 0.4537, y_center = –24.6765, z_center = –12.2435, x_size = 60, y_size = 50, z_size = 70) were used to generate the grid parameter file for the active site, which is located in chain-A of the protein spanning residues 72–165, illustrated in the figure which depicts the active site residues responsible for binding lepiotaprocerin D and peptidoglycan ligands. These residues include Gln58, Tyr81, Tyr89, Thr85, Tyr139, Ser157, and Glu161.
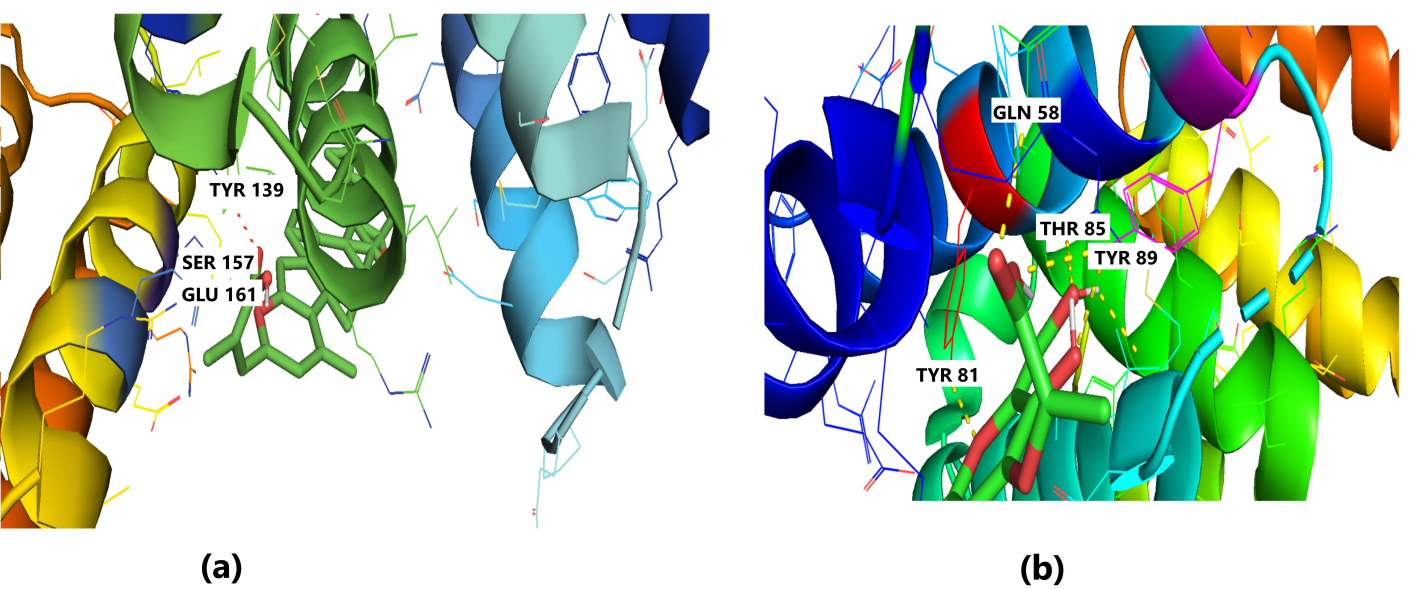
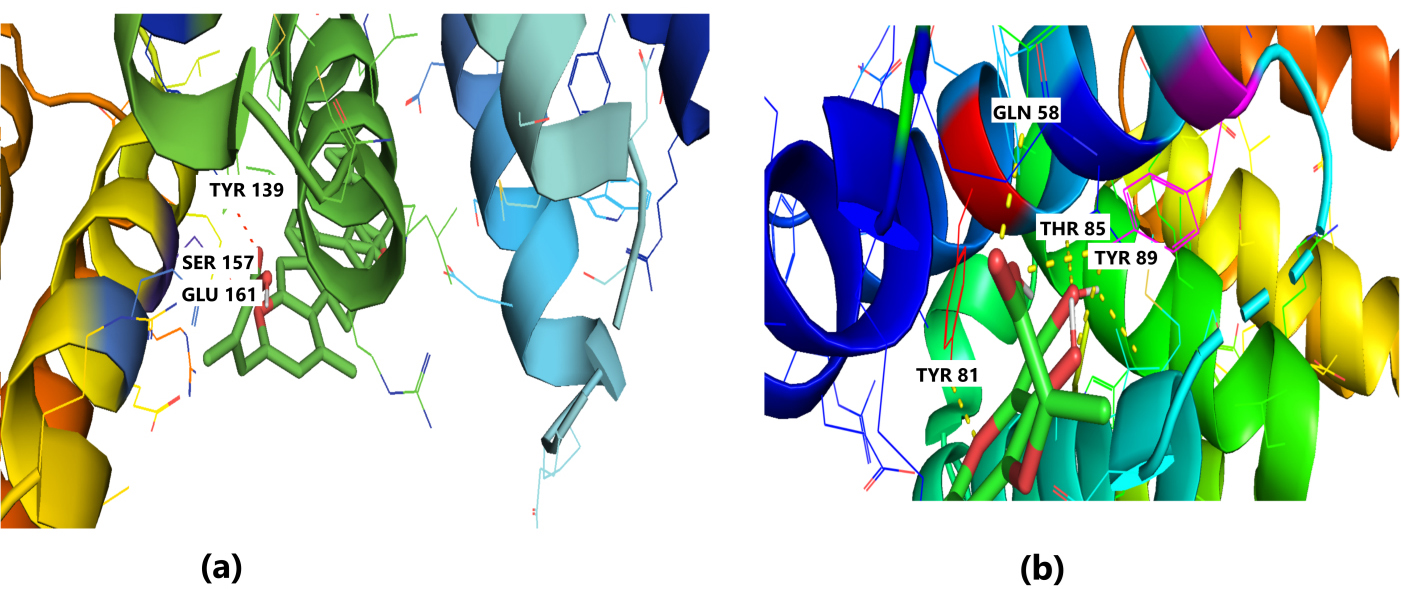 Fig. 4.
Fig. 4.Molecular Docking of 2WVI with the top two ligand molecules. (a) Lepiotaprocerin D forms a hydrogen bond with 2WVI and the binding site residues Ser157 and Glu161 and water bridges along Tyr139, Ser157, and Glu161. (b) Peptidoglycan formed ionic interactions along Tyr81, hydrogen bond along Gln58, Tyr81, Thr85, and Tyr89, and water bridges along Gln58, Tyr81, and Tyr89.
The in silico molecular docking revealed that the newly identified compounds exhibit excellent binding energy towards the target receptor, as shown in Table 3.
| S. no. | Compound name | Binding energy | RMSD | Free energy (kcal/mol) | Internal energy (kcal/mol) |
| 1. | Lepiotaprocerin D | –8.91 | 37.33 | –2325.98 | –8.18 |
| 2. | Peptidoglycan | –8.70 | 37.58 | –2325.20 | –7.40 |
| 3. | Grifolin | –8.14 | 35.37 | –2325.00 | –7.20 |
| 4. | Ganderiol | –7.73 | 38.77 | –2324.62 | –6.82 |
| 5. | Illudin S | –7.08 | 35.19 | –2324.86 | –7.06 |
BUB1B, BUB1 mitotic checkpoint serine/threonine kinase B; RMSD, root-mean-square deviation.
Docking only provides a static view of the interaction between the receptor and compound in the active site of the protein receptor, it does not provide dynamic observation of the interaction. Molecular dynamic simulation offers to compute the binding atom movements with the time using Newton’s motion. The molecular dynamic (MD) simulation was performed to determine the stability, confirmation, and intermolecular interaction of the top two ligand molecules (obtained from Autodock) with the 2WVI protein, as shown in Fig. 4a,b. The time-dependent modification of the complexes was calculated over 100 ns using the Desmond package. The MD simulation was performed under thermodynamics conditions (applied volume, density, pressure, and temperature). The complete system was annealed and equilibrated using ensembles. Moreover, the final production step was performed to investigate the structural modification of the complex.
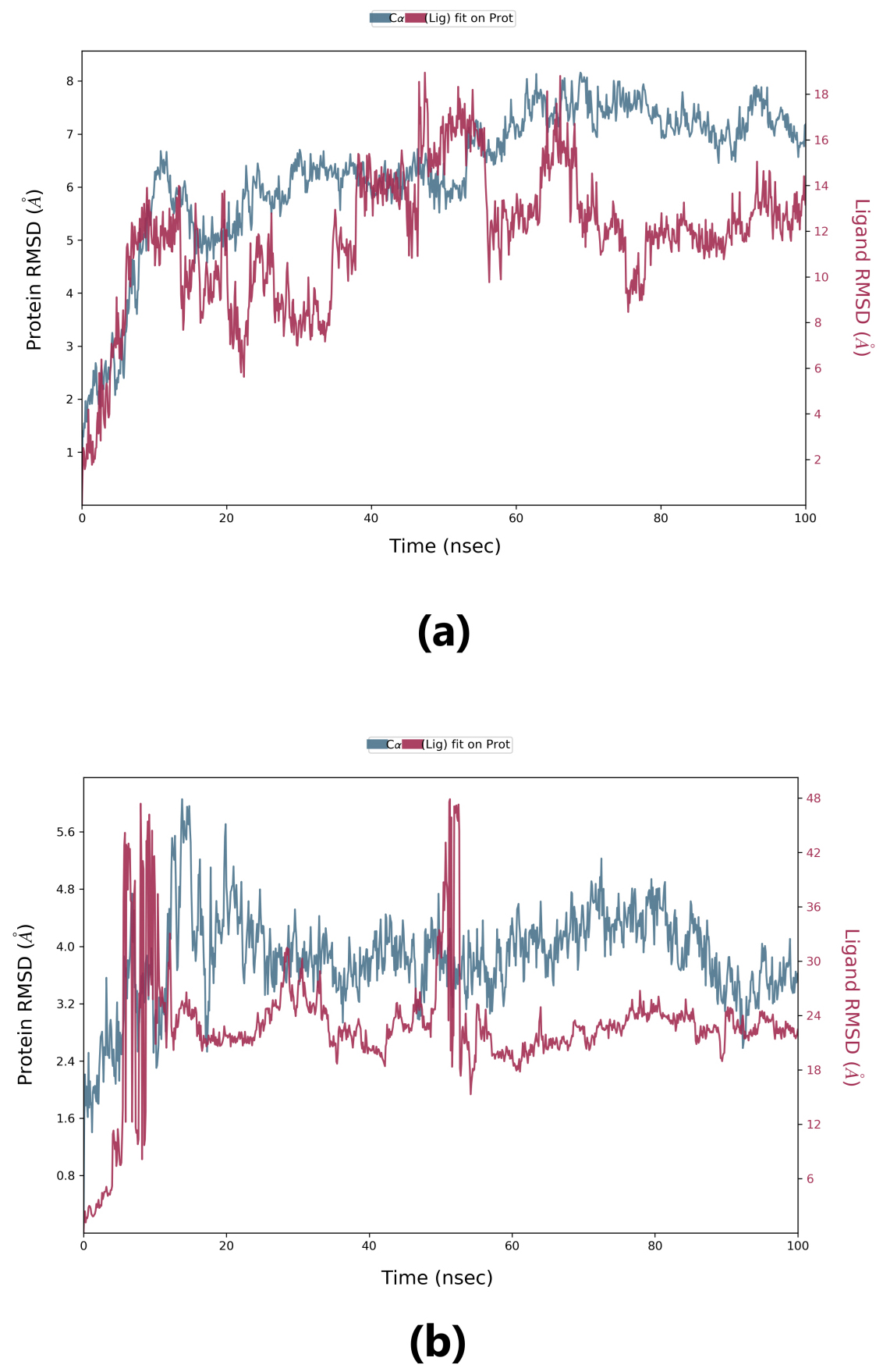
Root-mean-square deviation (RMSD) is used to measure the deviation in the backbone of a protein from its initial structure conformation to its final position. This deviation, which is produced during MD simulation, determines the protein’s stability relative to its conformation. A smaller deviation indicates a more stable protein structure. Ligand RMSD indicates the stability of the ligand with respect to the receptor (protein) and its respective binding pocket. The RMSD value for the ligand should be significantly lower than the protein RMSD because if this value is large, it is possible that the ligand may diffuse away from its initial binding pocket. From Fig. 5, it can be observed that for lepiotaprocerin D, the system equilibrated after 40 ns, after which the fluctuations were stable until 6 Å. The ligand was stable with respect to protein and confined to the binding pocket of the protein until approximately 48 ns, after which the ligand diffused away from the binding pocket owing to its RMSD value becoming larger than that of the receptor but again returning within the binding pocket after 52 ns. Similarly, for peptidoglycan, the system equilibrated after 10 ns, after which fluctuations stabilize for a short span, and then again, equilibration is obtained. We analyzed the conformer at specific times (t = 10 ns and t = 55 ns) and evaluated the conformation. Our analysis suggests that although the ligands were within the active site of the protein, there were some fluctuations between this interval which could explain the sudden rise of the amplitude of fluctuations.
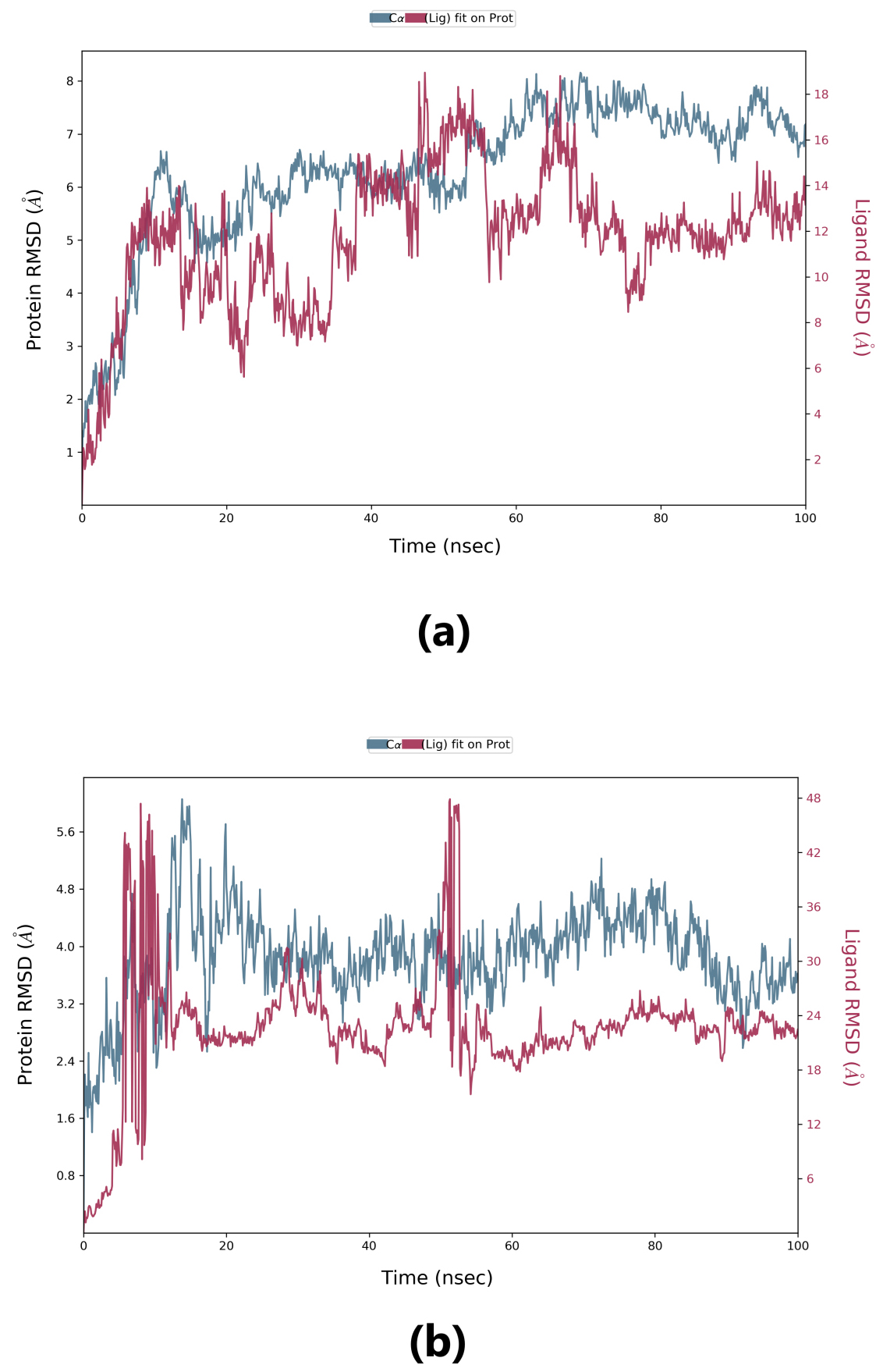 Fig. 5.
Fig. 5.Root-mean-square deviation (RMSD) plot. (a) RMSD plot for lepiotaprocerin D and 2WVI. X-axis on the left shows the RMSD for the 2WVI protein. The x-axis on the right shows the ligand (lepiotaprocerin D) stability. (b) The RMSD plot for peptidoglycan and 2WVI. X-axis on the left shows the RMSD for the protein (2WVI). The x-axis on the right shows the ligand (peptidoglycan) stability.
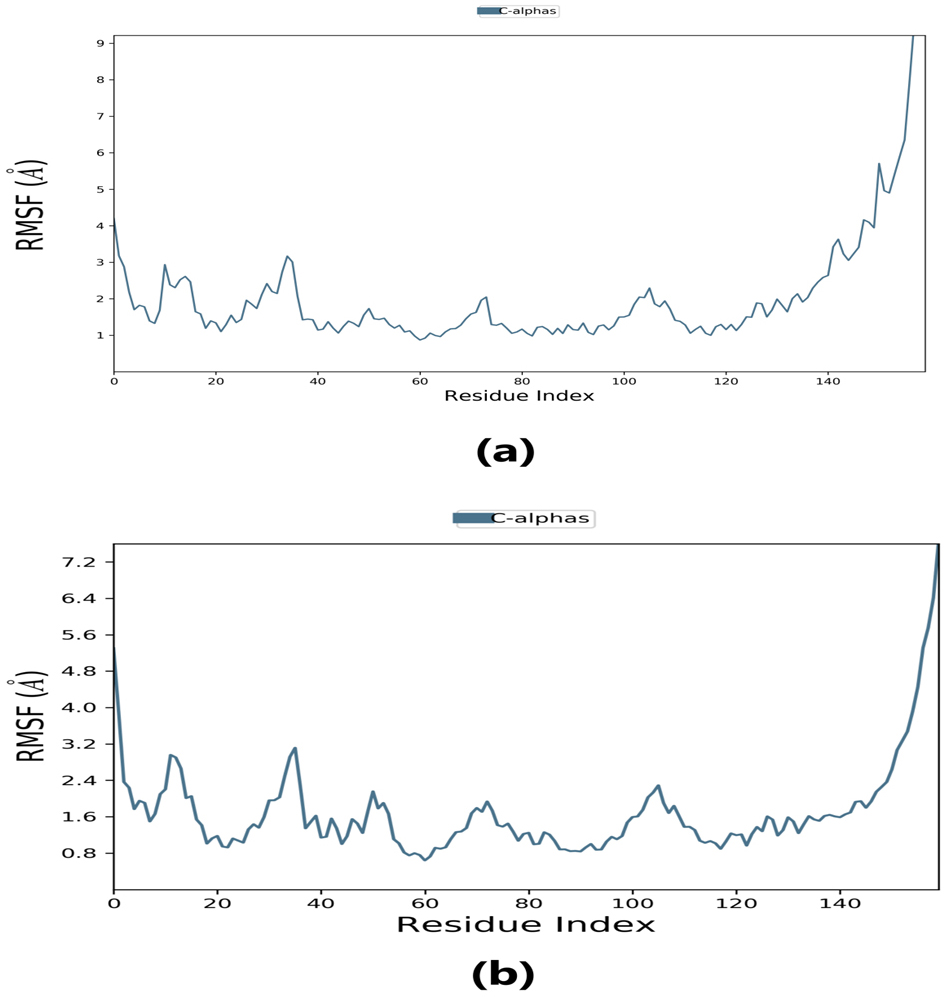
Root-mean-square fluctuation (RMSF) provides information about regions with higher flexibility levels. It measures the average deviation of atoms from their equilibrium positions over time, reflecting protein flexibility. Changes in the RMSF pattern can be correlated with protein function or activity. Specifically, the flexibility in the region between 120–140 in the protein structure can affect its interaction with the ligand and thus impact the protein’s activity. Understanding these dynamics may provide insight into the protein-ligand binding mechanism and inform the design of more effective drugs.
The RMSF plot peaks display the areas of maximum fluctuation of the protein. The
C
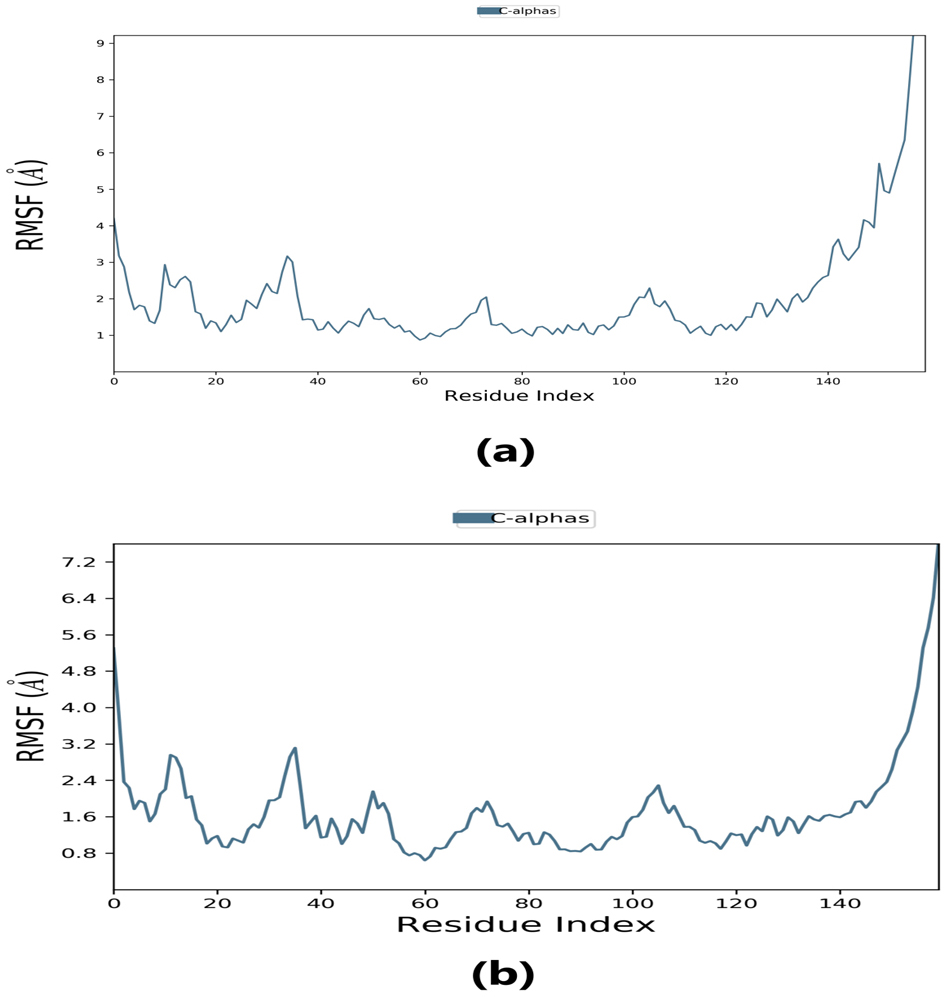 Fig. 6.
Fig. 6.Root-mean-square fluctuation (RMSF) plot. (a) RMSF plot of lepiotaprocerin D represents the flexibility level near residue number 30. (b) RMSF plot of peptidoglycan represents the flexibility level around residue numbers 120 and 140.
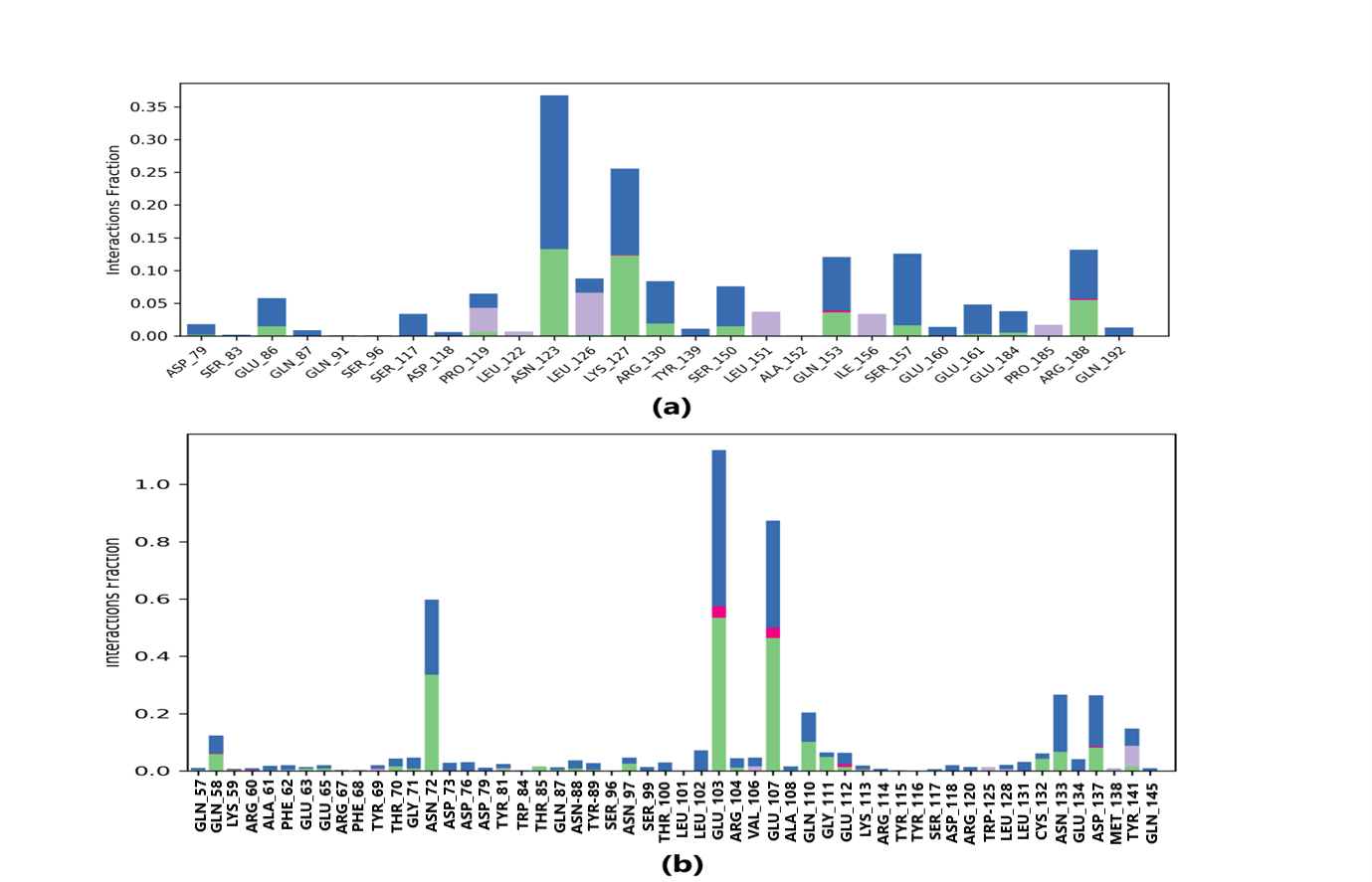
Protein-ligand interaction was observed throughout the simulation. There are four types of contact: hydrophobic, hydrogen bond, ionic, and water bridge. These intermolecular interactions play a vital role at an atomic level in predicting the binding mode of lepiotaprocerin D and peptidoglycan with the 2WVI receptor. Lepiotaprocerin D forms a hydrogen bond with 2WVI along with binding site residues of Ser157 and Glu161, and water bridges along Tyr139, Ser157, and Glu161, shown in Fig. 4a. Peptidoglycan interacts with the BUB1B protein through ionic interactions with Tyr81, hydrogen bonding with Gln58, Tyr81, Thr85, and Tyr89, and water bridges with Gln58, Tyr81, and Tyr89. The hydrophobic interactions are relatively small in terms of interaction fraction as compared to those of lepiotaprocerin D, shown in Fig. 4b. Also, lepiotaprocerin D contained three binding site residues where interactions occurred more than 10% viz. Tyr139, Ser157, and Glu161. However, there were no interactions of more than 10% in peptidoglycan (Fig. 7a,b).
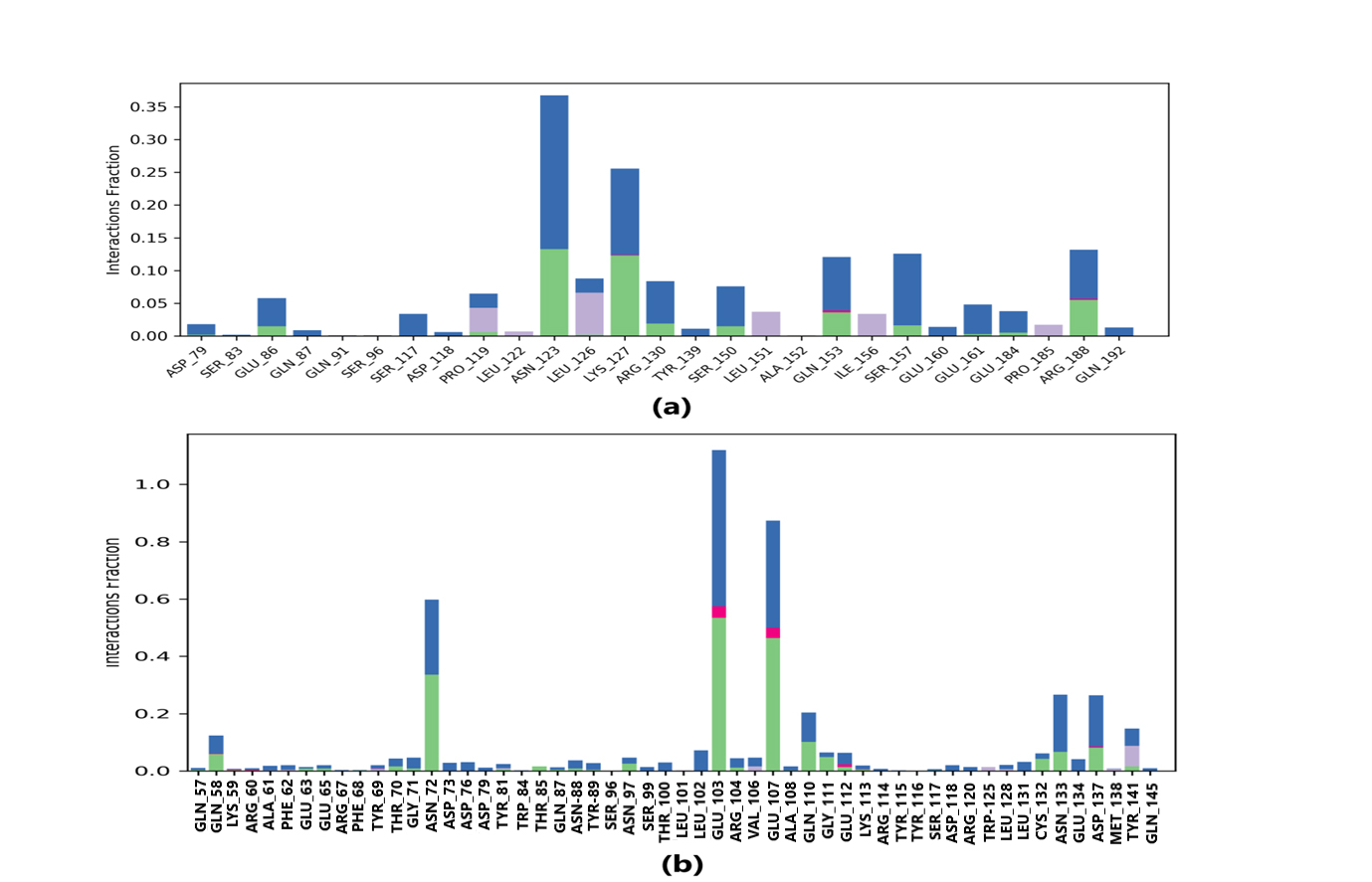 Fig. 7.
Fig. 7.Protein-ligand contacts. Blue indicates water bridges, green indicates H-bonds, grey indicates hydrophobic bonds, and pink indicates ionic bonding. (a) PL contacts showing amino acid residues corresponding to the interaction between 2WVI and lepiotaprocerin D. (b) PL contacts showing amino acid residues corresponding to the interaction between 2WVI and peptidoglycan.
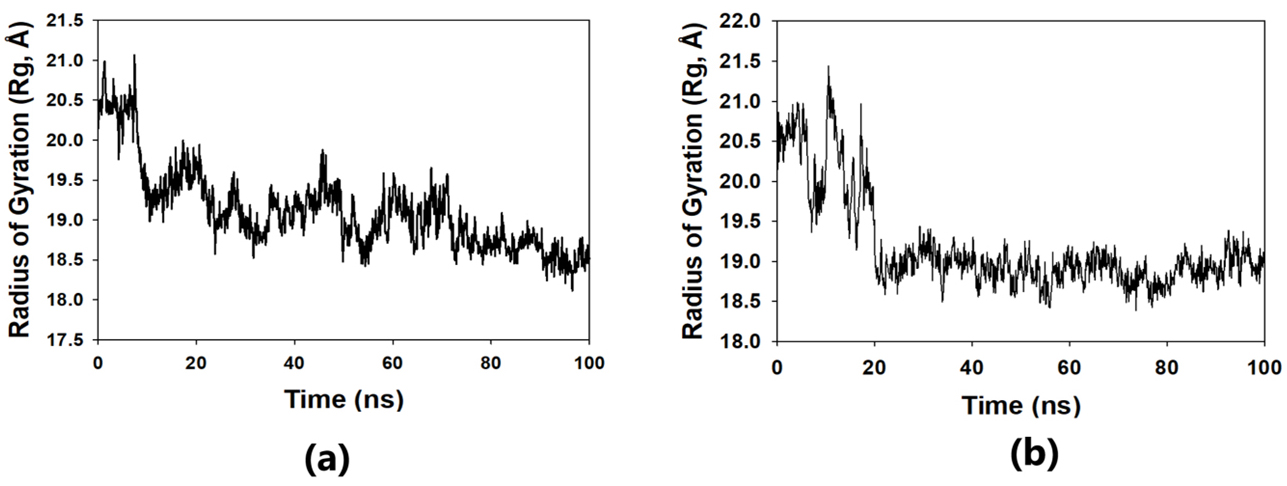
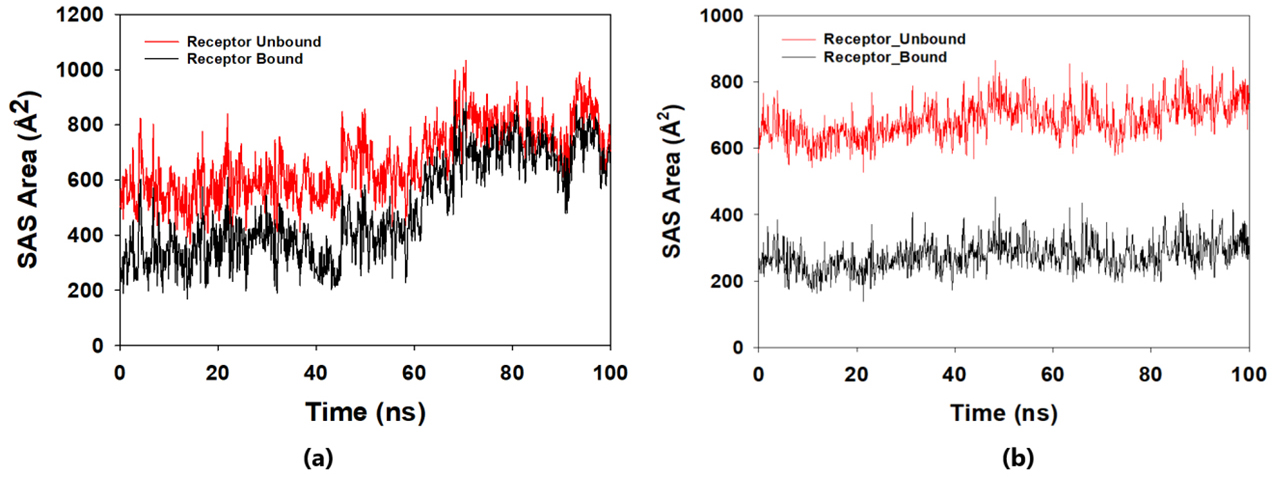
The solvent-accessible surface area analysis and radius of gyration analysis were carried out to ascertain the level of compactness in the structure and the accessibility of the solvent to both lepiotaprocerin D and peptidoglycan. Solvent accessible surface area (SASA) analysis was performed using Desmond to investigate the binding affinity and potential inhibitory activity of two ligands (lepitaprocerin D and peptidoglycan) with BUB1B. The SASA values of the protein-ligand complex were calculated for both bound and unbound forms. We focused on the SASA analysis to investigate the solvent-accessible surface area changes upon binding of each ligand to BUB1B. The SASA plots of the protein-ligand complex in both bound and unbound forms were generated to analyze the changes in the solvent-accessible surface area of the protein upon ligand binding. The radius of gyration of lepiotaprocerin D was observed from the figure as 19.8 Å (Fig. 8a), whereas peptidoglycan was 19.2 Å (Fig. 8b). The SASA plot also showed a higher value for lepiotaprocerin D as compared to peptidoglycan, shown in Fig. 9a,b.
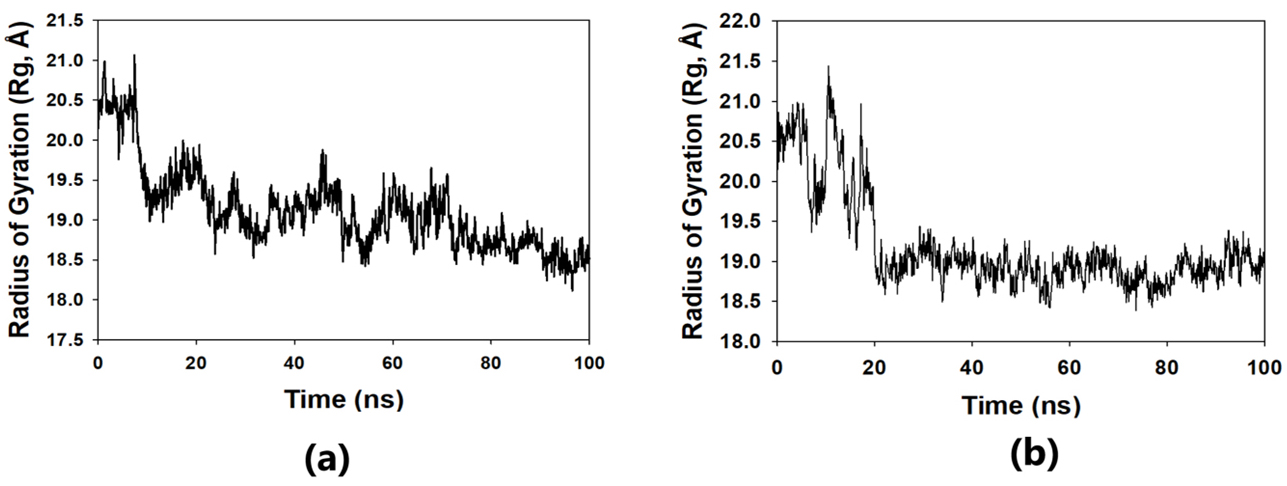 Fig. 8.
Fig. 8.Radius of gyration plot. (a) R
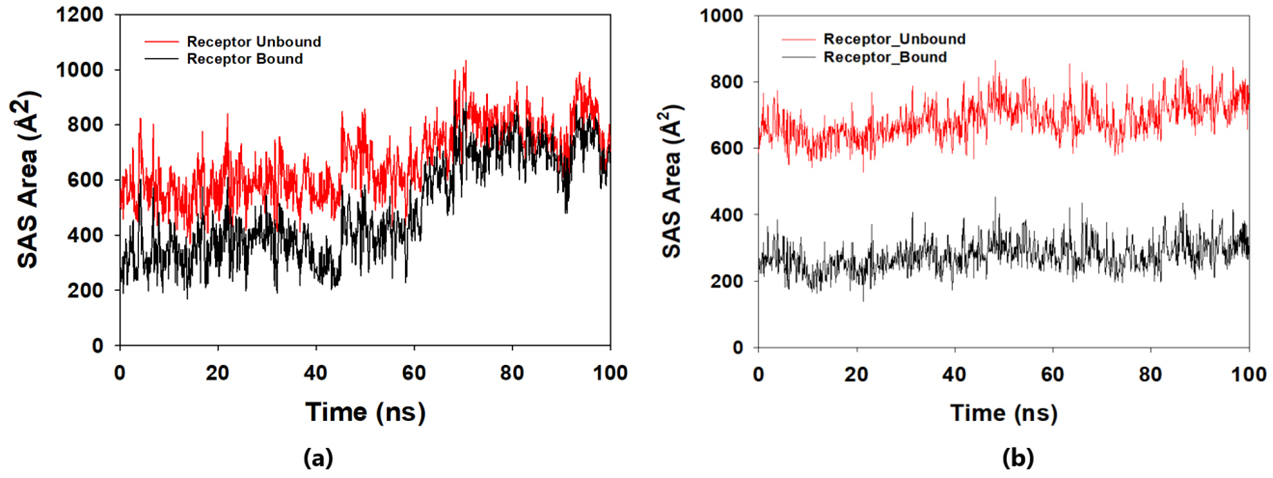 Fig. 9.
Fig. 9.Solvent accessible surface area (SASA) plot. The red fluctuation represents unbound receptor. The black fluctuation represents bound receptor. (a) The SASA plot shows a higher value for lepiotaprocerin D. (b) The SASA plot shows a lower value for peptidoglycan compared to lepiotaprocerin D.
The present study focused on identifying naturally occurring edible mushroom
compounds, such as P. ostreatus, with anticancer potential and the
ability to inhibit BUB1B (2WVI). The leading cause of tumor progression is
defective chromosomal segregation guarded by mitotic checkpoints [29, 30, 31]. This
chromosomal instability causes aneuploidy which plays a vital role in
tumorigenesis and cancer metastasis. The BUB1B protein, the primary component of
the SAC, is responsible for corroborating proper chromosomal segregation [4, 29, 30, 31, 32]. Compared to normal cells, the over-expression of BUB1B in cancer cells
regulates the tumorigenesis process. Hence, 2WVI is considered a potential
therapeutic target for the treatment of cancers as its inhibition could reduce
the process of aneuploidy, tumorigenesis and metastasis. The extract obtained
from edible mushrooms, P. ostreatus, reportedly suppressed cell
proliferation in breast and colon cancers through p53-dependent and
p53-independent pathways [10, 31, 32]. In this study, we predicted the
drug-likeness properties based on the ADMET properties of 70 bioactive compounds
extracted from edible mushrooms [33]. All 70 bioactive compounds qualified using
Lipinski’s rule of 5 (RO5). Lepiotaprocerin D, with a molecular weight of 480.60
daltons (
In this study, lepiotaprocerin D was bound with 2WVI along SER157, and this binding proposed this compound as the most suitable for inhibitor design. A novel discovery of this study was the formation of the hydrogen bond of ASN123 with lepiotaprocerin D which further strengthened the binding of 2WVI and lepiotaprocerin D as a potential inhibitor. Due to the highly helical nature of 2WVI in the case of lepiotaprocerin D, the binding between 2WVI and lepiotaprocerin D was found to be more stable. The radius of gyration that evaluates the compactness of protein structure was shown to decrease in lepiotaprocerin D, which gives an insight into the more stable conformation of the protein 2WVI. The decrease in the lepiotaprocerin D SASA supported our observation of the radius of gyration [36]. Hence, the molecular dynamic simulations parameters supported the efficacy of lepiotaprocerin D for the treatment of different cancers and could be used for the design of a suitable inhibitor that could reduce the effect of over-expressed 2WVI, thereby reducing tumorigenesis and metastasis of breast cancer and also the progression of glioblastoma multiforme (GBM) and hepatocellular carcinoma (HCC) [37].
The limitations of this study are that the above-discussed research work was carried out using an in silico approach. Hence, the results will need to be validated using a wet lab approach to obtain the significance of the obtained results.
BUB1B is a primary element of the SAC. As overexpression of BUB1B is related to
various cancers there is a need for suitable bioactive compounds to inhibit it
with minimal toxic side effects. Thus, 70 bioactive compounds from edible
mushrooms, which possess medicinal qualities and have proven anti-cancerous
properties, have been screened against BUB1B. Structure-based in-silico
approaches have been used, i.e., virtual screening, docking, and MD simulation.
Two potential bioactive compounds lepiotaprocerin D and peptidoglycan, exhibited
good drug-likeness and static affinity within the binding cavity of BUB1B through
docking. Further dynamic studies using MD simulation (100 ns; RMSD, RMSF, radius
of gyration (R
GBM, glioblastoma multiforme; HCC, hepatocellular carcinoma; ADMET, absorption,
distribution, metabolism, excretion; SAC, spindle assembly checkpoint; R
Datasets used and/or analyzed for this study are available from the corresponding author upon appropriate request.
All authors made substantial contributions to the conception and design of the work, as well as the acquisition, analysis, and interpretation of data. The specific contributions are as follows: Formal drafting, SNR, and SKS; Investigation and equal contribution to this work DM and AM; Project administration, MPS; Funding acquisition, EV. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest. EV is serving as one of the Guest editors of this journal. We declare that EV had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to AA.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.