


 , Sonia Fantone 1, Daniela Marzioni 1, Roberta Mazzucchelli 2
, Sonia Fantone 1, Daniela Marzioni 1, Roberta Mazzucchelli 21 Department of Experimental and Clinical Medicine, Università Politecnica delle Marche, 60126 Ancona, Italy
2 Department of Biomedical Sciences and Public Health, Section of Pathological Anatomy, Università Politecnica delle Marche, 60126 Ancona, Italy
Abstract
Prostate cancer is the second most common malignancy in men worldwide. Prostate cancer can be treated by surgery, radiotherapy and hormone therapy. The latter, in the form of androgen-deprivation therapy is needed to reduce prostate cancer progression at an advanced stage. Several studies demonstrated that oxidative stress is involved in cancer occurrence, development and progression and the Nuclear factor erythroid 2-related factor 2 (NRF2)/Kelch Like ECH Associated Protein 1 (KEAP1) pathway is affected by reactive oxygen species (ROS). Furthermore, the NRF2/KEAP1 signaling pathway has been investigated by several studies related to anti-androgen therapy, biochemical recurrence and radiotherapy. In this review we analysed the current literature regarding the indirect modulators involved in NRF2/KEAP1 pathway regulation and their role as possible therapeutic targets in prostate cancer cells.
Keywords
- NRF2
- chemotherapy
- prostate cancer
- signaling
- KEAP
- antioxidants
- modulator
Prostate cancer is the second most common malignancy in men [1]. Currently, the treatment options for prostate cancer include surgery, radiotherapy, hormone therapy and chemotherapy. Surgery and radiotherapy are the main treatment for localized prostate cancer, while androgen deprivation therapy (ADT) is the mainstay treatment for patients with advanced prostate cancer [2]. Although ADT initially shows good efficiency, the therapy often fails because patients become insensitive to the androgen blockade, and eventually develop castration-resistant prostate cancer (CRPC) [3, 4, 5]. Therefore, understanding the molecular mechanisms characterizing prostate cancer onset and progression may significantly improve patient treatment and outcomes.
The Nuclear Factor Erythroid 2-Related Factor 2 (NRF2 or NFE2L2)/Kelch-like
ECH-Associated Protein 1 (KEAP1) pathway plays a key role in protecting cells
from oxidative stress damage. In fact, it is affected by reactive oxygen species
(ROS) such as superoxide (O
Since NRF2/KEAP1 signaling pathway promotes the survival and proliferation of cancer cells, it is emerging as a potential therapeutic target in many neoplasms including ovarian, cervical, endometrial and prostate cancer [11, 21, 23]. In fact, NRF2 expression is significantly higher in chemo- and radio-resistant cancer tissues protecting cancer cells from the oxidative damage caused by chemotherapy and radiotherapy [11, 23, 24, 25]. Furthermore, NRF2 induces the expression of ATP-binding cassette (ABC) transporters to protect cancer cells from oxidative damage mediated by efflux chemotherapeutics [24]. NRF2 also plays a key role in regulating the self-renewal of cancer stem cells (CSCs), promoting the tumorigenicity, high rate of metastases and chemoresistance capacity of these cells [11].
The NRF2/KEAP1 signaling pathway plays an important role in modulating androgen receptor (AR) expression under ADT treatment. NRF2 can decrease AR expression in prostate cancer cells improving ADT therapy response and reducing cancer cells growth [26, 27].
Thus, therapeutic drugs targeting NRF2/KEAP1 signaling pathway in prostate cancer patients may significantly improve the therapy efficiency, improving the outcome of this disease.
Funes and colleagues [28] reported that NRF2 mRNA expression significantly decreased in prostate cancer tissues compared to normal tissues and was accompanied by a decreased expression of the NRF2-dependent genes Glutamate-Cysteine Ligase Modifier Subunit (GCLM), Glutamate-Cysteine Ligase Catalytic Subunit (GCLC) and NAD(P)H Quinone Dehydrogenase 1 (NQO1). Moreover, decreased NRF2 mRNA expression in prostate cancer tissue was associated with biochemical cancer recurrence (BCR) [28]. This study is in disagreement with two studies evaluating NRF2 protein expression. Kawata et al. [29] showed that cytoplasmic NRF2 and NQO1 protein expression were significantly upregulated after androgen-deprivation therapy (ADT) in both human prostate gland and carcinoma specimens suggesting that ADT induces NRF2 activation in prostate tissues and that NRF2 is a candidate molecular target for the prevention of castration-resistance progression of prostate cancer. Moreover, Raatikainen and colleagues [30] reported that cytoplasmic NRF2 expression was more abundant in prostate cancer tissues compared to normal tissues. Cytoplasmic NRF2 expression was associated with the elevation in Prostate-Specific Antigen (PSA) levels, which is the first sign of BCR in the prostate. Furthermore, in the multivariate analysis, increased cytoplasmic NRF2 expression predicted shortened BCR and worse overall survival [30].
The different results reported by Funes and colleagues [28] may be due to the only evaluation of NRF2 mRNA expression. The expression of mRNA can be posttranscriptionally modulated and stabilized by non-coding RNAs, thereby promoting its expression [31].
The involvement of the NRF2/KEAP1 signaling pathway in cancer progression was also evaluated in prostate cancer cell lines. It has been reported that ROS levels were significantly higher in PC3 cells than in DU145 cells. DU145 cells also showed higher levels of basal glutathione (GSH) content than PC3 cells. Moreover, DU145 cells had increased levels of NRF2 and NRF2-dependent genes such as GCLM, GCLC, Heme oxygenase 1 (HO-1) and Thioredoxin Reductase 1 (TXNRD1) leading to the radio-resistance of these cells. The sensitivity to radiation increased after NRF2 silencing [25]. Thus, NRF2 plays a key role in the radio-resistance of prostate cancer cells.
In this review, we analyzed the current literature regarding the indirect modulators involved in NRF2/KEAP1 pathway regulation and their role in prostate cancer cells as possible therapeutic targets.
The polybromo-associated BAF (PBAF) chromatin remodeling complex belongs to the Switch/Sucrose Non-fermentable (SWI/SNF) family and is essential for mammalian gene transcription and development [32, 33, 34]. The PBAF complex includes BRG1, PBRM1, ARID2, and BRD7, and is involved in regulating cell differentiation and genomic integrity [32]. In addition, polybromo-1 (PBRM1) is involved in DNA damage-induced transcriptional repression and DNA repair regulating the expression of genes involved in the oxidative stress response and apoptosis [35, 36, 37]. Mucin 1 C subunit (MUC1-C) is an oncogenic protein that promotes the progression of CRPC to neuroendocrine prostate cancer (NEPC) promoting self-renewal capacity, epithelial–mesenchymal transition (EMT), cancer stem cell (CSC) state, and tumorigenicity [38, 39, 40].
SLC7A11/xCT is a cystine-glutamate antiporter involved in mediating the uptake of extracellular cystine which is reduced to cysteine, a precursor in GSH synthesis. SLC7A11 is widely expressed in human tissues and protects cells from oxidative stress, thereby promoting GSH production, an important peptide with antioxidant effects [41, 42]. SLC7A11 plays also a key role in cancer favouring drug resistance [43].
Hagiwara and colleagues [44] reported that MUC1-C forms a complex with NRF2 and PBRM1 on the NRF2 target SLC7A11 gene, which encodes the xCT cystine-glutamate antiporter, increasing chromatin accessibility and inducing SLC7A11/xCT expression. Silencing MUC1-C or PBRM1 in DU-145 and LNCaP prostate cancer cells led to a significant downregulation of NRF2 and GSH levels increasing oxidative stress. Moreover, the authors found that MUC1-C and PBRM1 are necessary for the induction of other NRF2 target genes such as G6PD and PGD that are involved in regulating the pentose phosphate pathway. Thus, MUC1-C/PBRM1 signaling interacts with NRF2 to promote the expression of NRF2-dependent genes [44].
Pannexin 2 (PANX2) is a member of the pannexin family of membrane channel proteins [45]. High PANX2 expression promotes cancer cells proliferation and is associated with poor survival in renal cell carcinoma (RCC) [46]. Ferroptosis is an iron-dependent form of cell death caused by an accumulation of ferric iron and lipid peroxidation [47, 48, 49]. the NRF2/KEAP1 signaling pathway plays a significant role in preventing ferroptosis through the activation of antioxidant enzymes that counteract ROS production during ferroptotic cell death [50, 51].
PANX2 expression was significantly higher in prostate cancer cell lines (PC3, DU145, and LNCaP) compared to the normal immortalized human prostate epithelial cells (RWPE1). Moreover, PANX2 silencing significantly decreased cell proliferation, migration, and invasion, while increasing ferrous iron and malondialdehyde (MDA) levels in PC3 cells. However, these effects were rescued by oltipraz (an NRF2 activator), while PANX2 silencing significantly reduced NRF2, HO-1, and Ferritin heavy chain (FTH1) protein expression. Therefore, PANX2 may play a key role in prostate cancer pathogenesis since it regulates cancer cell proliferation, migration, invasion, and ferroptosis modulating the NRF2 signaling pathway [52].
The p62 protein, also known as sequestosome-1 (SQSTM-1), binds ubiquitinated proteins, targeting them for autophagy degradation [53]. Autophagy is a cellular mechanism involved in maintaining cellular homeostasis through the degradation and recycling of organelles, proteins, lipids, and nucleic acids [53]. High levels of p62 are associated with several diseases including inflammation, metabolic dysfunction, and cancer [54, 55]. An interesting study by Jiang and colleagues [56] found that p62 expression was significantly higher in prostate cancer cells compared to normal prostate tissues. Moreover, silencing p62 in DU145 cells significantly increased KEAP1 expression, while downregulating NRF2 expression and activation (nuclear expression). NRF2 silencing significantly decreased cell proliferation and invasion, whilst increasing apoptosis in DU145 cells. Furthermore, authors found that p62 activated the NRF2 pathway by binding KEAP1 and by inducing its proteasomal degradation, subsequently promoting NRF2 expression and the transcription of NRF2‑dependent genes such as HO-1, NQO1, GCLC and GLCM, resulting in decreased ROS levels. This study showed that p62 promotes prostate cancer progression by activating the KEAP1/NRF2 pathway. Thus, p62 may be a therapeutic target to improve prostate cancer outcomes [56]. Another study investigated the role of Speckle-type BTB/POZ protein (SPOP) in regulating p62 and NRF2 activity in prostate cancer. SPOP is a substrate recognition receptor for the CUL3/RING ubiquitin ligase complex that leads to the ubiquitination of its target substrates. Point mutations have been reported in the MATH domain (the substrate binding domain) of SPOP in prostate cancers [57, 58]. In addition, it has been reported that SPOP induces the non-degradative ubiquitination of p62, decreasing its ubiquitin-binding capacity, thereby suppressing p62-dependent autophagy in DU-145 cells. Moreover, SPOP attenuated p62-mediated KEAP1 sequestration facilitating NRF2 proteasomal degradation and decreasing the transcriptional activation of NRF2-dependent antioxidant genes. Furthermore, authors reported that SPOP mutants lose the capacity to ubiquitinate p62 by promoting autophagy and NRF2-induced antioxidant response in prostate cancer cells. Thus, SPOP plays a significant role in inhibiting autophagy through the p62/NRF2 signaling [59].
LncRNA taurine-upregulated gene 1 (TUG1) is a long non-coding RNA
(lncRNA) located on chromosome 22q12.2. Non-coding RNAs (ncRNAs) represent
approximately 98% of human genome transcripts which are classified as small
ncRNAs (
LncRNA TUG1 showed significant oncogenic effects in several cancers including colorectal, gastric, and breast cancers [73, 74, 75]. An interesting study found that lncRNA TUG1 expression was significantly increased in prostate cancer cells (DU145 and PC3 cell lines) compared to normal prostate epithelial RWPE1 cells. TUG1 silencing significantly decreased cell proliferation, migration, and invasion in PC3 cells but, these effects were reversed by using Oltipraz, a NRF2 activator. Authors also found that TUG1 silencing significantly decreased NRF2 expression, leading to a decreased expression of NRF2-dependent genes HO-1, FTH1, and NQO1. Thus, lncRNA TUG1 plays a significant oncogenic role in promoting cell proliferation and invasion through the modulation of NRF2 pathway in the PC3 cellular model [76]
Brain-type glycogen phosphorylase (PYGB), is the brain form of glycogen phosphorylase, a key enzyme involved in glycogen catabolism which converts glycogen into glucose-1-phosphate (G-1-P) providing energy to the body in an emergency state [77, 78]. High PYGB expression has been reported in several cancer types including gastric [79], lung [77] and colorectal [80] cancer.
The expression of PYGB has been reported to be significantly upregulated in prostate cancer tissues and was associated with disease progression. Moreover, PYGB silencing in PC3 cells significantly inhibited cell viability, thereby inducing apoptosis. Interestingly, these authors found that PYGB silencing increased the ROS content, which inhibited NRF2 protein in PC3 cells. Thus, PYGB can modulate prostate cancer cell growth, altering the NRF2 signaling pathway and increasing ROS content [81].
BReast CAncer gene 1 (BRCA1) is a tumor suppressor gene involved in
important cell functions such as DNA repair, cell-cycle regulation and
maintenance of genomic integrity. BRCA1 mutations confer an
increased risk for breast, ovarian and prostate cancers [82, 83]. The expression of
BRCA1 has been shown to protect cells from oxidant agents, inducing
antioxidant genes expression and subsequently activating/cooperating with important
transcription factors such as NRF2 [11, 84]. Labanca and colleagues [85] reported that BRCA1
binds to the HO-1 promoter increasing its expression in PC3 cells. BRCA1
overexpression significantly increased HO-1 expression while BRCA1 silencing had
the opposite effect. These authors also reported that BRCA1 overexpression
significantly decreased the expression of HO-1 suppressors such as MMP-9, uPA,
and Cyclin D1 in PC3 cells. Moreover, HO-1 was more expressed when both BRCA1 and
NRF2 were overexpressed compared to the single overexpression, indicating that
BRCA1 cooperates with NRF2 to induce HO-1 expression in PC3 cells [85]. These data are in
agreement with the study of Bae and colleagues [86] that found increased
expression of important antioxidant genes such as glutathione S-transferases (GSTs),
glutathione peroxidase 3 (GPX3), NQO1 and alcohol dehydrogenase 5 (ADH5) in DU145
cells that overexpress BRCA1. Consistently, BRCA1 overexpression conferred
resistance, while BRCA1 deficiency conferred sensitivity to oxidant agents such as
hydrogen peroxide and Paraquat. BRCA1 also stimulated the NRF2 antioxidant response
protecting cells against oxidative stress (induced by H
Inflammation plays a key role in the development and progression of several diseases including pregnancy complications [87, 88, 89, 90], endothelial dysfunction [13, 91, 92], skeletal disorders [93] and cancer [94, 95, 96].
It has been reported that mesenchymal stem cells (MSCs) can be recruited to the
tumor microenvironment due to local inflammation promoting angiogenesis and
tumor growth [97]. Yang and colleagues showed that MSCs pre-treatment with Tumor
necrosis factor alpha (TNF-
The studies discussed in this section are summarized in Table 1 (Ref. [44, 52, 56, 59, 76, 81, 85, 86, 100]), while a schematic representation of NRF2/KEAP1 signaling pathway regulation by cellular modulators is shown in Fig. 1.
| Modulator | Model studied | Effect | Ref. |
| MUC1-C/PBRM1 signaling | DU-145 and LNCaP cells | MUC1-C forms a complex with NRF2 and PBRM1 on the NRF2 target SLC7A11 gene increasing chromatin accessibility and inducing SLC7A11/xCT expression. MUC1-C or PBRM1 silencing led to the downregulation of NRF2 and GSH levels increasing oxidative stress. | [44] |
| PANX2 | PC3, DU145, LNCaP and RWPE1 cells | Higher PANX2 expression in PC3, DU145, and LNCaP cells compared to the normal prostate epithelial RWPE1 cells. PANX2 silencing in PC3 cells reduced NRF2, HO-1, and FTH1 expression decreasing cell proliferation, migration, and invasion while increasing ferrous iron and MDA levels. | [52] |
| p62 | DU145 cells | Higher p62 expression in prostate cancer cells compared to normal cells. p62 silencing increased KEAP1 expression, decreasing NRF2 expression and activation. NRF2 silencing decreased cell proliferation and invasion while increasing apoptosis. p62 binds KEAP1 inducing its proteasomal degradation and promoting NRF2 expression and the transcription of NRF2‑dependent genes HO-1, NQO-1, GCLC and GLCM, subsequently decreasing ROS. | [56] |
| SPOP | DU-145 cells | SPOP binds and induces p62 decreasing p62-dependent autophagy. SPOP attenuated p62-mediated KEAP1 sequestration facilitating NRF2 proteasomal degradation and decreasing NRF2-dependent antioxidant genes expression. SPOP mutants lose the capacity to ubiquitinate p62, thereby promoting autophagy and NRF2-induced antioxidant response in prostate cancer cells. | [59] |
| LncRNA TUG1 | DU145, PC3 and RWPE1 cells | Increased lncRNA TUG1 expression in DU145 and PC3 cell lines compared to normal prostate epithelial RWPE1 cells. TUG1 silencing decreased cell proliferation, migration, invasion, and NRF2, HO-1, FTH1, and NQO1 expression. | [76] |
| PYGB | PC3 cells | PYGB silencing inhibited NRF2 protein expression increasing ROS content and inducing apoptosis. | [81] |
| BRCA1 | PC3 cells | HO-1 was more expressed when both BRCA1 and NRF2 were overexpressed compared to the single overexpression. BRCA1 facilitated HO-1 expression interacting with NRF2. | [85] |
| BRCA1 | DU145 cells | BRCA1-overexpression increased GPX3, NQO1 and ADH5 expression. BRCA1 overexpression conferred resistance while BRCA1 deficiency conferred sensitivity to oxidant agents. BRCA1 stimulated the NRF2 antioxidant response, protecting cells against oxidative stress (which was attenuated by NRF2 silencing). | [86] |
| TNF- |
Mesenchymal stem cells (MSCs) and murine RM-1 prostate cancer cells | MSCs treatment with TNF- |
[100] |
MUC-1C, Mucin 1 C subunit; PBRM1, Polybromo-1; PANX2, Pannexin 2; SPOP,
Speckle-type BTB/POZ protein BRCA1, BReast CAncer gene 1; PYGB, Brain‑type
glycogen phosphorylase; TNF-
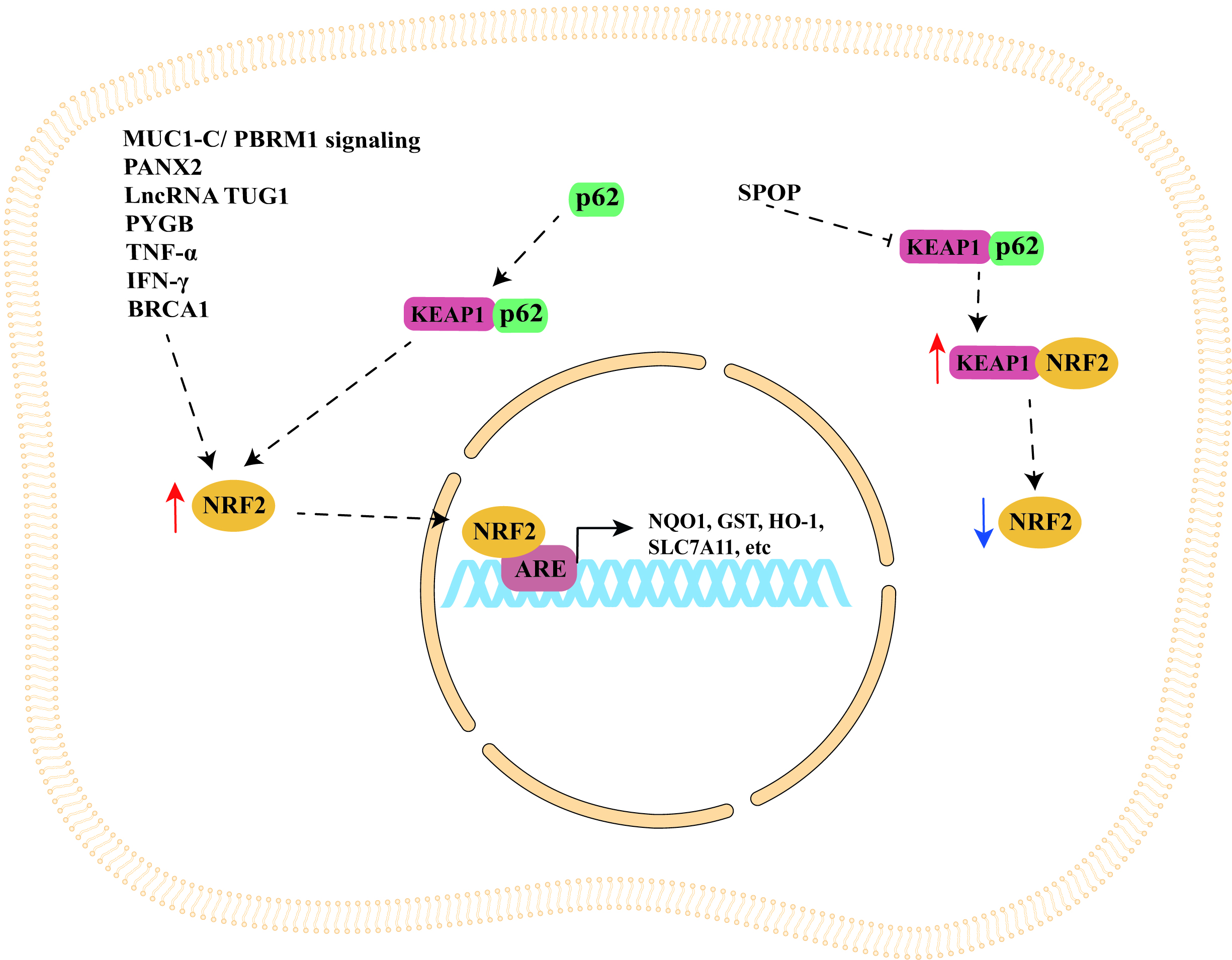 Fig. 1.
Fig. 1.Schematic representation of NRF2/KEAP1 signaling pathway
regulation by cellular modulators. NRF2/KEAP1 signaling pathway can be
indirectly upregulated by important cellular modulators such as MUC1-C/PBRM1
signaling, PANX2, p62, SPOP, lncRNA TUG1, PYGB, TNF-
Somatic mutations of NRF2 can impair NRF2 recognition by KEAP1 leading to an upregulation of NRF2 expression in some [101, 102, 103], but not in prostate cancer [103, 104]. Somatic mutations of KEAP1 have also been reported in lung and gallbladder cancers and were associated with increased NRF2 expression and chemotherapy resistance [105, 106, 107].
Yoo and colleagues investigated KEAP1 mutations in prostate cancer and found that 1/75 (1.3%) of prostate cancer tissues analyzed processed a G1069A mutation that leads to a D357N amino acid substitution [104]. Another study by Zhang and colleagues [108] sequenced the KEAP1 gene in six prostate cancer cell lines (DU-145, PC3, LNCaP, C42B, LAPC4, and CWR22RV1) and in normal prostate epithelial cells (PrEC). The study identified a C/T transition in CWR22Rv1 cells which leads to a threonine to methionine substitution at the 314th amino acid (T314M) in KEAP1 protein. Moreover, they found a T/C transition in LNCaP and C42B cells that causes a tyrosine to histidine substitution at the 255th amino acid position (Y255H). No mutation was indentified in KEAP1 in DU-145, LAPC4, PC3, and PrEC cell lines. Furthermore, the authors demonstrated that both T314M and Y255H mutations significantly inhibited NRF2 degradation, causing a significant increase in NRF2 activity that leads to an overexpression of NQO1 and HO-1 genes. Additionally, the study reported an hypermethylation of CpG islands in the KEAP1 promoter and an aberrant splicing of KEAP1 in DU-145 cells which leads to low levels of KEAP1 mRNA and an increased NRF2 activity with consequent expression of antioxidant genes. NRF2 silencing decreased GSH levels in DU-145 cells making them sensitive to chemotherapeutic drugs (paclitaxel, cisplatin, and etoposide) and radiation [108]. Thus, the authors demonstrated that in addition to KEAP1 mutations, hypermethylation of CpG island and aberrant splicing of KEAP1 also play a key role in the antioxidant response of prostate cancer cells influencing their sensitivity to chemotherapy and radiotherapy.
Nevertheless, although quite rare (1.3%), mutations have been reported in the NRF2 repressor, KEAP1 gene, in prostate cancer tissues. KEAP1 mutations have also been indentified in three prostate cancer lines: CWR22Rv1, LNCaP, and C42B cell lines. Finally, hypermethylation of the KEAP1 promoter and an aberrant splicing of KEAP1 have been reported in the DU-145 prostate cancer cell line [108].
The NRF2/KEAP1 signaling pathway plays a key role in prostate cancer onset and
progression and thus is a promising target for the treatment of this disease. In
this review, we discussed many studies highlighting the pleiotropic role of
the NRF2/KEAP1 signaling pathway in modulating important cellular processes such as
chemotherapy and radiotherapy response, cell proliferation, apoptosis, and
autophagy. In particular, we highlighted that NRF2/KEAP1 signaling pathway can be
indirectly regulated by several cellular modulators (see Table 1)
including MUC1-C/PBRM1 signaling, PANX2, p62, SPOP, lncRNA TUG1, PYGB, and BRCA1.
Moreover, it came up that NRF2/KEAP1 signaling can be modulated by TNF-
In conclusion, NRF2/KEAP1 signaling pathway modulation plays a pivotal function in protecting normal cells from oxidative stress and modulating cancer cell response to radiotherapy and chemotherapeutic agents. Although several studies are focused on directly modulation of the NRF2/KEAP1 signaling pathway, here we highlighted cellular modulators of the NRF2/KEAP1 signaling pathway that could be indirect therapeutic targets of this signaling in the treatment of prostate cancer.
GT contributed to writing original draft. GT, SF, DM and RM participated in conceptualization and writing review & editing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest. Given the role as Guest Editor, Giovanni Tossetta had no involvement in the peer-review of this article and has no access to information regarding its peer-review. Full responsibility for the editorial process for this article was delegated to Graham Pawelec.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.