
- Academic Editors
†These authors contributed equally.
Background: Muscle atrophy
resulting wholly or partially from disuse represents a serious medical
complication that decreases quality of life and increases morbidity and
mortality. The accumulation of misfolded/unfolded proteins disrupts endoplasmic
reticulum (ER) homeostasis and thus causes ER stress. Growing evidence indicates
that ER stress plays an essential role in skeletal muscle remodeling under
various physiological or pathophysiological conditions. However, whether ER
stress is involved in disuse-induced muscle atrophy remains unclear.
Methods: To induce muscle atrophy, 8-week-old C57BL/6JNifdc male mice
were subjected to 3, 7, or 14 days of hindlimb unloading (HU), and rhesus
macaques (Macaca mulatta) were subjected to 10
Skeletal muscle is the largest organ of the human body, accounting for almost 40% of the total body weight. The mass and myofiber type composition of skeletal muscle are essential for its functions, including postural control, exercise, and metabolism [1]. However, prolonged periods of disuse, such as those occurring during spaceflight, bed rest, hindlimb unloading (HU), and immobilization or denervation, induce significant muscle atrophy, with the greatest loss of muscle mass observed in antigravity muscles (e.g., the soleus muscle) [2, 3, 4]. In addition, disuse-induced muscle atrophy is accompanied by a transition from oxidative (type I and IIa myosin heavy chain isoforms, MyHC-I and MyHC-IIa, respectively) to glycolytic (type IIx and IIb myosin heavy chain isoforms, MyHC-IIx and MyHC-IIb, respectively) myofiber type [2, 3]. Consequently, disuse-induced muscle atrophy leads to weakness, reduces whole-body insulin sensitivity, and increases the risk of falls and mortality [2, 4]. Despite these broad negative impacts on human health, the molecular mechanisms responsible for disuse-induced muscle atrophy are not well understood.
As a dynamic tissue, skeletal muscle undergoes extensive adaptations in response
to a large variety of stimuli [1]. One of the main structural components involved
in the rapid skeletal muscle response to environmental changes is the highly
developed endoplasmic reticulum (ER) [5, 6]. The ER plays a pivotal role in
protein folding, the structural maturation of cellular proteins, and calcium
homeostasis [7]. However, the accumulation of misfolded and unfolded proteins,
caused by environmental or genetic factors, can ultimately lead to stress in the
ER lumen. In response to stress and to restore ER homeostasis, cells initiate the
unfolding protein response (UPR). The UPR is mediated by three major ER-resident
stress sensors: inositol-requiring protein 1 (IRE-1), protein kinase R-like ER
kinase (PERK), and activating transcription factor 6 (ATF-6) [8]. Binding
immunoglobulin protein (BiP), one of the most abundant ER chaperones, dissociates
from these three sensors upon stress, leading to their activation through either
oligomerization or export. Activated PERK phosphorylates eukaryotic initiation
factor 2 (eIF2), which increases the translation of ATF4 and expression of
stress-responsive genes such as C/EBP homologous protein (CHOP) [8]. The
endoribonuclease activity of IRE-1
Accumulating evidence has highlighted the essential role of ER stress and the
UPR pathway in skeletal muscle remodeling under various conditions [9, 10, 11]. ER
stress is activated in skeletal muscle by exercise and regulates exercise-induced
adaptations through the peroxisome proliferator-activated receptor gamma
coactivator-1
In this study, we examined changes of ER stress markers in the soleus muscle for different periods of HU. Our results showed that most ER stress components were upregulated in a trend similar to atrophy-related genes, showing close association with muscle atrophy. Inhibition of ER stress alleviated HU-induced muscle atrophy and concomitant myofiber type transition. ER stress activation was confirmed in head-down tilted bed rest (HDBR)-induced rhesus soleus muscle atrophy. These data suggest that ER stress activation mediates disuse-induced muscle atrophy and inhibiting ER stress represents an attractive therapeutic strategy in combating this muscle wasting disorder.
Male C57BL/6JNifdc mice (8 weeks old) were obtained from Vital River Laboratories (Beijing, China) and acclimatized to their new surroundings for 1 week before the start of the study. To induce HU, mice were elevated sufficiently to prevent their hindlimbs from touching the cage floor or sides for 3, 7, or 14 days as previously described [18]. To inhibit ER stress, tauroursodeoxycholic acid (TUDCA) (500 mg/kg body weight) was administered daily by gavage during the 14 days of HU [19], as it has been demonstrated to display potential therapeutic benefits in various models of many diseases at this dose [20, 21, 22]. Control mice were given an equal volume of vehicle (phosphate-buffered saline [PBS]). At the end of the experimental periods, hindlimb muscles were immediately removed, weighed, and frozen in liquid nitrogen or embedded in tissue-freezing medium for subsequent morphological and immunohistochemical analyses. All animal procedures were conducted in accordance with standard ethical guidelines and approved by the Institutional Animal Care and Use Committee of the China Astronaut Research and Training Center (ACC-IACUC-2021-021).
A total of nine healthy male rhesus macaques, aged 5–7 years and weighing 9–10 kg, were purchased from the Beijing Institute of Xie’erxin Biology Resource (Beijing, China). The animals were acclimatized for 3 months including preliminary caretaker handling, confinement jacket fitting, and tilt-table acclimation training at the Laboratory Animal Center of the China Astronaut Research and Training Center prior to being used in experiments. The rhesus macaques were randomly divided into control (n = 3) and 42-day HDBR groups (n = 6) with matching weights. Rhesus macaques in the HDBR group were subjected to 42 days of –10° HDBR as previously described [23]. At the end of the 42-day HDBR experiment, approximately 100 mg muscle was harvested from the belly of the soleus muscle for subsequent analyses. All procedures were performed in accordance with the standard ethical guidelines and approved by the Institutional Animal Care and Use Committee of the China Astronaut Research and Training Center (ACC-IACUC-2019-002).
For each muscle sample, total RNA was extracted with TRIzol according to the manufacturer’s instructions (15596-026; Invitrogen, Carlsbad, CA, USA). The PrimeScript RT Reagent Kit (RR037A; Takara, Shiga, Japan) was used to reverse transcribe total RNA (500 ng) into complementary DNA according to the manufacturer’s instructions. Quantitative PCR (qPCR) was performed in triplicate using the StepOnePlus Realtime PCR System (4376357; Invitrogen). Each PCR mixture (final reaction volume 50 µL) contained 21 µL sterile water, 25 µL SYBR Green (4309155; Invitrogen), 2 µL cDNA (50 ng/µL), 1 µL forward primer (10 pmol/µL), and 1 µL reverse primer (10 pmol/µL). PCR was performed with initial denaturation at 95 °C for 10 min, followed by 40 cycles of 95 °C for 10 s, 62 °C for 15 s, and 72 °C for 20 s. Gene amplification was performed according to a melting program of 70 °C for 15 s, and fluorescence was monitored continuously during the change in temperature from 60 °C to 95 °C at 0.3 s. Target gene expression levels were calculated to that of the 18S rRNA gene and normalized to the control group. Experiments were repeated at least three times. The primer sequences are listed in Supplementary Tables 1 and 2.
Skeletal muscle was homogenized in lysis buffer (in mM) containing: 25 Tris-HCl
(pH 7.6), 150 NaCl, 1 EDTA, 1% Triton X-100, 10% glycerol, and 1% protease
inhibitor cocktail. Supernatants were collected, and the protein concentration
was determined using Bradford protein assay reagents (500-0203; Bio-Rad,
Hercules, CA, USA). Equal amounts of extracted proteins (30 µg per
lane) were denatured in sodium dodecyl sulfate (SDS) loading buffer and separated
on SDS-polyacrylamide gels. Proteins were electrotransferred to a nitrocellulose
membrane. The membranes were blocked in 5% nonfat milk or bovine serum albumin
(BSA) for 1 h and then incubated overnight at 4 °C with primary
antibodies against phospho-IRE1
Cryosections of soleus muscle were fixed in 4% paraformaldehyde for 10 min at 4
°C and washed with PBS. After incubating for 30 min with 0.3% Triton
X-100, sections were blocked for 1 h in 5% goat serum. The cross-sectional area
(CSA) of soleus muscle fibers was determined by immunostaining with anti-laminin
antibody (ab11575; Abcam). To distinguish different myofiber types, soleus muscle
sections were double-immunostained by combining rabbit-anti-laminin with one of
the following antibodies (all from Hybridoma Bank, Iowa City, IA, USA): BA-D5
that recognizes the MyHC-I isoform, SC-71 for the MyHC-IIa isoform, 6H1 for the
MyHC-IId isoform, or BF-F3 for the MyHC-IIb isoform. Primary antibodies were
detected with Alexa Fluor
Data are presented as the mean
To elucidate the role of ER stress in disuse-induced muscle atrophy, a time
course of changes in ER stress markers in the soleus muscle of mice was
determined in response to HU. As expected, the soleus muscle displayed the
greatest muscle mass loss during HU compared to other hindlimb muscles (Fig. 1A).
Atrophy-related genes including Fbxo32 (atrogin-1),
Trim63 (MuRF1), and Fbxo30 (MUSA1) were
induced after 3 days of HU and reached their highest expression after 7 days of
HU (Fig. 1B). Of the nine ER stress marker genes measured, four (PERK,
IRE1
To confirm the correlation between ER stress and muscle atrophy, a correlation assay was performed between the relative expression of ER stress markers and the loss of soleus muscle mass during HU. As shown in Fig. 1D, four of the nine ER stress markers (ATF4, growth arrest and DNA damage-inducible protein 34, Xbp1s, and BiP) were positively correlated with the loss of soleus muscle mass caused by HU. These data indicate that ER stress is significantly activated by HU and may contribute to HU-induced muscle atrophy.
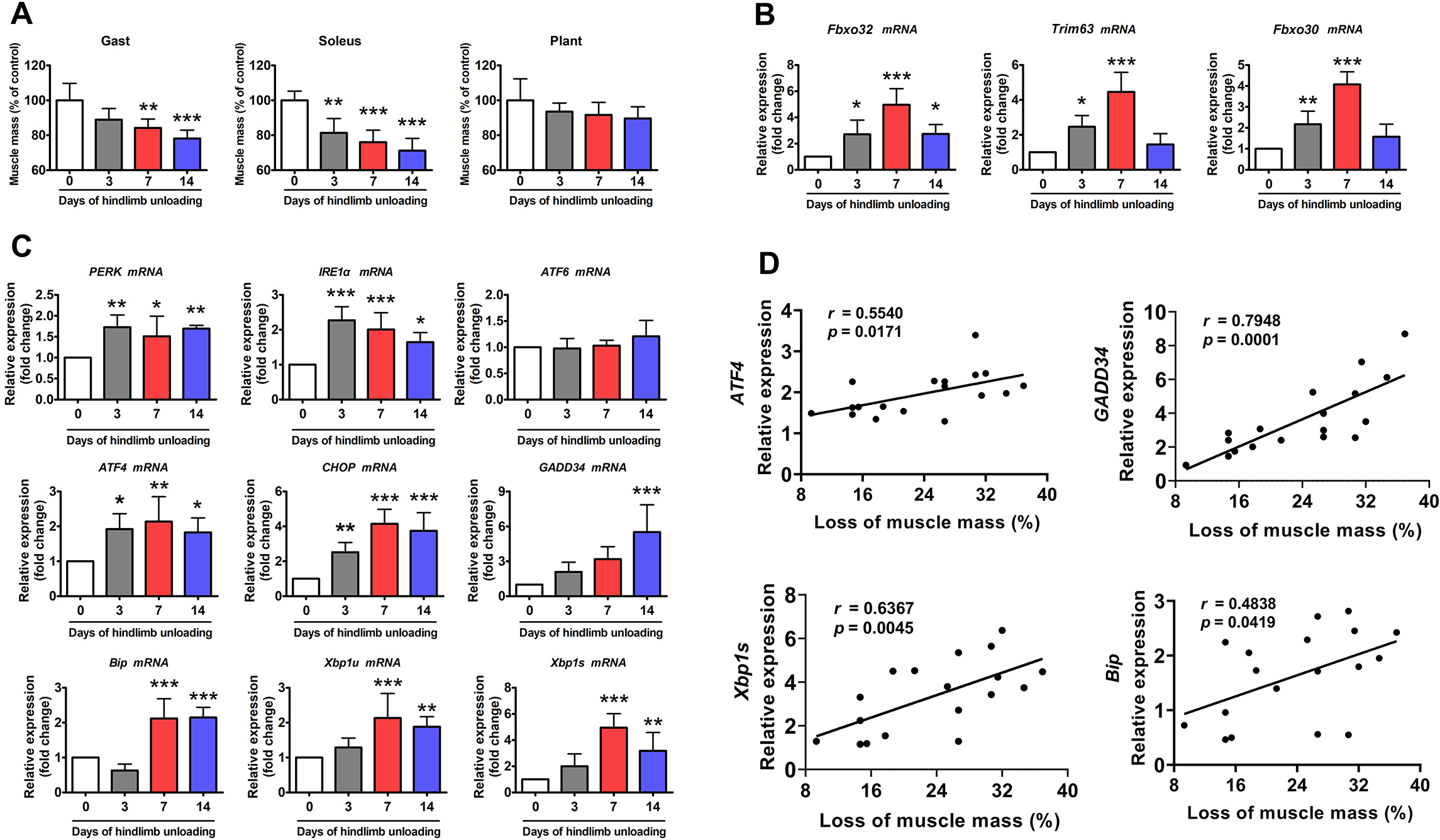 Fig. 1.
Fig. 1.ER stress was activated in the soleus muscle of mice during HU.
(A) Changes in skeletal muscle mass during 14 days of HU (n = 6). Gast,
gastrocnemius; Plant, plantaris. (B) qPCR analysis of Fbxo32, Trim63 and
Fbxo30 in the soleus muscle of mice during 14 days of HU (n = 6). (C)
qPCR analysis of the indicated ER stress marker genes in the soleus muscle of
mice during 14 days of HU (n = 6). (D) Correlation between the expression of ER
stress marker genes and loss of soleus muscle mass. Regression lines are
displayed. r, correlation coefficient. Pearson’s correlation analysis was used to
determine the correlations. Data are shown as the mean
To determine whether the ER mediates disuse-induced muscle atrophy, the effect of ER stress inhibition on HU-induced muscle atrophy was determined. TUDCA, a pan-ER stress inhibitor, was used to inhibit HU-induced ER stress activation (Fig. 2A). HU resulted in significant loss of soleus muscle mass in vehicle-treated mice, which was less evident in TUDCA-treated mice (Fig. 2B). Anti-laminin immunostaining analysis clearly delineated a decrease in myofiber CSA in unloaded soleus muscle, indicative of severe myofiber atrophy (Fig. 2C). By contrast, TUDCA treatment attenuated the reduction in myofiber CSA caused by HU (Fig. 2C). Western blot analysis showed the marked upregulation of Atrogin-1 and MuRF1 in unloaded soleus muscle (Fig. 2D). This upregulation was significantly reduced in TUDCA-treated mice (Fig. 2D), which further confirmed the inhibitory effect of TUDCA on HU-induced muscle atrophy.
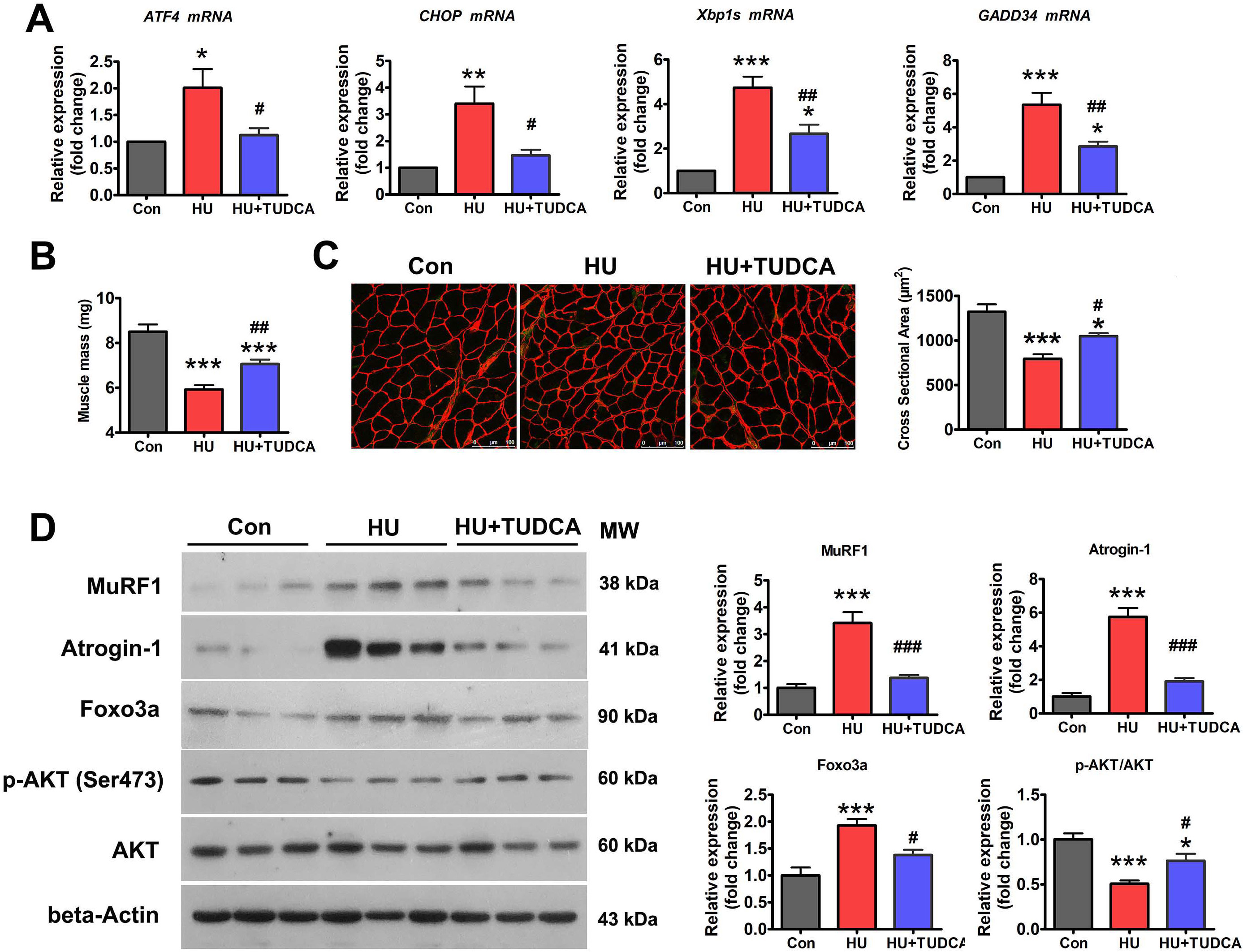 Fig. 2.
Fig. 2.TUDCA inhibited HU-induced muscle atrophy via
regulation of the p-AKT/FoxO3a pathway. (A) qPCR analysis of the indicated ER
stress markers in the soleus muscle of control, HU, and HU+TUDCA mice (n = 5).
(B) Soleus muscle mass from control, HU, and HU+TUDCA mice (n = 10). (C)
Representative immunostaining for laminin in the soleus muscle cross-sections of
control, HU, and HU+TUDCA mice and quantification of CSA (n = 4). Scale bars: 100
µm. (D) Western blot analysis of MuRF1, Atrogin-1, FoxO3a, p-AKT (Ser473),
and total AKT in the soleus muscle of control, HU, and HU+TUDCA mice (n = 6). MW,
molecular weight. Data are shown as the mean
Because AKT/FoxO3a is a key signaling pathway that regulates the expression of atrophy-related genes, we next determined whether the inhibition of ER stress with TUDCA affected the activity of the AKT/FoxO3a pathway during HU-induced muscle atrophy. Western blotting showed that the levels of phosphorylated AKT (Ser473) (p-AKT) were decreased and those of FoxO3a were increased in the soleus muscle of vehicle-treated mice after HU (Fig. 2D). However, treatment with TUDCA restored the expression of p-AKT and decreased the expression of FoxO3a in unloaded soleus muscle (Fig. 2D), suggesting that TUDCA inhibited HU-induced muscle atrophy by regulating the AKT/FoxO3a pathway.
Disuse-induced muscle atrophy is characterized by oxidative-to-glycolytic myofiber type transition. Thus, the effect of ER stress inhibition on HU-induced myofiber type transition was determined. As shown in Fig. 3A, HU markedly downregulated the mRNA expression of MyHC-IIa and upregulated the mRNA expression of MyHC-IIx and MyHC-IIb. Immunostaining for MyHC isoforms in unloaded soleus muscle sections also exhibited a reduced proportion of MyHC-IIa isoform and an increased proportion of MyHC-IIb isoform with HU (Fig. 3B). TUDCA treatment partially prevented the downregulation of MyHC-IIa and the upregulation of MyHC-IIb caused by HU (Fig. 3A,B). Protein expression of slow-twitch troponin (Tn I-SS) and fast-twitch troponin (Tn I-FS) in the soleus muscles was also determined by western blotting. Tn I-SS was decreased, and Tn I-FS was increased after 14 days of HU in vehicle-treated mice, and these changes were attenuated in TUDCA-treated mice (Fig. 3C). Collectively, these results suggested that inhibition of ER stress with TUDCA prevented the myofiber type transition caused by HU.
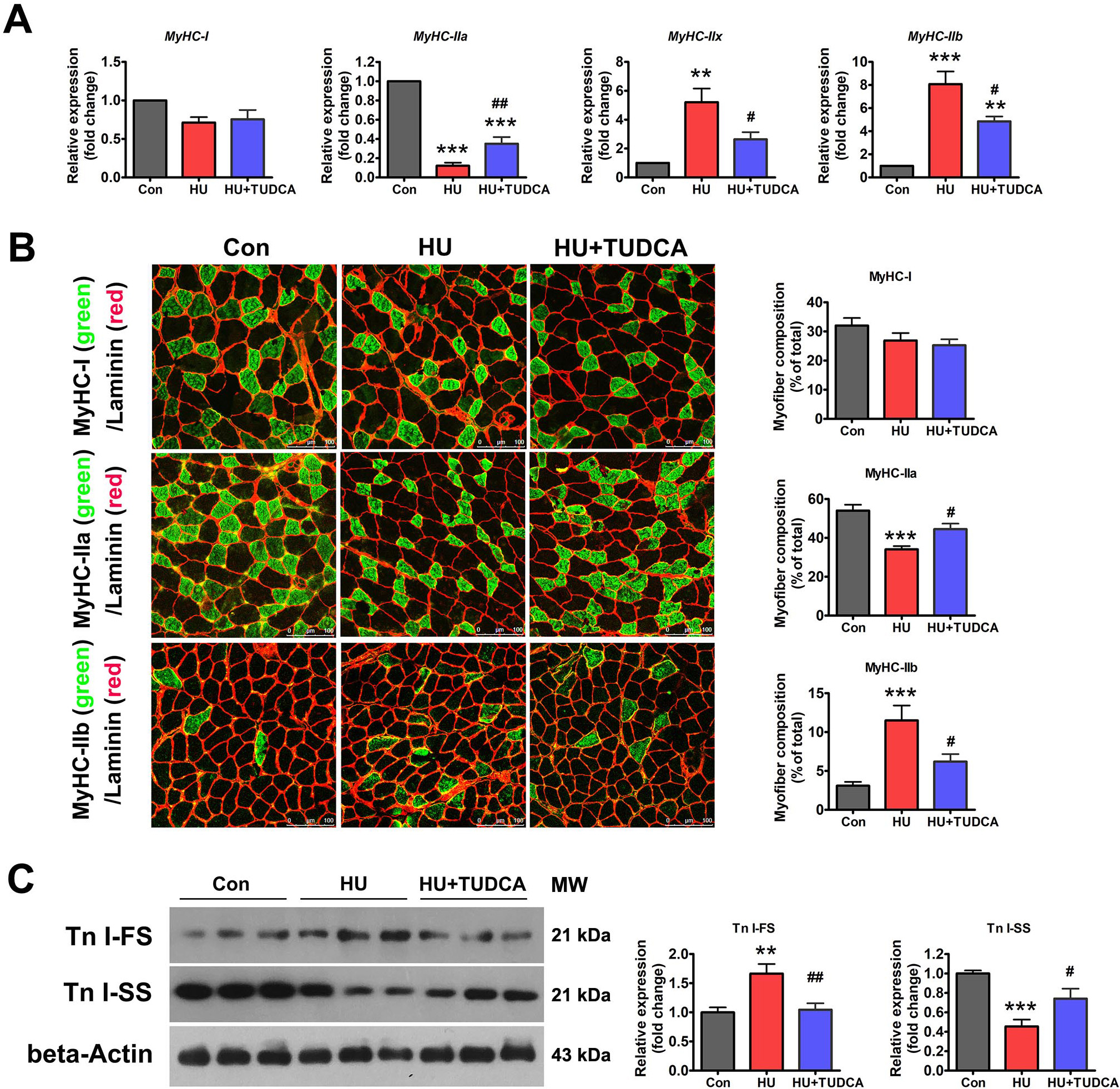 Fig. 3.
Fig. 3.TUDCA inhibited the oxidative-to-glycolytic myofiber type
transition induced by HU. (A) qPCR analysis of MyHC isoforms in the
soleus muscle of control, HU and HU+TUDCA mice (n = 5). (B) Representative
immunostaining of different MyHC isoforms in combination with anti-laminin in the
soleus muscle cross-sections of control, HU and HU+TUDCA mice and quantification
of the myofiber composition (n = 6). Scale bars: 100 µm. (C) Western blot
analysis of Tn I-SS and Tn I-FS in the soleus muscle of control, HU, and HU+TUDCA
mice (n = 6). MW: molecular weight. Data are shown as the mean
Finally, ER stress was measured in atrophied primate muscle caused by disuse.
Rhesus macaques were subjected to 42 days of –10° HDBR to induce muscle
atrophy. HDBR resulted in a 44.7% decrease in the soleus muscle myofiber CSA
(Fig. 4A), which was accompanied by increased expression of both the
atrophy-related gene Fbxo30 and glycolytic myofiber isoform
MyHC-IIb (Fig. 4B,C). Regarding ER stress markers, ATF6 and
ATF4 mRNA levels were increased by HDBR, whereas other genes remained
unchanged (Fig. 4D). At the protein level, phospho-IRE1
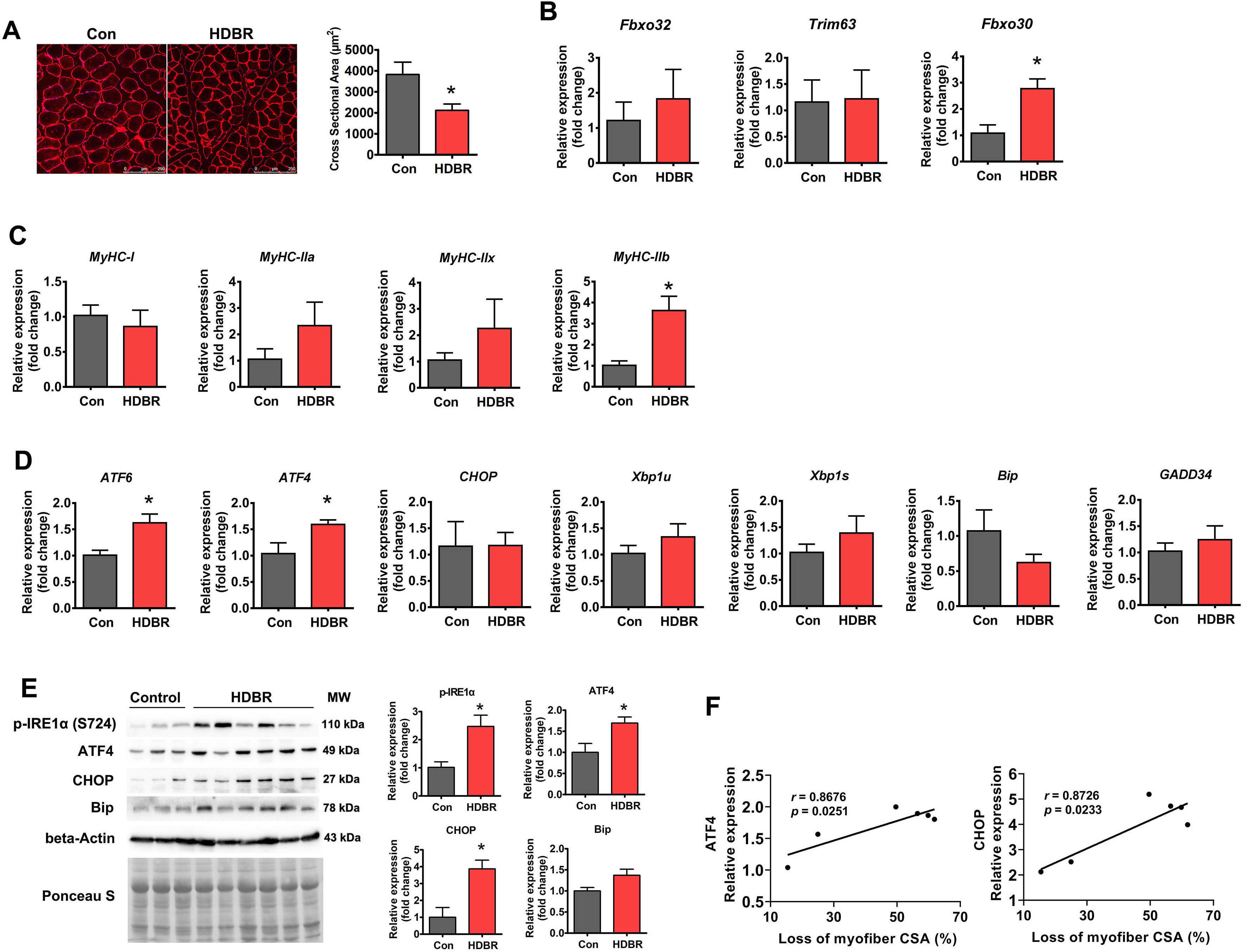 Fig. 4.
Fig. 4.ER stress was activated in HDBR-induced muscle atrophy. (A)
Representative immunostaining for laminin in the soleus muscle cross-sections
from rhesus macaques with (HDBR) or without (Con) HDBR and quantification of CSA.
Scale bars: 250 µm. (B) qPCR analysis of atrophy-related genes in the
soleus muscle of Con and HDBR rhesus macaques. (C) qPCR analysis of MyHC
isoforms in the soleus muscle of Con and HDBR rhesus macaques. (D) qPCR analysis
of the indicated ER stress marker genes in the soleus muscle of Con and HDBR
rhesus macaques. (E) Western blot analysis of p-IRE1
Loss of skeletal muscle mass, commonly referred to as muscle atrophy or wasting, results from various physiological and pathological conditions such as disuse, aging, cancer, diabetes, inflammation, and starvation [1, 2, 3, 4]. Skeletal muscle atrophy not only leads to muscle weakness and compromises quality of life but also increases the risk of both morbidity and mortality in numerous pathologies. Thus, the development of treatments to combat skeletal muscle atrophy is essential for the maintenance of human health [1, 2, 3, 4]. However, a comprehensive understanding of the molecular mechanisms of skeletal muscle atrophy has not been fully defined, which has somewhat hindered the development of effective treatments for muscle atrophy.
Over the past decade, studies have increasingly recognized the importance of ER
stress and UPR in the regulation of muscle mass [11]. ER stress and UPR are
activated in most wasting disorders induced by various stimuli including
starvation, sarcopenia, cancer cachexia, denervation, and myopathies [9].
However, research on the expression of ER stress markers during disuse-induced
muscle atrophy remains controversial [15, 17, 24]. For example, an early study
reported no significant changes in the expression of UPR markers such as BiP,
calreticulin, CHOP, and eIF2
TUDCA is one of the most commonly used pan-inhibitors of ER stress, which acts as a chemical chaperone to alleviate ER stress and UPR by stabilizating improperly folded proteins [25]. To confirm the involvement of ER stress in disuse-induced muscle atrophy, mice were treated with TUDCA to block ER stress activation in response to muscle disuse. ER stress inhibition with TUDCA attenuated the muscle atrophy and myofiber type transition caused by HU, indicating that ER stress activation contributes to disuse-induced muscle atrophy. In agreement with these results, overexpression of ATF4 has been found to be sufficient to induce muscle atrophy by inhibiting global protein synthesis [26], and targeted knockout of the ATF4 gene in skeletal muscle partially prevents acute muscle atrophy during fasting and disuse [26, 27]. By contrast, ER stress is activated in cachexia-induced muscle atrophy, and inhibition of ER stress with 4-phenyl butyric acid (another pan-ER stress inhibitor) aggravates the loss of muscle mass caused by cachexia [13], indicating that ER stress plays distinct roles in different types of muscle atrophy. We could not rule out the possibility that this discrepancy may result from different ER stress inhibitors used in these two different models, but it is more likely that ER stress may crosstalk with distinct signaling pathways that regulate muscle mass in different types of muscle atrophy. Indeed, in the muscle of Lewis lung carcinoma (LLC)-bearing mice, p-AKT was not altered, whereas phosphorylated mammalian target of rapamycin (p-mTOR) was significantly reduced [13]. Additionally, inhibition of ER stress in LLC-bearing mice further reduced p-mTOR [13]. However, p-mTOR was not altered in disuse-induced muscle atrophy [28], whereas p-Akt was reduced, and its downstream target, FoxO3a, was increased in unloaded muscles, as was also shown in this study. Inhibition of ER stress with TUDCA reversed HU-induced changes in the expression of p-AKT and FoxO3a in unloaded muscles. Thus, it is plausible that induction of ER stress activation by various atrophic stimuli may result in different outputs by interacting with distinct signaling pathways.
One limitation of this study is that we did not determine the role of ER stress in the atrophy of other hindlimb muscles (such as the gastrocnemius muscle) induced by disuse. Although the atrophy of the gastrocnemius muscle occurs to a relatively lesser extent than that of the soleus muscle during HU, this muscle still shows a significant loss of muscle mass under disuse conditions. Whether ER stress is activated and contributes to the loss of gastrocnemius muscle mass needs to be investigated in future studies. Interestingly, ER stress was activated during the recovery process following disuse and showed different expression patterns between young and aged skeletal muscle [18]. Whether this discrepancy is associated with differences in the speed of recovery in young and aged skeletal muscle also warrants further study [16]. The second limitation of this study is that we performed expreiments only in male animals. Recent research have begun to determine whether sex difference affect muscle response to disuse conditions. One study reported that male rats experience greater muscle atrophy in response to HU than females [29], but another study come the opposite conclusion that female rats showed greater loss of muscle mass than males in response to HU [30]. Therefore, whether sex could influence different muscle response to disuse still remain controversial. Whether ER stress is differentially activated in male and female mice and whether TUDCA also display protective effects on female muscle atrophy upon HU deserve further investigation in our future research. The third limitation of this study is that the underlying mechanisms that trigger ER stress during disuse conditions are still unclear, but we speculate that this may be for the following reasons. The first may be the disturbance in calcium homeostasis. Previous studies have confirmed that during disuse, the cytosolic free calcium concentration is elevated [31], and endogenous ER calcium content in myofibers is decreased [32]. Of note, when calcium levels are lowered in the ER (such as caused by thapsigargin), calcium-dependent ER chaperones lose their chaperone activity, leading to the accumulation of unfolded proteins and UPR [33]. Therefore, in disuse-induced muscle atrophy, the decreased concentration of calcium in the ER may also lead to the accumulation of unfolded proteins and ER stress. Second, ER stress may be secondary to oxidative stress. It has been reported that oxidative stress disrupts ER homeostasis and activates ER stress [34]. Given that reactive oxygen species (ROS) production is increased in disuse-induced muscle atrophy [35], it is intriguing to determine whether ROS induced by disuse contributes to the activation of ER stress in atrophied muscle. Third, the accumulation of fatty acids may contribute to ER stress. The capacity to oxidize long-chain fatty acids in skeletal muscle is significantly reduced in rats after 9 days of spaceflight [36]. Pagano [37] also observed that 3 days of dry immersion promotes fatty acid infiltration into the skeletal muscle of healthy adult men. As saturated fatty acids, especially palmitate, can induce ER stress in a variety of cells including skeletal muscle [38], the accumulation of palmitate in skeletal muscle during disuse may also be a potential factor triggering ER stress. The above-mentioned factors may not be sufficient to induce ER stress activation alone, but the combined action of these factors makes it possible.
Collectively, our results provide clear evidence that ER stress is involved in disuse-induced muscle atrophy. Particularly, the high expression of ER stress markers at basic physiological levels in primate muscles and their significant upregulation during HDBR-induced muscle atrophy suggest that ER stress may play a more pronounced role in muscle atrophy in humans than in mice. Pan-inhibition of ER stress with TUDCA represents an attractive therapeutic strategy for disuse muscle atrophy. It has also shown efficacy and tolerability in patients with amyotrophic lateral sclerosis [39]. Moreover, if we translate TUDCA dosage from mice to humans by using the body surface area normalization method [40], the dose used in this study is equivalent to that has been used in humans [39, 41]. Thus, TUDCA has great potential for the treatment of muscle atrophy in individuals suffering from long-term disuse such as bedrest, denervation, or aging.
ATF, activating transcription factor; BiP, binding immunoglobulin protein; CHOP, C/EBP homologous protein; eIF2, eukaryotic initiation factor 2; ER, endoplasmic reticulum; FoxO3a, forkhead box O3a; HDBR, head-down tilted bed rest; HU, hindlimb unloading; IRE1, inositol requiring enzyme 1; MuRF1, muscle RING finger 1; MUSA1, muscle ubiquitin ligase of SCF complex in atrophy-1; MyHC, myosin heavy chain; PERK, PKR-like endoplasmic reticulum kinase; qPCR, quantitative PCR; TUDCA, tauroursodeoxycholic acid; UPR, unfolded protein response; XBP1s, X-box binding protein 1 spliced; XBP1u, X-box binding protein 1 unspliced.
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
PZ and XC designed the research study. LW, XP, SL, and WL performed the research. LW, XP and PZ analyzed the data. PZ, LW, XP and XC wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
All experiments involving animals were conducted in accordance with standard ethical guidelines and approved by the Institutional Animal Care and Use Committee of China Astronaut Research and Training Center (Approval: ACC-IACUC-2021-021 for mice experiments and ACC-IACUC-2019-002 for rhesus experiments).
Not applicable.
This work was supported by grants from the National Natural Science Foundation of China (81871522, 32171173) and the State Key Laboratory Grant of Space Medicine Fundamentals and Application (SMFA18B01, SMFA20A02).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.