
- Academic Editor
Background: Coronary artery disease is a leading public health problem.
However, the mechanisms underlying mitochondrial damage remain unclear. The
present study verified and explored the novel mechanisms underlying ischemic
injury based on a metabolomic analysis. Methods: Mouse models of acute
myocardial infarction were established, and serum samples were collected for
targeted liquid chromatography with tandem mass spectrometry analysis. Based on
metabolomic analyses, the N-methyl-d-aspartic acid receptor
(NMDAR)–related calcium transporting signaling pathway was selected. Primary
cardiomyocyte cultures were used, and N-methyl-d-aspartic acid
(NMDA) was used as an agonist to confirm the role of NMDAR in ischemic injury. In
addition, Bax, Bcl-2, mitochondrial calcium, potential, and mitochondrial
reactive oxygen species accumulation were used to explore the role of NMDAR in
mitochondrial damage–induced apoptosis. Results: Glutamate-related
metabolism was significantly altered following in acute myocardial infarction.
NMDA induces apoptosis under hypoxic conditions NMDAR was translocated to the
mitochondrial-related membrane after activation, and its mitochondrial expression
was significantly increased (p
Coronary artery disease remains a leading public health problem in developed and developing countries [1]. Although evolutionary primary coronary intervention (PCI) significantly improves the survival of patients with acute myocardial infarction (AMI), many AMI patients do not receive PCI in the window for different reasons [2]. However, patients may still experience heart failure after revascularization of the major culprit vessels through PCI [3]. In ischemic attack, the supply of oxygen and nutrition is strictly limited [4]. A remarkable number of cardiomyocytes die because of the accumulation of free radicals, lipid peroxidation, unbalanced calcium dynamics, altered energy, mitochondrial damage, or DNA disruption [5, 6, 7, 8]. Our previous studies mainly focused on mitochondrial damage after ischemic injury [9, 10]. Mitochondria, as the energetic core of the body, play a critical role in this pathological process.
Metabolomics is an emerging “omics” science involving the comprehensive features of metabolites and metabolism in biological systems [11]. Metabolomics is a promising method for diagnosing diseases, understanding pathophysiology, exploring the inner mechanisms, identifying novel drugs and pharmacological targets, and monitoring therapeutic outcomes [12]. Therefore, the present study used metabolomics to scan metabolic changes in a AMI murine model, which provided a detailed interrogation and several findings on the altered metabolism in this model. We further verified the target key protein and explored its mechanism of action in ischemic injury.
Fifteen male 10-week-old C57BL/6 mice (Chengdu DOSSY Experimental Animals Co., Ltd., China) from the same batch were divided into a sham-control group (n = 5) and an AMI model group (n = 10). As described by Gao et al. [13], the mice were anesthetized with an intraperitoneal injection of ketamine (50 mg/kg) and pentobarbital sodium (50 mg/kg) without ventilation support. A small incision was made left of the sternum, and a purse-string suture was prepared. The thorax was punctured and dilated using a mosquito clamp at the left 4th intercostal space. The beating heart was gently “popped out” and an 8-0 silk suture was quickly applied at a site on the left free wall approximately 3 mm below the atrium to block the left anterior descending coronary artery. Ligation was confirmed when the anterior left ventricular wall became pale. Following ligation, the heart was returned to the intrathoracic space and intrathoracic air was evacuated manually. The previous purse-string suture was tied and the mouse monitored during the recovery period. Mice in the sham-control group underwent the same surgical procedure and the beating hearts “popped out” without any ligation. All animal experiments were strictly performed following the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Publication no. 8023, revised in 1978) and registered with the Animal Ethics Committee of West China Hospital, Sichuan University (no. 20220221020).
All serum samples were collected at 24 h postoperative. As in our previous study
[11], each serum sample was precipitated using methanol spiked with two internal
standards. The supernatant was then collected. The dried supernatant was
reconstituted with 10 mM ammonium acetate in 30% water/70% acetonitrile + 0.2%
acetic acid containing 12.6 µM
The final sample reconstitution was added to the ultra-high-performance liquid
chromatography with tandem mass spectrometry under positive and negative ion
modes. The quality control (QC) sample was set as the pooled sample with a mixture of all serum
samples. The targeted metabolomics analysis was conducted using an LC-40A Ultra-Performance Liquid Chromatography (UHPLC)
system coupled with an AB Sciex triple quadrupole 5500 mass spectrometer (Nexera,
Framingham, MA, USA). Chromatographic separation was performed on a BEH Amide
column (2.1
Myocardiocytes are isolated rat cardiomyocytes, and the cell type was further verified with IF staining of α-actinin (Supplementary Fig. 1). Neonatal cardiomyocytes were prepared as described previously [14]. In brief, the hearts were collected from neonatal mice within 24–48 h and cut into pieces in D-Hank’s balanced salt solution. The cardiac tissues were digested in 22.5 µg/mL liberase blendzyme IV (Roche, Mannheim, Germany) at 37 °C for 40 min. Isolated cardiomyocytes in the pellet after centrifugation were resuspended in M199 medium with 10% fetal bovine serum and cultured in pre-coated plates. After 48 h, the cardiomyocytes were subjected to various treatments and measurements after 48 h of culture.
The oxygen–glucose deprivation (OGD) model was defined as a glucose-free medium under a deoxygenated atmosphere. In the present study, the OGD model was set with no glucose and 0% oxygen for 6 h of treatment before the sample collection.
NMDA (HY-17551; MedChemExpress, NJ, USA) was used as an agonist of NMDAR. NMDA was administered 30 min before the ODG or normoxic condition. In the preliminary study, NMDA concentrations were set at 10 µM, 50 µM, and 100 µM, and the final working dose was 10 µM. MK-801 (HY-15084B; MedChemExpress), which was used as a selective NMDAR antagonist, was pre-administered for 30 min at a concentration of 10 µM.
Cardiomyocyte lysates were subjected to western blotting analysis. Specific
target proteins (Caspase-3, NMDAR1A, Bax, and Bcl-2) were separated from the cell
lysates, transferred to polyvinylidene difluoride membranes, and visualized by
chemiluminescence. The primary antibodies included rabbit anti-
Total RNA from cultured cardiomyocytes was extracted using TRIzol reagent (Sigma, Poole, Dorset, UK). Circular DNA was synthesized using M-MLV reverse transcriptase (Invitrogen, Burlington, IA, USA). SYBR Green PCR master mix was used according to the manufacturer’s instructions (Eurogentec, San Diego, CA, USA), while 28S rRNA was used as a loading control. The primer sequences were as follows: nmdar1a 5′-ACT TGA AGG GCT TGG AGA-3′ (forward) and 5′-GGA GTG GAA CGG AAT GAT-3′ (reverse); nmdar2b 5′-GCT GGA GGC GTT GGA TGT-3′ (forward) and 5′-AAT GTG GAT TGG GAG GAC-3′ (reverse); 28S rRNA 5′-TTG AAA ATC CGG GGG AGA G-3′ (forward) and 5′-ACA TTG TTC CAA CAT GCC AG-3′ (reverse). A total of 34 amplification cycles were conducted on an Eppendorf Real-Time PCR machine. The expression of nmdar1a/nmdar2b in relation to the 28S rRNA control was determined.
The samples were fixed with 3% glutaraldehyde for 24 h, fixed in 1% osmium tetroxide, dehydrated in a series of acetone, infiltrated in Epon 812 for 30–60 min each time, and finally embedded. The semi-thin sections were stained with methylene blue, and ultrathin sections were cut with a diamond knife and stained with uranyl acetate and lead citrate. The sections were examined using a transmission electron microscopy (TEM, JEM-1400 Flash, JEOL Ltd., Tokyo, Japan).
A cell counting kit-8 (CCK-8) assay kit was purchased from Dojindo (Dojindo Molecular Technologies, Inc., Kumamoto, Japan). Cardiomyocytes were cultured in 96-well plates according to the manufacturer’s instructions. Following the different treatments, 10 µL of CCK-8 solution was added to each well. After 2 h of incubation, the absorbance was measured at 450 nm by a microplate reader.
Calcium concentration (ab142780; Abcam), mitochondrial ROS (M36008; Thermo Fisher Scientific), and mitochondrial membrane potential (M34152; Thermo Fisher Scientific) were measured using specific assay kits according to the manufacturer’s instructions.
The statistical analysis was performed using SPSS software version 19 (SPSS
Inc., Chicago, IL, USA). The results are presented as mean
To identify the altered profiles of metabolites in myocardial infarction, a targeted liquid chromatography with tandem mass spectrometry comprehensive analysis of 238 metabolites in positive and negative modes was performed on serum samples. A principal component analysis indicated that the metabolic profiles of mice in the AMI groups deviated from those of the sham-control group, showing alterations in metabolism profiles in AMI (Fig. 1).
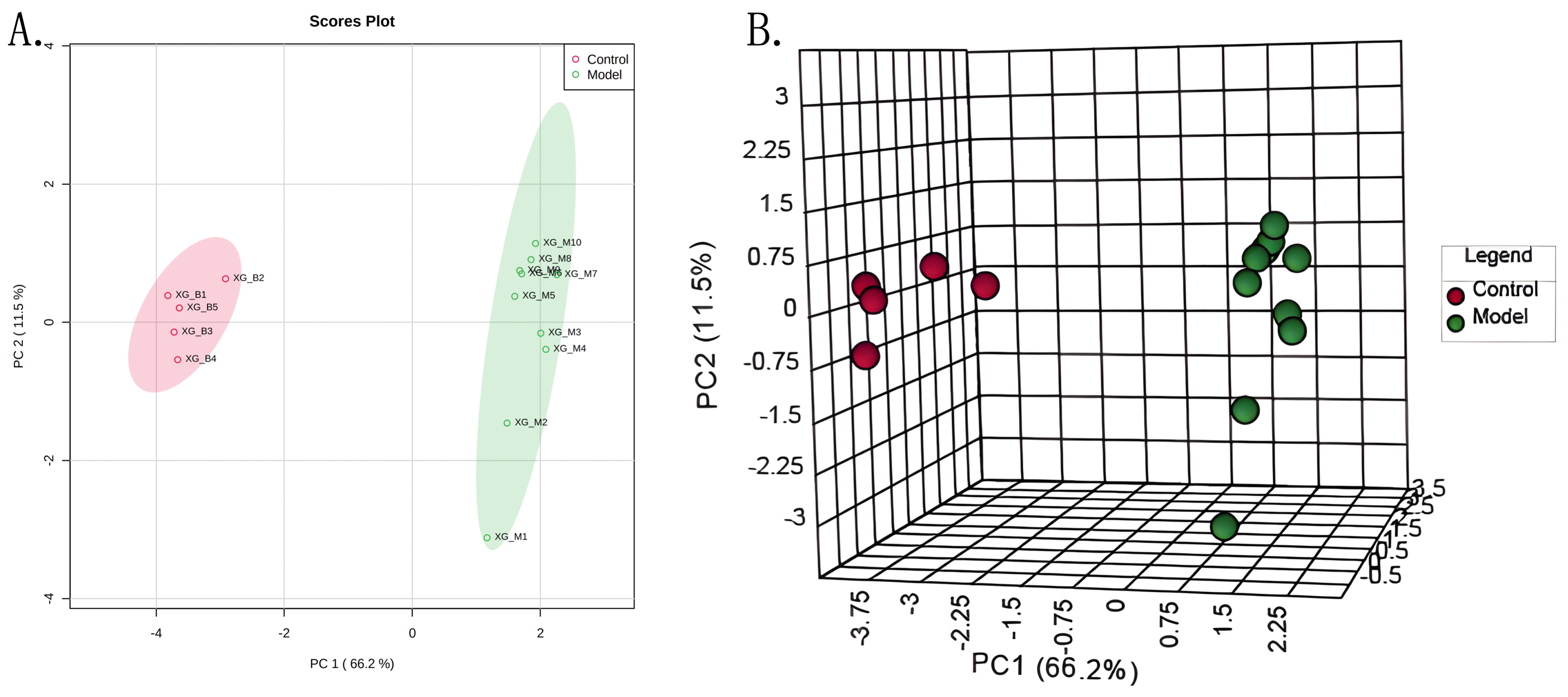 Fig. 1.
Fig. 1.PCA Scores plot (A) and 3D scores plot (B) for control and model groups. Abbreviations: PC1, principle component 1; PC2, principle component 2.
A heat map is shown in Fig. 2A. The most affected pathways were identified
according to the Kyoto Encyclopedia of Genes and Genomes signaling pathway
analysis based on the metabolites (impact values
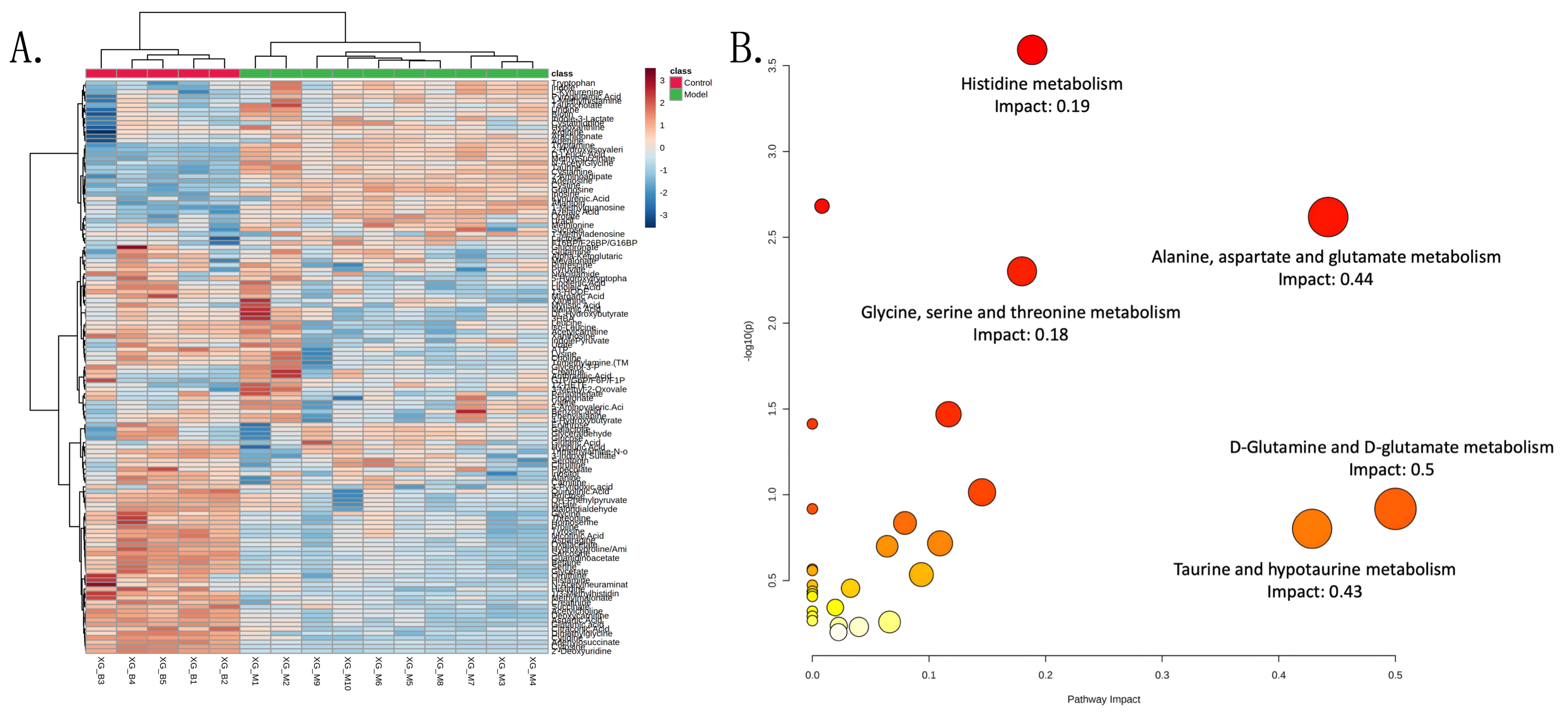 Fig. 2.
Fig. 2.Heatmap and impact pathways. (A) Heatmap of 128 metabolites in control and model groups. The colour bar from blue to red represents low to high mean intensity after normalization using the Z-score. (B) KEGG pathway analysis on the metabolites of two groups. The bubble diagram displayed selected pathways with an impact value larger than 0.2. The size of the bubbles represented impact values ranging from 0.2 to 1.0, and the colour of the bubbles means the p value after the negative Log transform.
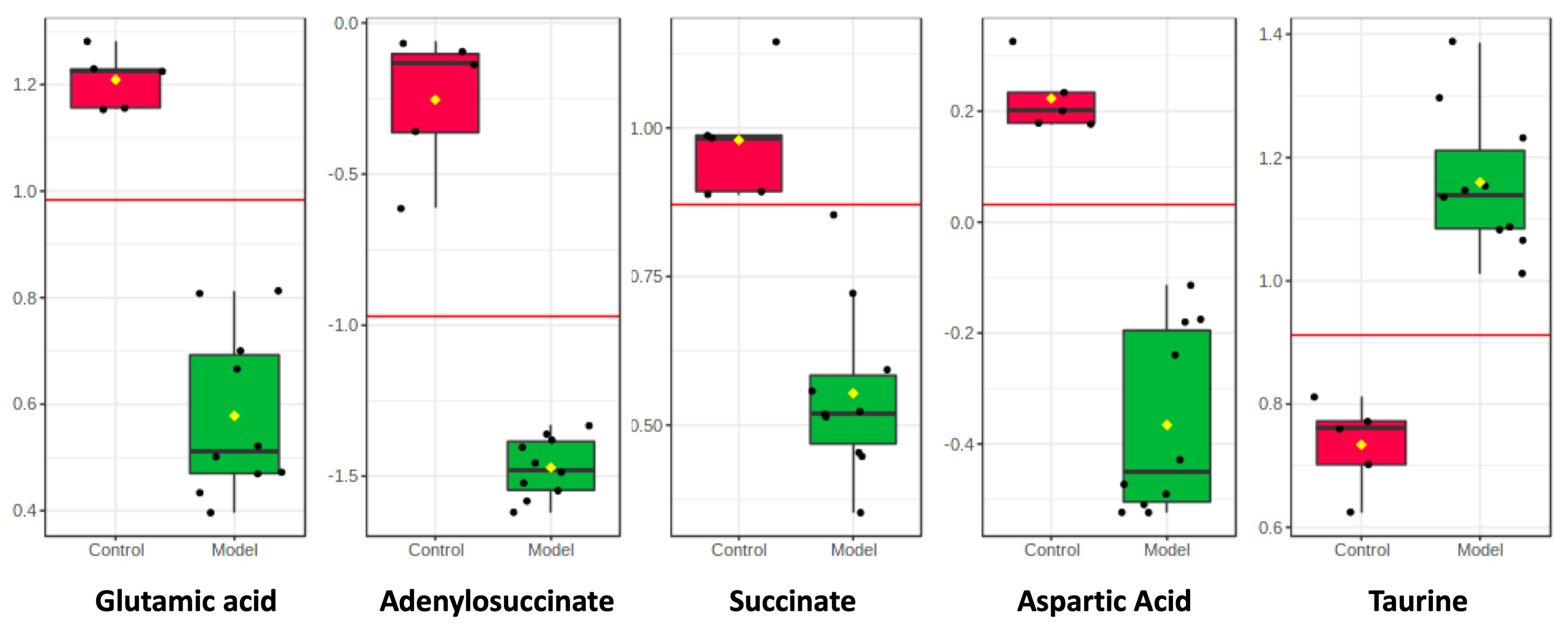 Fig. 3.
Fig. 3.Box plots showing integrated intensities for 5 selected LC-MS-identified biomarkers in two groups. The black dots represent the concentrations of the selected feature from all samples. t-test was used to identify the difference.
According to the metabolomics results, glutamate was selected as an essential
excitatory neurotransmitter, which reportedly exerts multiple regulatory effects
through its specific ligand (N-methyl-d-aspartic acid receptor
[NMDAR]) [15]. Thus, to confirm the role of NMDAR in ischemic injury–induced
cardiomyocyte apoptosis, we performed further studies in cell culture. NMDA is a
highly selective agonist of NMDAR. Different doses (10 µM, 50
µM, and 100 µM) of NMDA were used for pre-treatment for 30
min. As shown in Fig. 4A, NMDA did not exert any adverse effects under normal
conditions (p
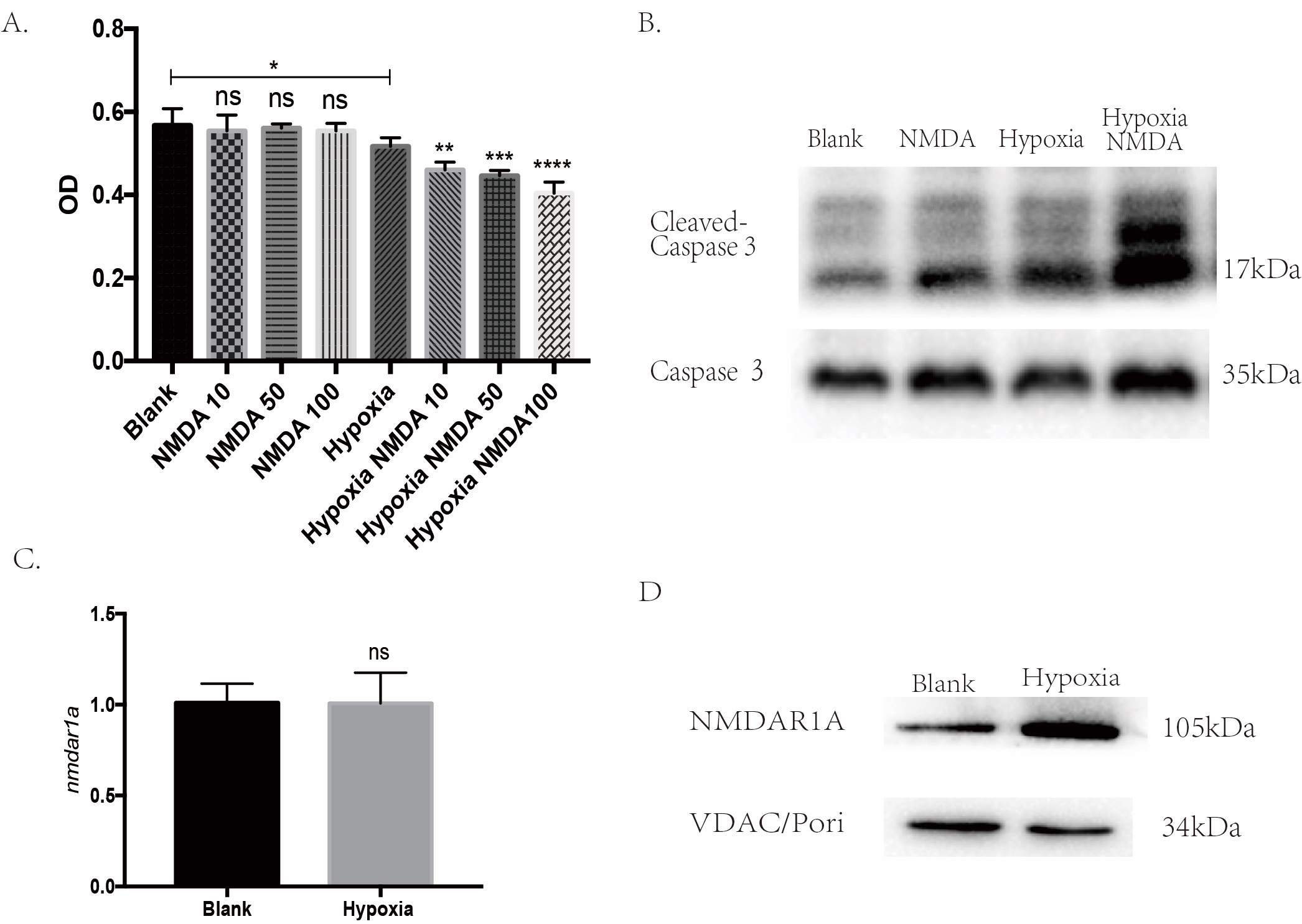 Fig. 4.
Fig. 4.NMDAR participates in the ischemic injury-induced cardiomyocyte
apoptosis. (A) CCK-8 assay showed NMDA could aggravate the hypoxic injury in a
dose-dependent manner (*p
Given the translocation of NMDAR into the mitochondria under hypoxic injury, we
further explored NMDAR-mediated mitochondrial damage in hypoxic injury–induced
apoptosis. MK-801, a selective NMDAR antagonist, was administered. With NMDAR
inhibition, apoptosis under hypoxic injury was significantly blocked, and the
CCK-8 assay showed improved cell viability (p
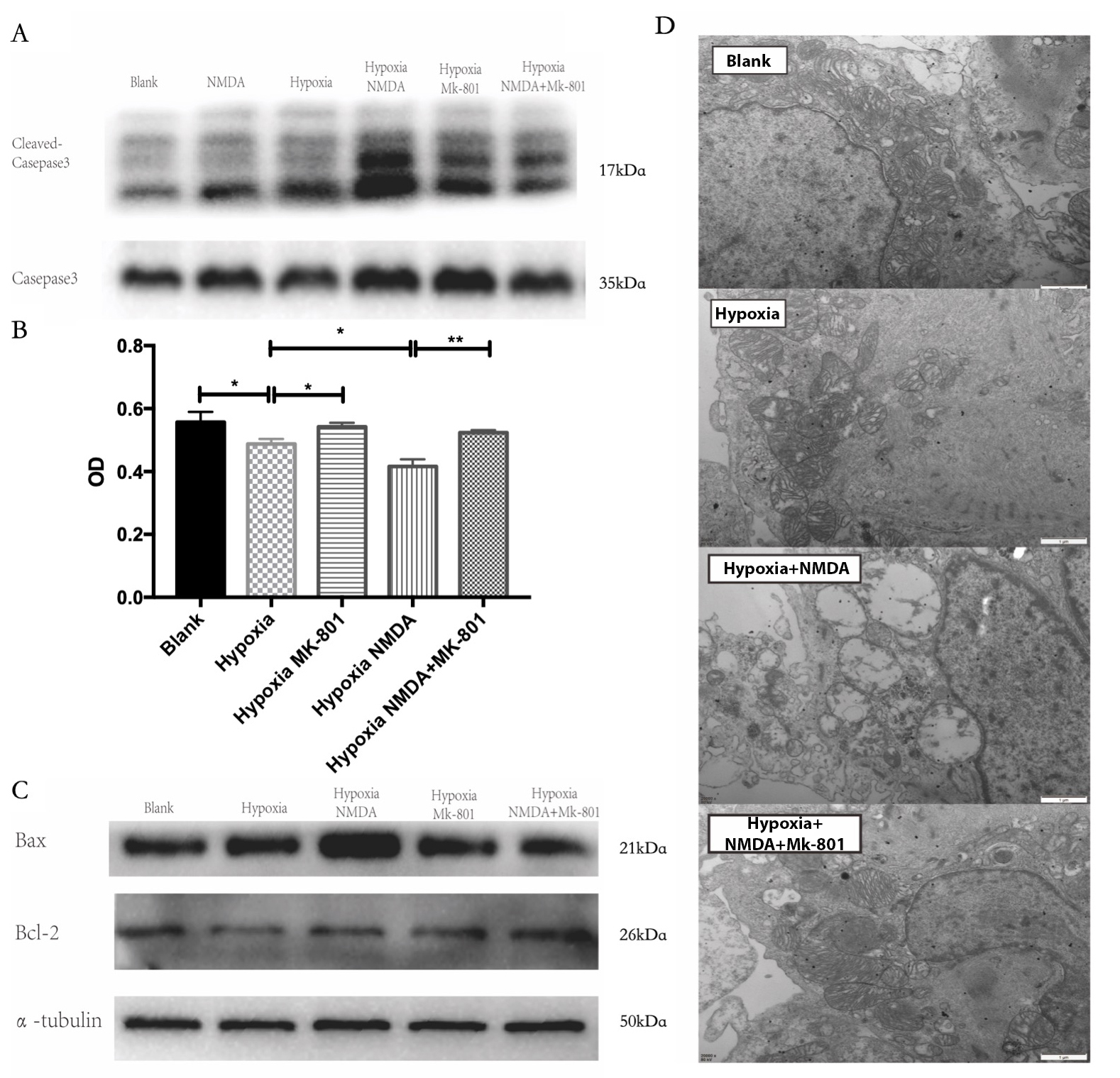 Fig. 5.
Fig. 5.NMDAR-mediated mitochondrial damage induces cardiomyocyte
apoptosis. (A,B) Inhibition of NMDAR with MK-801 significantly limited apoptosis
and improved survival (*p
Calcium accumulation or overload is an essential phenomenon in ischemic
injuries. The Rhod-2 calcium indicator was used to detect calcium levels in
mitochondria under different circumstances. Fluorescent images showed remarkable
calcium accumulation under hypoxic and non-hypoxic conditions with NMDA.
Treatment with MK-801 significantly reduced mitochondrial calcium accumulation in
cardiomyocytes (p
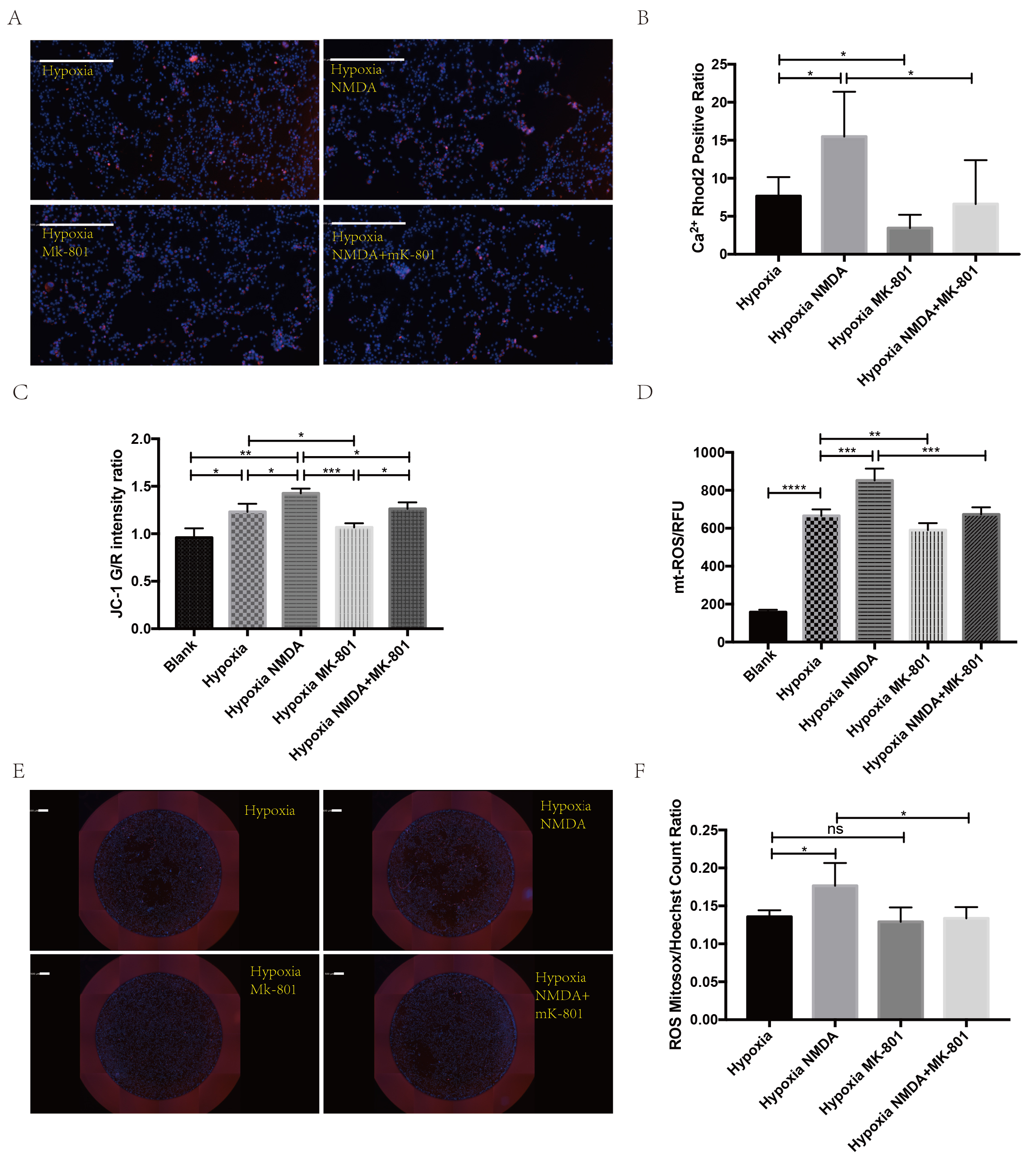 Fig. 6.
Fig. 6.NMDAR promotes calcium and mitoROS accumulation in the
mitochondria. (A,B) Representative images of calcium indicator and
quantification showed less calcium accumulation after MK-801 treatment
(white bar =500 µm; *p
In the present study, serum from AMI mice was subjected to liquid chromatography with tandem mass spectrometry–based metabolomic analysis, which showed significantly disturbed metabolic pathways and metabolite alterations. With further analysis, a glutamate-related metabolism pathway was identified in the process of AMI and NMDAR as a calcium channel, and its role in AMI was further explored.
As an emerging metabolomics technology, many studies have examined patients with AMI or AMI in animal models [16, 17]. However, given the high sensitivity of metabolomic analyses, intragroup consistency has been difficult in many studies, particularly human studies. In our study, the C57BL/6 mouse strain was chosen since it has a genome-stable background. A novel and efficient mouse model of left anterior descending artery ligation–induced AMI, first reported by Gao et al. [13], was used to ensure that all mice in the same group were derived from the same batch and established under the same conditions. Additionally, serum samples were used instead of cardiac tissue. The area supplied by the culprit vessels was difficult to identify, and normal cardiac tissue may significantly affect the metabolomic results. Therefore, as shown in our results section, 10 samples from the model group and five from the control group had good consistency, which ensured the validity of further metabolic pathway analyses.
The impact of the pathways was analyzed by pathway analysis using MetaboAnalyst 5.0 (http://www.metaboanalyst.ca) to evaluate their importance after AMI. Among them, glutamate and taurine metabolism are involved in the pathological changes associated with AMI. Taurine is a sulfur amino acid present in cardiomyocytes at high concentrations. It is well known that taurine exerts antioxidative activity, protects mitochondria, and inhibits calcium overload [18, 19, 20]. More importantly, taurine can modulate calcium channels to reduce calcium overload [21].
Glutamate has multiple cellular functions, including antioxidative,
pro-inflammatory, and membrane stabilization [22]. Many metabolites are
synthesized from glutamate, which serves as a precursor for amino acid
biosynthesis. Moreover, the excitatory neurotransmitter
(N-acetyl-l-aspartyl-l-glutamate) and inhibitory
neurotransmitter (
NMDAR was recently reported in the cardiovascular system, and its extensive activation drives calcium influx to promote ischemic injury or ischemia–reperfusion injury [26, 27, 28, 29, 30]. Liu et al. [28] reported that NMDA could promote calcium influx and calcium-channel blockers could rescue NMDAR-induced apoptosis through the p38 MAPK signaling pathway.
In the present study, we identified the effects of NMDAR activation under normoxic and hypoxic conditions. NMDA-stimulated NMDAR showed no difference in cell death under normoxic conditions, whereas NMDA showed significant adverse effects on hypoxic cardiomyocytes. In addition, under hypoxic conditions, no significant difference in NMDAR mRNA levels was found, a finding that was inconsistent with Hu’s research [29]. However, we observed significantly higher mitochondrial protein levels. Therefore, we hypothesized that NMDAR might translocate to the mitochondrial membrane to transport calcium under hypoxic stress.
Mitochondrial damage–induced apoptosis is a classic cell death pathway in ischemic heart disease [31]. Calcium overload and ROS accumulation in the mitochondria are two key pathological milestones in ischemic injury [32]. The present study was the first to identify mitochondrial damage and alterations in related signaling pathways. NMDA significantly deteriorated mitochondrial damage and apoptosis, whereas MK-801 inhibited NMDAR-mediated mitochondrial damage and apoptosis. Furthermore, the measurement of intracellular calcium, mitoROS, and mitochondrial potential revealed the detailed adverse effects of NDMAR on mitochondrial damage and apoptosis.
Although our study presented the metabolomic results of a highly consistent myocardial infarct mouse model and a promising target based on this metabolomic analysis, it has some limitations. First, only cellular studies were performed to explore the mechanism. Primary cardiomyocytes and a classic OGD cell model were used to mimic myocardial infarction. The mechanism will be further tested in animal models in our lab. Second, the translocation of NMDAR from the endoplasm to the mitochondrial or mitochondria-associated endoplasmic reticulum membranes requires confirmation. Overall, NMDAR, as a functional receptor in the neural system, requires further exploration in the cardiovascular system.
Here we reported the results of our metabolomics study using a highly consistent myocardial infarction mouse model. According to a metabolomic study, NMDAR was identified as a promising target that could induce mitochondrial damage and cell apoptosis by mediating calcium influx and ROS formation. In conclusion, our study provides solid data for further study of the role of NMDAR in cardiovascular diseases and a promising target to interfere with apoptosis in myocardial infarction.
The datasets used and/or analyzed during the currentstudy are available from the corresponding author upon reasonable request.
YW and LH performed most experiments. DD and ZC performed metabolic experiments. YW performed computational data analyses. CQ provided samples. CQ contributed protocols, made some constructs, and provided consultation. CQ established collaborations and allocated funding for this study. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity.
All animal work was registered in the ethical committee of Sichuan University (No. 20220221020) and strictly performed following the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978).
Not applicable.
The present study was financially supported by the National Natural Science Foundation of China (Grant No. 81900311) and the Science and Technology Agency Foundation of Sichuan province (Grant 2022YFS0364).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.