
 , Manuel Martínez-Sellés 1,2,3,*
, Manuel Martínez-Sellés 1,2,3,*
1 Cardiology Department, Hospital General Universitario Gregorio Marañón, CIBERCV, 28007 Madrid, Spain
2 School of Health and Biomedical Sciences, Universidad Europea, 28670 Madrid, Spain
3 School of Medicine, Universidad Complutense, 28040 Madrid, Spain
Abstract
Pharmacotherapy is the cornerstone treatment for patients with heart failure (HF) that uses drugs targeting the renin-angiotensin-aldosterone system (RAAS), including angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), and the angiotensin receptor-neprilysin inhibitor (ARNI) sacubitril/valsartan. This article reviews the pathophysiology of the RAAS and the neurohormonal changes seen in patients with HF as well as the targets and the mode of action of these drugs. We also assess the role of ACE in ventricular remodeling and summarize the main evidence for the use of ACE-related drugs in HF patients.
Keywords
- angiotensin-converting enzyme
- heart failure
- mortality
The renin-angiotensin-aldosterone system (RAAS) plays an essential role in the mechanism of congestive heart failure. Current guidelines recommend drugs aimed at this system, particularly in patients with reduced ejection fraction, to improve survival and stabilize or reverse cardiac remodeling [1]. These drugs include angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), and the angiotensin receptor-neprilysin inhibitor (ARNI) sacubitril/valsartan [1, 2].
This review aims to explore the pathophysiology of heart failure (HF), particularly its relation to the RAAS, as well as the mechanism of action of ACE-related drugs and the evidence for their use in HF.
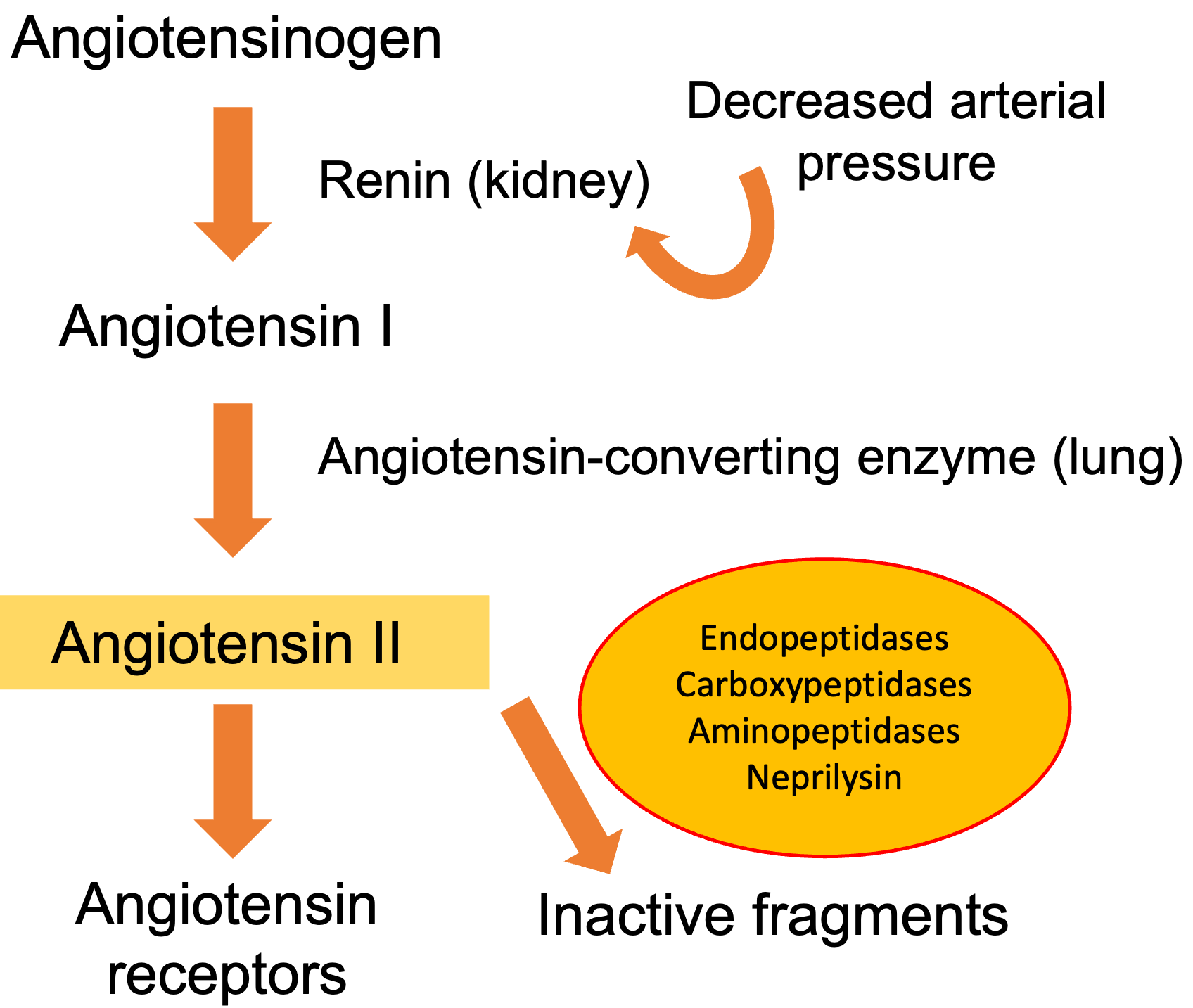
The compensatory mechanisms triggered in HF include the activation of the adrenergic nervous system and the RAAS. Short-term hemodynamic effects are mainly due to the influence of the adrenergic nervous system on total peripheral vascular resistance and capacitance, along with its effects on inotropism. In the long term, the RAAS is of greater importance in maintaining cardiac output through sodium and water retention, peripheral arterial vasoconstriction, and increased myocardial contractility [2, 3]. In HF, several factors increase renin liberation by renal juxtaglomerular cells, including the drop in renal perfusion, the reduction in the amount of sodium in the distal tubule, and the intensified sympathetic stimulation (Fig. 1).
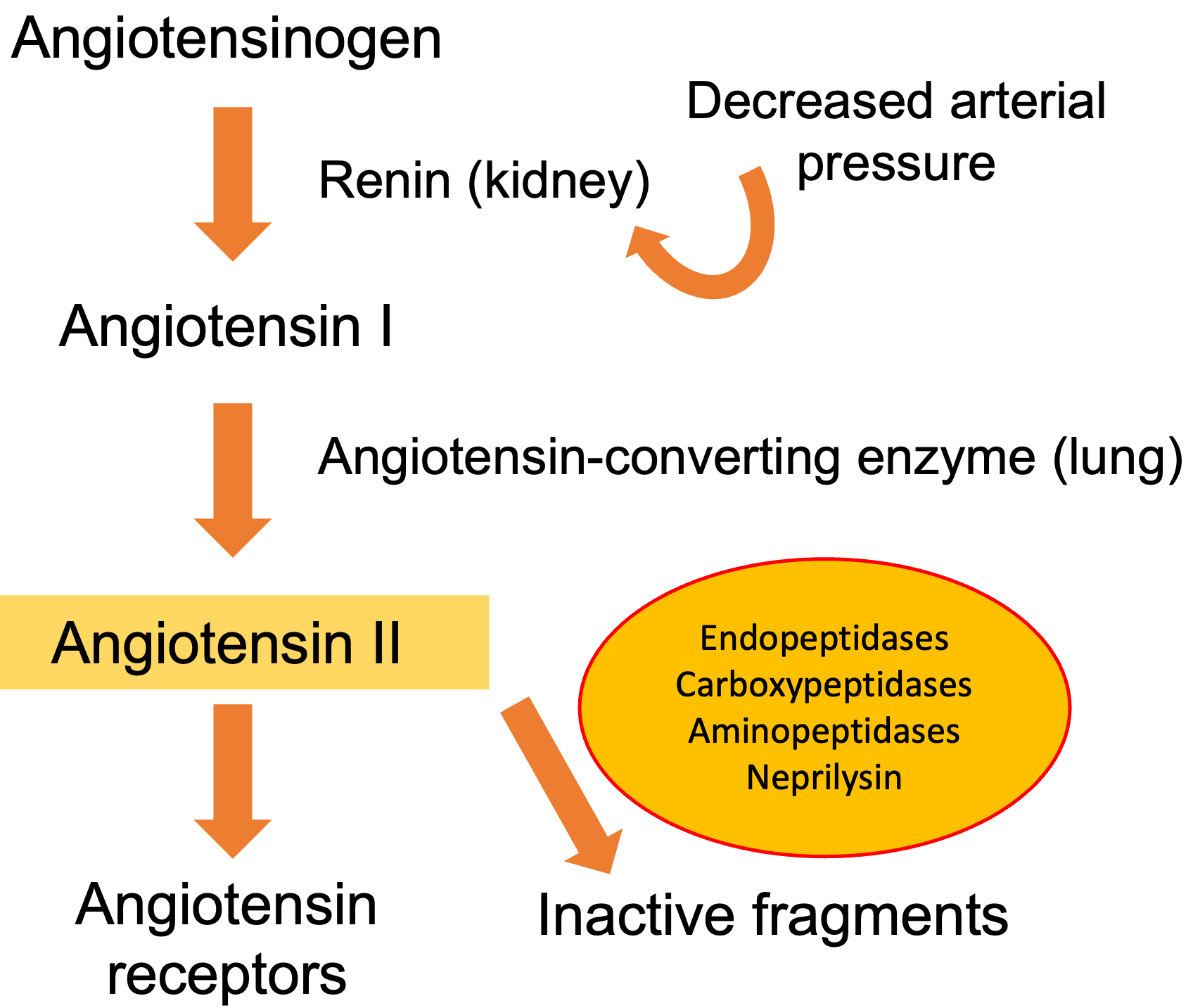 Fig. 1.
Fig. 1.The renin-angiotensin-aldosterone system. Angiotensin II plays a key role in the pathophysiology of heart failure. Angiotensin II is digested by neprilysin, endopeptidases, and carboxy-peptidases into peptide fragments.
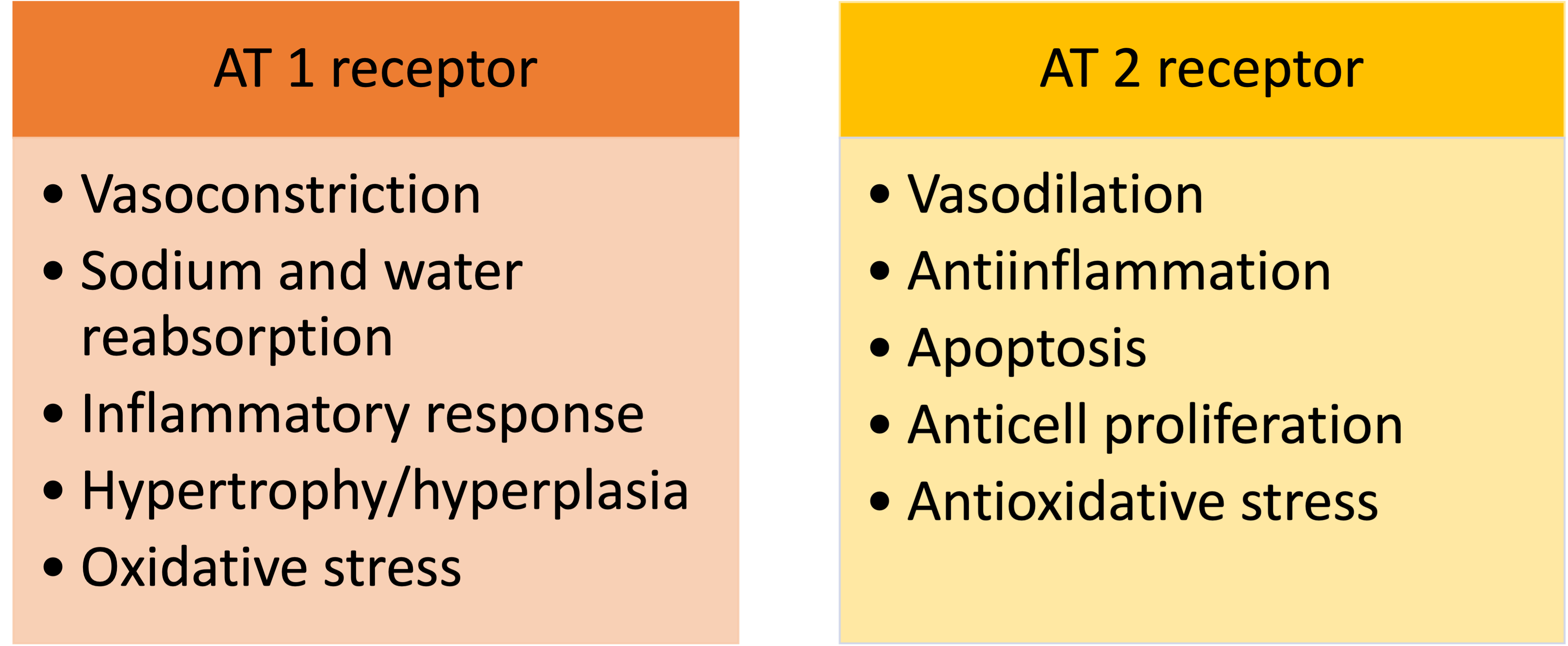
Renin catalyzes the conversion of a plasma protein synthesized in the liver, angiotensinogen, to form angiotensin I, a peptide with only mild vasoconstrictor properties. The ACE is a nonspecific enzyme, present mainly in the endothelium of lung vessels, involved in the conversion of angiotensin I into angiotensin II. Other tissues, such as the kidneys and blood vessels, contain the ACE, allowing local production of angiotensin II. Angiotensin II is a strong vasoconstrictor critical in maintaining circulatory homeostasis that is usually rapidly inactivated by different angiotensinases [3]. It binds to two receptors, angiotensin type I (AT 1) and angiotensin type 2 (AT 2), with opposite effects (Fig. 2). In the human myocardium, the ratio of AT 2 to AT 1 is 2:1, however, the expression of AT 1 receptors rises in HF, leading to vasoconstriction, aldosterone secretion, cell growth, and catecholamine release [2].
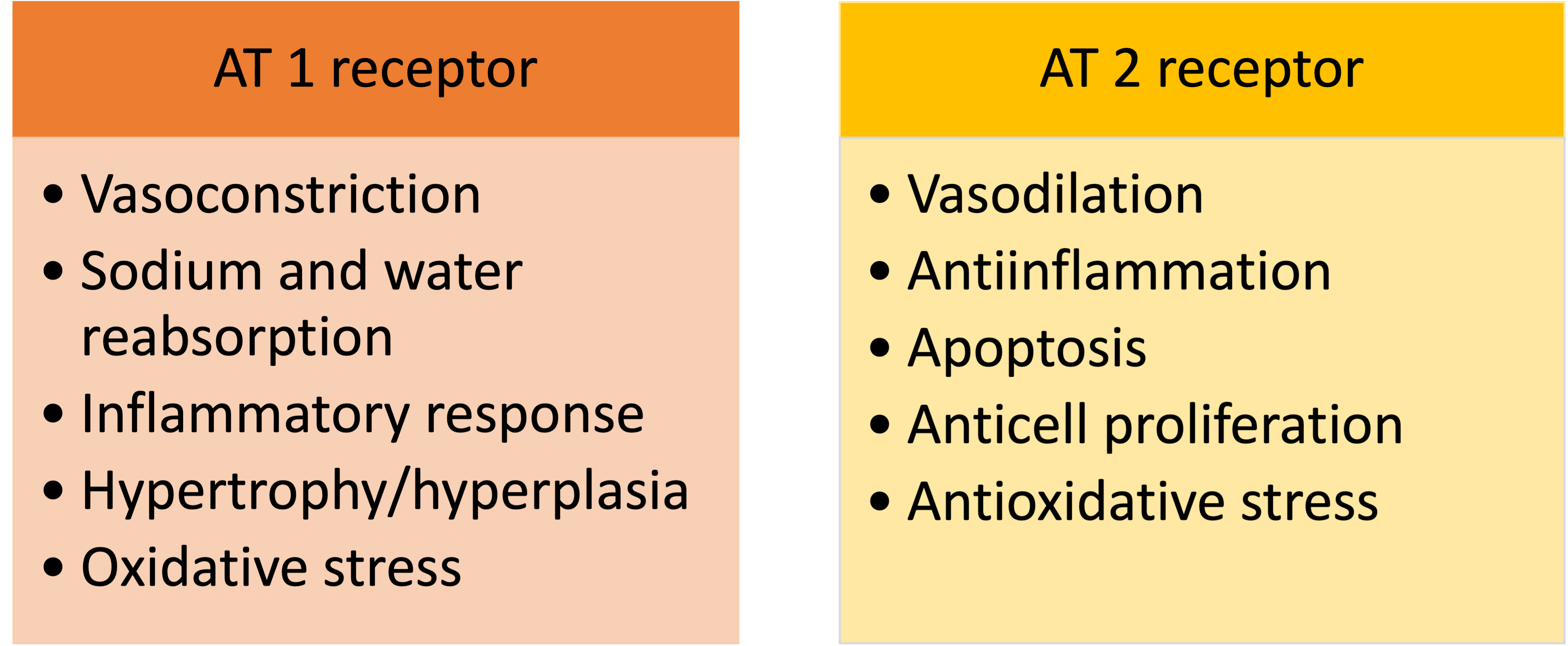 Fig. 2.
Fig. 2.Angiotensin II (AT) receptors. Main effects related to receptor activation.
During its persistence in the tissues, angiotensin II elevates arterial blood pressure by two mechanisms [3]:
1. Vasoconstriction, with the increase in peripheral resistance and venous return.
2. Reduction of renal excretion of sodium and water, increasing extracellular fluid volume; directly and through the stimulus that promotes aldosterone secretion from the adrenal glands.
In HF, the production of angiotensin II is detrimental, producing heart and kidney fibrosis. Angiotensin II also results in worsening neurohormonal activation, as it increases the release of norepinephrine and stimulates the adrenal cortex to produce aldosterone. Aldosterone has independent effects on extracellular volume regulation, binding to the mineralocorticoid receptor in the distal tubule and promoting sodium retention and potassium loss. In addition, aldosterone causes myocardial and vascular fibrosis, direct vascular damage, baroreceptor dysfunction, and prevents the uptake of norepinephrine by the myocardium [4].
Several counter-regulatory systems become activated in HF patients to compensate for the effects of the vasoconstricting neurohormones. Natriuretic peptides are released in response to increases in atrial and myocardial stretch, unloading the heart by increasing kidney excretion of salt and water, and inhibiting the release of renin and aldosterone. Natriuretic peptides promote vasodilation by enhancing cyclic guanosine monophosphate-mediated smooth muscle relaxation and increasing capillary permeability. Despite great increases in circulating natriuretic peptides, as HF progresses, their renal effect becomes progressively dulled, allowing RAAS effects to persist [5]. Natriuretic peptides are broadly synthesized in multiple tissues and degraded mainly by internalization, followed by lysosomal and enzymatic degradation by neutral endopeptidase neprilysin [2]. The autonomic nervous system and other local auto-regulatory mechanisms interact to preserve blood flow in the brain and heart while causing intense vasoconstriction to decrease flow to other organs during exercise or injury. This process is mediated by vasoconstricting neurohormones, which in turn activate counter-regulatory vasodilator responses to offset their deleterious effects. Bradykinin is one of these vasodilators released at sites of inflammation and coagulation. It creates powerful arteriolar dilation as well as increased capillary permeability. The enzymes ACE and neprilysin mediate the breakdown of bradykinin as well as the formation of the potent vasoconstrictor angiotensin II [3, 6].
Cardiac remodeling is defined as a sum of molecular, genetic, cellular, and interstitial changes that are generated after cardiac burden or injury, which produces an increase in heart volume and changes from elliptical to a spherical shape, resulting in ventricular systolic and diastolic dysfunction [7]. In patients with HF, cardiac remodeling is associated with a poor prognosis; conversely, its reversal is associated with improved outcomes [8, 9]. Therefore, it is essential to avoid the hemodynamic load and the neurohormonal mechanisms that produce cardiac remodeling. Early HF treatment might prevent cardiac remodeling development and slow disease progression [10].
ACE inhibitors are competitive inhibitors of ACE that reduce the levels of angiotensin II by diminishing the conversion of angiotensin I to angiotensin II, therefore modulating the RAAS [11]. In addition, ACE inhibitors reduce the secretion of aldosterone and vasopressin, lower sympathetic nerve activity, and inhibit kininase II, leading to the up-regulation of bradykinin, subsequently increasing the effects of angiotensin suppression. ARBs block the effects of angiotensin II on the AT 1 receptor, the subtype responsible for the maladaptive effects in the remodeling of the heart. ARBs exert similar effects to ACE inhibitors on blood pressure, renal function, and potassium. More recently, the ARNI that antagonizes the RAAS and inhibits the neutral endopeptidase has emerged. The mixture of a neprilysin inhibitor (sacubitril) and an AT 1 receptor antagonist (valsartan) inhibits the neurohormonal response, avoiding vasoconstriction, sodium retention, and maladaptive remodeling [12]. By decreasing the degradation of natriuretic peptides, bradykinin, and adrenomedullin, an ARNI can enhance diuresis, natriuresis, and myocardial relaxation. In addition, the ARNI inhibits renin and aldosterone secretion, while selectively blocking the AT 1 receptor [2, 12], implying reduced vasoconstriction, salt and water retention, and myocardial hypertrophy.
Most adverse effects of ACE inhibitors are linked to their action in the RAAS, including hypotension, mild azotemia, and potassium retention (Table 1) [2]. Side effects of kinin potentiation include nonproductive cough and angioedema. ARBs have no effect on the kinin systems and may be used in patients intolerant to ACE inhibitors due to cough, skin rash, or angioedema [2]. In the Prospective Comparison of ARNI with ACE inhibitor to Determine Impact on Global Mortality and Morbidity in Heart Failure (PARADIGM-HF) trial, patients treated with sacubitril/valsartan were more likely to have symptomatic hypotension than those treated with enalapril, although this adverse effect usually did not require drug withdrawal. On the other hand, cough, worsening of renal function, and hyperkalemia were less frequent in the sacubitril/valsartan group [12]. The incidence of angioedema was low and did not differ between groups.
| Adverse effects | ACE-I | ARB | ARNI |
| Hypotension | 5–10% | 1.3–5% | 14% |
| Hyperkalemia | 4–6% | 2–3% | 4% |
| Worsening renal function | 4–7% | 1–7% | 3% |
| Cough | 10–15% | 0.2% | 11% |
| Angioedema | 1% | 0.1% | 0.2% |
ACE-I, Angiotensin-converting enzyme inhibitor; ARB, Angiotensin receptors blocker; ARNI, angiotensin receptor-neprilysin inhibitor.
Antagonism of the RAAS has been associated with the so-called “aldosterone breakthrough” (i.e., a rebound increase in circulating aldosterone), a phenomenon in which the aldosterone levels are reduced in the initial treatment phases, but may later increase, even surpassing their initial values [13]. Aldosterone breakthrough might affect the beneficial effects of ACE-related drugs, as the expression of aldosterone increases fluid retention and has deleterious effects, including enhancement of inflammation, fibrosis, and oxidant-mediated cell injury. The incidence of aldosterone breakthrough ranges from 10% to 53% in the literature and is similar to ACE inhibitors and ARBs but seems to be uncommon with the ARNI [14], though clinical data supporting aldosterone breakthrough mitigation with ARNIs are scarce [15, 16]. In addition, spironolactone and eplerenone can diminish aldosterone rebound [4, 17, 18].
Sodium-glucose cotransporter type 2 (SGLT2) inhibitors may also interact with ACE-related drugs. SGLT2 inhibitors decrease urinary glucose and sodium reabsorption in the proximal tubule, increasing osmotic diuresis, natriuresis, and glycosuria. Their effects on the RAAS are still unclear. On one hand, the reduction in plasma volume and blood pressure and the increase of sodium delivery in the macula densa may potentially lower the levels of renin [19]. On the other, an increase in renin activity in the early stages of SGLT2 inhibitor treatment has been described, but it seems to disappear in the long term without an effect on aldosterone levels [20]. Nevertheless, clinical evidence supports the fact that SGLT2 inhibitors decrease the risk of hyperkalemia, and slow the progression of renal dysfunction, without increasing the risk of symptomatic hypotension [21].
Two neurohormonal systems, the sympathetic nervous system, and the RAAS, are intricately involved in the progression of HF. These systems interact in a positive feedback manner, whereby sympathetic activation results in increased renin secretion leading to RAAS activation, and RAAS activation leads to sympathetic overactivity by increasing noradrenaline release. There is considerable rationale for combining beta-blockers, which target the sympathetic nervous system, with ACE-related drugs, and robust evidence of the benefits of beta-blocker and ACE-related drugs in patients with HF and reduced ejection fraction. In combination, these two classes provide a comprehensive neuroendocrine blockade targeting both the heart, where beta-blockade reduces cardiac output, and the vessels, where ACE inhibition induces vasodilation [22].
ACE inhibitors have been associated with both hemodynamic and symptomatic improvements in patients with congestive HF [23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35] (Table 2, Ref. [12, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56]). The COoperative North Scandinavian ENalapril SUrvival Study (CONSENSUS) trial was the first to show that an ACE inhibitor, enalapril, reduced mortality in HF patients with reduced ejection fraction and severely reduced functional class [36]. In 1991, the Studies of Left Ventricular Dysfunction (SOLVD) trials extended the indication to patients in New York Heart Association Classes I–III [37, 39]. Although these trials showed a considerable reduction of mortality with enalapril, the endpoint seems to be consistent with other agents [38, 57], suggesting an ACE-inhibitor class effect. They reduce mainly deaths attributed to HF progression, although they have also been shown to reduce the incidence of myocardial infarction, arrhythmic deaths, and fatal stroke, suggesting that there are multiple mechanisms of benefit, including prevention of ventricular remodeling, anti-ischemic mechanisms, and reduction of neurohormonal activation [58, 59]. Furthermore, ACE inhibitors have also consistently demonstrated ameliorating symptoms and increasing quality of life in HF patients with reduced ejection fraction [1].
| Trial | Drug | Inclusion criteria | N | Outcome’s improvement |
| Angiotensin-converting enzyme inhibitors | ||||
| Magnani and Magelli, 1986 [23] | Captopril | HF, NYHA II–III | 94 | NYHA class, exercise capacity, and LEVF |
| Bussman et al., 1987 [24] | Captopril | HF, NYHA III–IV | 23 | Hemodynamic parameters and NYHA class |
| Captopril-Digoxin Multicenter Research group, 1988 [25] | Captopril | HF, NYHA I–II | 300 | Exercise capacity and NYHA class |
| Captopril Multicenter Research Group, 1988 [26] | Captopril | HF, NYHA II–IV | 105 | Mortality |
| Barabino et al., 1991 [27] | Captopril | HF |
150 | NYHA class and 6-min walking test |
| Munich Mild Heart Failure Trial, 1992 [28] | Captopril | HF, NYHA II | 170 | HF progression |
| Cleland et al., 1985 [29] | Enalapril | HF, II–IV | 20 | NYHA class, symptoms, and exercise capacity |
| CONSENSUS, 1987 [36] | Enalapril | Congestive HF, NYHA IV | 253 | Mortality |
| Enalapril CHF investigators, 1987 [30] | Enalapril | Congestive HF, NYHA II–III | 36 | NYHA class and exercise capacity |
| Dickstein et al., 1991 [31] | Enalapril | Congestive HF, NYHA II–III, previous MI | 41 | - |
| SOLVD-Treatment, 1991 [37] | Enalapril | NYHA I–IV, LVEF |
2569 | Mortality |
| SOLVD-Prevention, 1992 [39] | Enalapril | NYHA I–II, LVEF |
4228 | Composite of death and HF admission |
| Chalmers et al., 1987 [32] | Lisinopril | Congestive HF, NYHA II–IV | 130 | Exercise capacity and NYHA class |
| ATLAS, 1999 [38] | Lisinopril | NYHA II–IV, LVEF |
3164 | Composite of death and HF admission |
| Lechat et al., 1993 [33] | Perindopril | Congestive HF, NYHA II–III | 125 | Exercise capacity and NYHA class |
| Riegger et al., 1990 [34] | Quinapril | Congestive HF, NYHA II–III | 225 | Exercise capacity and NYHA class |
| Gundersen et al., 1994 [35] | Ramipril | Congestive HF, NYHA II–III | 223 | NYHA class |
| Angiotensin receptor blockers | ||||
| Crozier et al., 1995 [44] | Losartan | NYHA II–IV, LVEF |
134 | Pulmonary capillary wedge pressure |
| Dickstein et al., 1995 [45] | Losartan | NYHA II–IV, LVEF |
166 | - |
| ELITE, 1997 [46] | Losartan | NYHA II–IV, LVEF |
722 | Mortality |
| Weber et al., 1997 [47] | Losartan | NYHA II–IV | 154 | - |
| Lang et al., 1997 [48] | Losartan | NYHA II–IV, LVEF |
116 | - |
| Mazayev et al., 1998 [49] | Valsartan | HF, NYHA II–IV | 116 | Pulmonary capillary wedge pressure |
| Val-HeFT, 1999 [41] | Valsartan | NYHA II–IV, LVEF |
5010 | Mortality/morbidity |
| STRETCH, 1999 [50] | Candesartan | NYHA II–III, LVEF 30%–45% | 844 | Exercise capacity and NYHA class |
| RESOLVD, 1999 [51] | Candesartan | NYHA II–IV, LVEF |
768 | - |
| SPICE, 1999 [52] | Candesartan | NYHA II–IV, LVEF |
270 | - |
| Tonkon et al., 2000 [53] | Irbesartan | NYHA II–IV, LVEF |
109 | - |
| ELITE II, 2000 [43] | Losartan | NYHA II–IV, LVEF |
3152 | - |
| ADEPT, 2001 [54] | Eprosartan | NYHA II–IV, LVEF |
36 | - |
| CHARM-Added, 2003 [40] | Candesartan | NYHA II–IV, LVEF |
2548 | Composite of death and HF admission |
| CHARM-Alternative, 2003 [42] | Candesartan | NYHA II–IV, LVEF |
2028 | Composite of cardiovascular death and HF admission |
| HEAAL, 2009 [55] | Losartan | NYHA II–IV, LVEF | 3846 | HF admission |
| Angiotensin receptor-neprilysin inhibitors | ||||
| PARADIGM-HF, 2014 [12] | Sacubitril-valsartan | NYHA II–IV, LEVF |
8442 | Composite of cardiovascular death and HF admission |
| PIONEER-HF, 2019 [56] | Sacubitril-valsartan | LVEF |
881 | Composite of cardiovascular death and HF admission |
ADEPT, Addition of the AT 1 Receptor antagonist Eprosartan to ACE Inhibitor Therapy in Chronic Heart Failure (trial); ATLAS, Assessment of Treatment with Lisinopril And Survival (trial); CHARM-Added, Candesartan Cilexitil in Heart Failure Assessment of Mortality and Morbidity (trial); CHARM-Alternative, Candesartan in Heart failure Assessment of Reduction in Mortality and Morbidity (trial); CONSENSUS, COoperative North Scandinavian ENalapril SUrvival Study (trial); ELITE, Losartan Heart Failure Survival Study (trial); HEAAL, Heart failure Endpoint evaluation of Angiotensin II Antagonist Losartan (trial), HF, heart failure; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association; PARADIGM-HF, Prospective Comparison of ARNI with ACE-I to Determine Impact on Global Mortality and Morbidity in Heart Failure (trial); PIONEER-HF, Comparison of Sacubitril/Valsartan versus Enalapril on Effect on NT-proBNP in Patients Stabilized from an Acute Heart Failure Episode (trial); RESOLVD, Randomized Evaluation of Strategies for Left Ventricular Dysfunction (trial); SOLVD, Studies of Left Ventricular Dysfunction (trial); SPICE, Study of Patients Intolerant of Converting Enzyme Inhibitors (trial); STRETCH, Response to Exercise Trial of Candesartan Cilexetil in Heart Failure (trial); Val-HeFT, Valsartan Heart Failure Trial.
ARBs are currently a second-line treatment for patients intolerant to ACE inhibitors or ARNIs due to cough, skin rash, or angioedema [1]. The Candesartan in Heart failure: Assessment of Reduction in Mortality and Morbidity (CHARM)-Added study showed only a modest effect in the addition of an ARB to an ACE inhibitor treatment in patients with stable HF [40], whereas no benefit on mortality was shown in the Valsartan Heart Failure Trial (Val-HeFT) study [41]. However, valsartan did show a reduction of the combined endpoint of mortality and morbidity in comparison with placebo in those patients not treated with an ACE inhibitor, whereas candesartan considerably reduced the composite of all-cause mortality, cardiovascular death, or hospital admission in the CHARM trial [42]. A direct comparison of ACE inhibitors and ARBs was assessed in the Losartan Heart Failure Survival Study (ELITE-II), showing no increased survival in older HF patients treated with losartan in comparison with captopril [43]. No ARB trial has shown a benefit in overall mortality regarding ACE inhibitors in HF patients [44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 60], so their indication remains for patients unable to tolerate ACE inhibitors. High doses of losartan were associated with a major reduction in HF admissions in another study [55], suggesting that up-titration of ARBs may add clinical benefit.
In PARADIGM-HF [12], sacubitril/valsartan, compared with enalapril, reduced the composite of mortality from cardiovascular causes or a first HF admission in HF patients with reduced ejection fraction. Sacubitril/valsartan was also associated with an improvement in quality of life and physical and social activities, particularly household chores and sexual activity [61]. The trials Comparison Pre- and Post-discharge Initiation of Sacubitril/Valsartan in HFrEF Patients After an Acute Decompensation Event (TRANSITION) [62] and Comparison of Sacubitril/Valsartan versus Enalapril on Effect on NT-proBNP in Patients Stabilized from an Acute Heart Failure Episode (PIONEER-HF) further showed that early initiation and up-titration of sacubitril/valsartan provided a benefit also after acute decompensated HF [56, 63]. Based on this evidence, current guidelines suggest that either ACE inhibitors or ARBs are replaced by sacubitril/valsartan in ambulatory patients with HF and reduced ejection fraction that persist symptomatic even though under optimized treatment (Class IA), lowering the indication for ACE inhibitor-naïve patients (Class IIb) [1].
Sacubitril/valsartan improves cardiac remodeling, increasing left ventricular ejection fraction [64] and improving most echocardiographic indexes of systolic and diastolic function [65, 66]. In the Prospective ARNI vs. ACE inhibitor trial to DetermIne Superiority in reducing heart failure Events after Myocardial Infarction (PARADISE-MI), sacubitril/valsartan reduced global longitudinal strain by 42% compared to ramipril [67]. Furthermore, the drug also improves renal dysfunction, an effect mainly driven by the increase in natriuretic peptides and the intracellular mediator cyclic guanosine monophosphate, mitigating the effectiveness characteristic of chronic HF [5, 68]. In the United Kingdom Heart and Renal Protection III study, sacubitril/valsartan had similar effects as irbesartan on kidney function and albuminuria at 12 months, and also the additional effect of lowering blood pressure and cardiac biomarkers in patients with chronic kidney disease [69, 70].
The ARNI has shown superiority over ACE inhibitors in reducing all-cause mortality and HF hospitalization in patients with reduced ejection fraction who had been previously treated with an ACE inhibitor/ARB in the PARADIGM-HF trial and in patients hospitalized with acute decompensated HF in the PIONEER-HF trial. Despite the lack of information on ACE inhibitor/ARB-naïve patients, subgroup analyses of clinical trials, including PIONEER-HF [71] and Prospective Study of Biomarkers, Symptom Improvement, and Ventricular Remodeling During Sacubitril/Valsartan Therapy for Heart Failure [72] have shown a consistent benefit of ARNIs in these patients. Real-world clinical practice data support this benefit [73, 74, 75] and the American College of Cardiology Expert Consensus Decision Pathway on HF Treatment recommends ARNI as the preferred RAAS inhibitor in ACE inhibitor/ARB-naïve patients [76]. As sacubitril/valsartan is an excellent drug for reducing HF hospitalizations and improving the overall quality of life in symptomatic patients with HF and reduced ejection fraction, our recommendation is to use it as soon as possible in these patients.
Current HF guidelines distinguish between HF with mildly reduced ejection fraction and HF with preserved ejection fraction [1]. However, most clinical trials have used the 40% cut-off for left ventricular ejection fraction. To date, none of the trials performed with ACE-related drugs in HF patients with preserved ejection fraction have met their primary endpoint (Table 3, Ref. [77, 78, 79, 80, 81]). The Perindopril in Elderly People with Chronic Heart Failure (PEP-CHF) trial did not report outcomes according to ejection fraction [77]. The Irbesartan in Heart Failure with Preserved Ejection Fraction Study (I-PRESERVE) [78] and Candesartan in Patients with Chronic Heart Failure and Preserved Left Ventricular Ejection Fraction (CHARM-Preserved) [79] trials with ARBs missed their primary endpoint of cardiovascular death or HF hospitalizations, although a subsequent analysis including recurrent hospitalizations suggested a significant reduction of the latter among the entire CHARM-Preserved cohort [82].
| Trial | Drug | Inclusion criteria | N | Outcome’s improvement |
| PEP-CHF, 2006 [77] | Perindopril vs. placebo | LVEF |
850 | NYHA class and 6-min walking test |
| I-PRESERVE, 2008 [78] | Irbesartan vs. placebo | NYHA II–IV, LVEF |
4128 | - |
| CHARM-preserved, 2003 [79] | Candesartan vs. placebo | NYHA II–IV, LVEF |
3023 | - |
| PARAMOUNT, 2012 [80] | Sac/Val vs. valsartan | NYHA II–IV, LVEF |
266 | NTproBNP |
| PARAGON-HF, 2019 [81] | Sac/Val vs. valsartan | NYHA II–IV, LEVF |
4822 | - |
CHARM-Preserved, Candesartan Cilexitil in Heart Failure Assessment of Mortality and Morbidity (trial); HF, heart failure; I-PRESERVE, Irbesartan in Patients with Heart Failure and Preserved Ejection Fraction (trial); LVEF, left ventricular ejection fraction; NTproBNP, NT-proB-type Natriuretic Peptide; NYHA, New York Heart Association; PARAGON, Prospective Comparison of ARNI with ARB Global Outcomes in HF with Preserved Ejection Fraction (trial); PARAMOUNT, LCZ696 Compared to Valsartan in Patients with Chronic Heart Failure and Preserved Left Ventricular Ejection Fraction (trial); PEP-CHF, Perindopril in Elderly People with Chronic Heart Failure (trial).
Sacubitril/valsartan significantly reduced natriuretic peptides in comparison with valsartan alone in the Prospective comparison of ARNI with ARB on Management Of heart failUre with preserved ejectioN fracTion (PARAMOUNT-HF) trial [80]. As for clinical outcomes, the Prospective Comparison of ARNI with ARB Global Outcomes in HF with Preserved Ejection Fraction (PARAGON-HF) trial failed to show significant differences in the primary endpoint of mortality and HF hospitalization [81]. However, a subgroup analysis showed a significant reduction in cardiovascular death and total HF hospitalizations in those with an ejection fraction under 57%. In addition, a combined analysis of PARADIGM-HF and PARAGON-HF trials showed a beneficial effect of sacubitril/valsartan on HF hospitalizations in patients with mildly reduced ejection fraction [83]. Current guidelines recommend that an ACE inhibitor, ARB, or ARNI may be considered for patients with HF and mildly reduced ejection fraction to reduce the risk of HF hospitalization and death (IIb indication) [1].
ACE-related drugs modulate the disproportionate activation of the RAAS and the adrenergic nervous system typically seen in HF patients. These drugs can stabilize and/or reverse cardiac remodeling, improve HF symptoms, and reduce mortality.
MMS designed the manuscript structure. SÁZ performed the literature research. Both authors wrote and approved the manuscript.
Not applicable.
Not applicable.
English revision supported by the funding from Proyecto de Investigación PI21/01501, Instituto de Salud Carlos III. Madrid. Spain.
The authors declare no conflict of interest. MMS had served as one of the Guest editors of this journal. We declare that MMS had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Graham Pawelec.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.