

 , George S. Baillie 1
, George S. Baillie 11 School of Cardiovascular & Metabolic Health, University of Glasgow, G12 8QQ Glasgow, UK
Abstract
The second messenger, cyclic adenosine monophosphate (cAMP), is a master regulator of signal transduction that maintains cell homeostasis. A fine balance between cAMP synthesis by adenylyl cyclase and degradation by phosphodiesterases (PDEs) underpins receptor-specific responses. As multiple receptors rely on cAMP for signaling, PDEs shape three-dimensional, localized gradients of the cyclic nucleotide to drive appropriate signaling cascades. Of the 11 PDE families, PDE4, which comprises long, short, and supershort isoforms and a dead-short isoform, is of great interest due to its implication in disease. Aberrant PDE4 expression and post-translational modifications are hallmarks of several clinical indications for which curative treatment is not yet available. While some PDE4-specific small molecule inhibitors directed against the active site are approved for clinical use, they are limited by severe side effects owing to the high degree of conservation of the catalytic domain between over 20 unique isoforms. Some attempts to use the different modular structure that exists between long and shorter isoforms are now bearing success. However, these inhibitors are exclusively aimed at PDE4 long isoforms, which have been the focus of the majority of research in this area. Here, we have summarised literature on the lesser-studied short PDE4 isoforms and provide a record of the discovery, regulation, and disease relevance of this class of enzymes that represent an untapped target for specific inhibition in the future.
Keywords
- phosphodiesterase
- cyclic AMP
- short isoform
- inflammation
- multiple sclerosis
- Alzheimer's disease
- traumatic brain injury
- cancer
- chronic obstructive pulmonary disease
- drug target
The first identified second messenger, 3
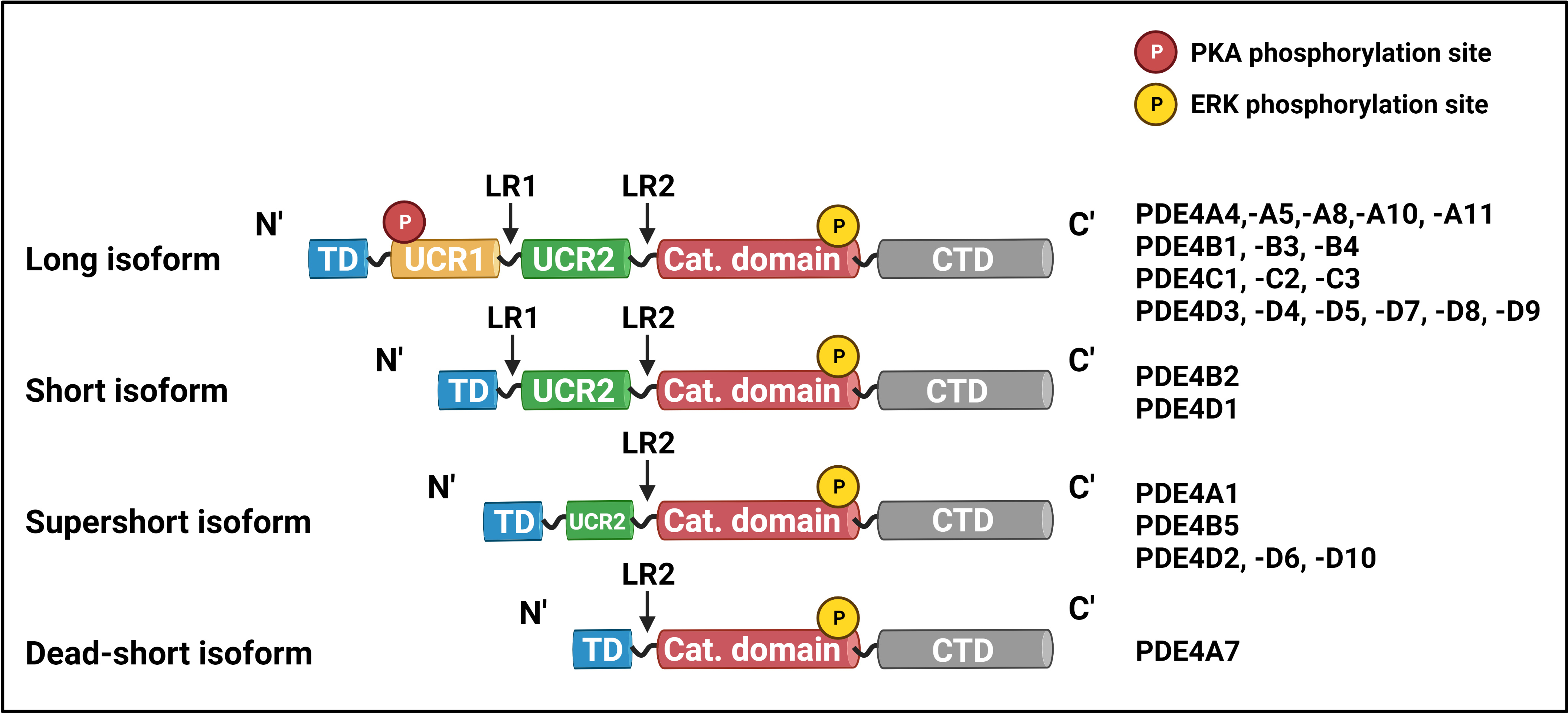
Phosphodiesterase activity is indispensable for maintaining discrete cellular responses to the multitude of signals a cell receives, and dysregulated PDE expression is associated with numerous pathologies of the cardiovascular, nervous, and immune systems. Importantly, altered PDE activity is also a contributing factor to the development and progression of malignant disease. Of the 11 families of PDEs, the PDE4 family is the largest: it comprises over 20 isoforms, encoded by genes A, B, C, and D, which are classified as long, short, supershort, or dead-short, depending on the presence and length of the upstream conserved region (UCR) 1 and 2 domains, unique to the PDE4 hydrolases (Fig. 1) [8].
 Fig. 1.
Fig. 1.PDE4 isoform structure and classification. Depending on the
length of their N-terminal region, members of the PDE4 family are long, short,
supershort, or dead-short. All PDE4 isoforms are listed on the right of their
respective class and structure. PDE4, phosphodiesterase 4; TD, targeting domain;
UCR, upstream conserved region; LR, linker region; Cat. Domain, catalytic domain;
CTD, C
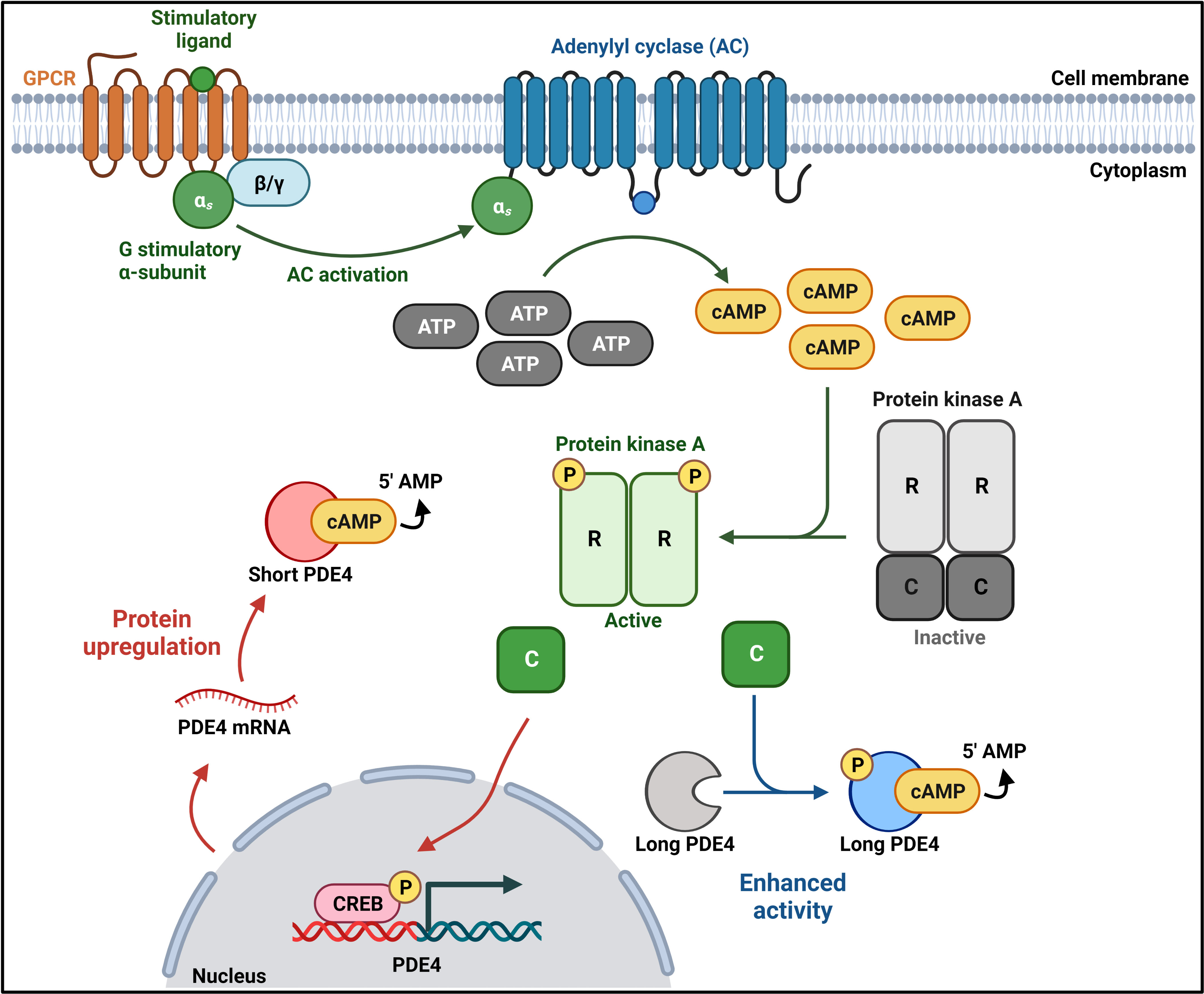
In long isoforms, the carboxy-terminus of UCR1 interacts with the amino-terminus of UCR2 to form a regulatory unit which, in the resting PDE4 state, occludes the catalytic domain of a second PDE4 (partner in a dimer) to downregulate its activity. Thus, UCRs not only regulate cAMP hydrolysis but they contribute to the quaternary, dimeric structure of all long PDE4 isoforms. Phosphorylation of a classical PKA consensus motif (RRxS) in UCR1 by PKA enhances PDE4 activity through the disruption of the UCR1-UCR2 inhibitory conformation [9, 10, 11, 12] (Fig. 2). Conversely, phosphorylation of the extracellular signal-regulated kinase (ERK2) consensus motif (PxS/TP) within the catalytic domain results in long PDE4B, PDE4C, and PDE4D inhibition [11, 13] (Fig. 2). In contrast to long isoforms, which have both UCRs, short isoforms only have UCR2, supershort isoforms lack UCR1 and have a truncated UCR2, and dead-short isoforms have no UCRs and a truncated catalytic domain [14, 15]. In complementary fashion to long-form activation, the activation of CREB by PKA can result in the enhanced transcription/translation of short and supershort isoforms (SASSI), ensuring only transient increases in cellular cAMP concentrations (Fig. 2). The function, expression, and role of long PDE isoforms in disease have been extensively reviewed elsewhere [6, 16], though no such undertaking has been completed for human PDE4 SASSI. This review aims to bring together current research on the roles of PDE4 SASSI in cellular homeostasis and disease.
 Fig. 2.
Fig. 2.Long and short PDE4 regulation. G-protein-coupled receptor (GPCR) activation induces adenylyl cyclase (AC) activation and the production of cyclic adenosine monophosphate (cAMP) from adenosine triphosphate (ATP). Increased cAMP levels lead to phosphorylation and activation of Protein kinase A (PKA). Conformational changes induced by the phosphorylation of PKA lead to the dissociation of the PKA catalytic (C) subunit from the regulatory (R) subunit dimer. The catalytic subunit can phosphorylate long PDE4 isoforms to enhance their activity and phosphorylate the cAMP-response element binding (CREB) protein to enhance short, supershort, and dead-short PDE4 isoform transcription, which in turn drives upregulated protein expression. Created with BioRender.com.
The Drosophila melanogaster (D. melanogaster) dunce
(dnc) gene, which encodes a PDE hydrolase, was the first one
identified as necessary for normal fly behavioral development: dnc
mutants are characterized by short-term memory and learning
deficiencies [17]. Screening of rat libraries with a probe representing
dnc enabled the identification and cloning of the rat dnc
homolog, ratdnc-1. Of the four cDNA clones isolated in the study, PDE4A1
(formerly RD1) was the first identified supershort isoform [18]. In an
investigation of the structure and function of the rat dnc homolog, a
rat testis cDNA library was screened using a cDNA clone of the D.
melanogaster dunce PDE. This study yielded four groups of clones
(ratPDE1-4) encoded by four different genes [19]. Shortly after, in a
study of hormonal regulation of PDEs, ratPDE3.1 and ratPDE3.2 (homologs of human
PDE4D1 and PDE4D2 below) were characterized [20]. In search of
human dnc homologs, Bolger and colleagues isolated the cDNA of the four
human genes from the PDE4 family: PDE4A, PDE4B, PDE4C,
and PDE4D (formerly DPDE1 through 4) [8]. Subsequently, the first human
short isoform to be uncovered was PDE4B2 (formerly hb-PDE1a): its cDNA was
isolated by screening a human frontal cortex cDNA library using a monocyte cDNA
fragment encoding a PDE4. DNA sequencing indicated that PDE4B2 has a
truncated open reading frame and is homologous only to the 3
The multitude of non-redundant isoforms within the PDE4 family results from alternative mRNA splicing and the use of different promoters within each of the four genes [29]. The structural differences between long and short isoforms underpin their differential regulation and, subsequently, the ability of cells to adapt to both short and long-term environmental stimuli [30]. Early work on SASSI revealed that they are subject to hormonal regulation: Sertoli cells treated with follicle-stimulating hormone had increased mRNA levels of rat PDE4D1 and PDE4D2 compared to unstimulated cells [20]. Characterization of the intronic promoter of PDE4D1/PDE4D2 confirmed its inducibility by hormonal stimulation, revealing further evidence for the versatility of the PDE4D gene [31]. In rat thyroid cells, PDE4D1 mRNA was shown to be upregulated in response to long-term treatment with thyroid-stimulating hormone [23]. PDE4D1 mRNA was also observed to change levels in Sertoli cells depending on their developmental state, with older, quiescent cells expressing more PDE4D1 mRNA than both younger cells and terminally differentiated ones [32]. Interestingly, differences in SASSI expression have also been recorded in pregnancy: PDE4B2 mRNA levels were reported to be higher in the myometria of pregnant women compared to non-pregnant women [33]. Treatment of cultured human myometrial cells with cAMP-elevating compounds induced specific upregulation of both PDE4B2 and PDE4D1, indicating that their expression is influenced by hormones known to fluctuate during pregnancy [34]. Notably, preterm birth following infection has been linked to contractions induced by cytokine activity with a concomitant increase in PDE4 SASSI, highlighting the role of these isoforms in inflammation, as discussed in the Inflammation section below [35].
Unlike long PDE4 isoforms, which are inhibited by ERK2 phosphorylation, SASSI PDE4B2 and PDE4D1 are activated [11, 36]. Interestingly, the supershort PDE4D2 has shown weak inhibition consequent to ERK2 phosphorylation: this has been attributed to its truncated UCR2, indicating that it is UCR2 that dictates the different outcomes of the conserved serine phosphorylation in the catalytic domain of SASSI [11]. At the time of writing, there are no published observations on the effect of ERK2 phosphorylation on PDE4B5 and PDE4D10 activity. It is, therefore, tempting to speculate that the effect would be similar to that on PDE4D2, as the aforementioned are supershort PDE4s [26, 27, 28]. Owing to its truncated catalytic domain and unique structure, the dead-short PDE4A7 does not seem to be regulated by ERK2 [11, 15].
The fine-tuning of the inflammatory response would be impossible without the regulation of cAMP synthesis, localization, and degradation. Ever since the discovery of PDE roles in the cAMP pathway and, consequently, in the innate and adaptive immune responses, the topic continues to be an active research subject. Alongside the growing understanding of PDE biochemistry, studies on the signaling pathways targeted by asthma therapies have demonstrated the potential of PDE inhibitors as anti-inflammatory agents [37]. The interplay between the cAMP and inflammatory pathways has been extensively reviewed elsewhere [38]. Briefly, as a function of PDE inhibition, increased intracellular levels of cAMP lead to reduced cytokine release and decreased immune cell recruitment and activation, and vice versa. For the purpose of this review, the focus will be on the current knowledge of PDE4 SASSI as drivers of damaging, auto-inflammatory signaling in disease and injury.
Some of the first evidence that PDE4 SASSI are differentially regulated at different stages of immune cell activation has been obtained through the treatment of human monocytes with cAMP-elevating compounds. Increased PDE4B2 mRNA expression in Mono Mac 6 cells has been reported within 1 hour of treatment with dibutyryl-cAMP. Expression of PDE4B2 mRNA was shown to peak after 3 hours of treatment, whereas PDE4D1 mRNA expression peaked between 2 and 8 hours of treatment. PDE4B2 mRNA levels were shown to be decreased after 4 hours, and both SASSI mRNA were reported to return to basal levels at 24 hours. Protein expression was shown to follow a similar pattern: peak expression was observed between 3 and 5 hours of treatment with a gradual decrease to basal levels at 24 hours [39]. Further studies on Jurkat T cells have demonstrated that PDE4B2, PDE4D1, and PDE4D2 are among the very first PDE4 isoforms to be upregulated upon cAMP elevation [40]. Together, these findings demonstrate that the upregulation of SASSI is a compensatory response to increased cAMP levels.
In a study aiming to elucidate the exact role of PDE4 in T-cell activation,
fully activated T cells (those with T-cell receptors (TCR) and CD28 receptors
co-stimulated) were shown to recruit SASSI PDE4B2, PDE4D1, and PDE4D2 together
with
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS) characterized by the progressive myelin sheath degradation of neuronal axons. Symptoms may be episodic or progressive and include cognitive impairment, fatigue, and muscle weakness [44]. It is estimated to affect 2.8 million people worldwide, with a mean age of diagnosis of 32 and female patients being twice as likely to live with MS compared to males [45]. Pathologically, MS results from myelin-reactive T- and B-cell infiltration of the CNS, which creates an inflammatory environment and drives neural degeneration. Current therapies aim to reduce inflammation, but curative treatment remains an unmet clinical need [44]. Studies on rat MS models have shown that PDE4B2 mRNA is upregulated in microglia residing near brain vessels, indicating the isoform’s involvement in disease progression [46]. In vivo studies have reported that PDE4B2 is, in fact, the only PDE4 upregulated in MS, and, importantly, it significantly correlates with disease score and the expression of some inflammatory markers [47]. The PDE4B inhibitor, A33, has been recently reported to reduce neuroinflammation by suppressing nitric oxide production in both human and murine macrophages, as well as lowering the levels of pathogenic T cells at the peak of disease in a mouse MS model [48]. Notably, CRISPR-Cas9 knockdown of PDE4D1 and PDE4D6 in primary mouse oligodendrocyte precursor cells was shown to increase myelin basic protein levels, a marker of oligodendrocyte differentiation and remyelination [48]. Collectively, these findings highlight the potential of PDE4B and PDE4D SASSI as pharmacological targets in MS.
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by a decline in memory and cognition which lead to behavioral changes and deterioration in speech, orientation, and motoric function. It is responsible for up to 80% of dementia cases, and its curative treatment remains an unmet clinical need [49]. Studies on mouse models of AD have shown that the administration of small molecule PDE4 inhibitors, including rolipram and roflumilast, improves cognition and reduces memory loss [50, 51]. Importantly, PDE4 inhibition has also been demonstrated to significantly improve the cognitive performance of healthy young and older adults [52, 53]. These findings strongly indicate that the PDE4 family of phosphodiesterases is a relevant clinical target for AD treatment.
In vivo studies on mouse AD models have shown that PDE4D but not PDE4B inhibition enhances cognitive capabilities [54]. What is more, an investigation into PDE4D regulation changes in AD has uncovered a significant association between PDE4D1 expression and cognitive impairment: in temporal lobe AD patient samples, increased PDE4D1 positively correlates with higher plaque pathology [55]. Together with a study proposing that PDE4D1 has a role in neuronal plasticity, these experimental outcomes have led to the conclusion that in AD, increased PDE4D1 expression is implicated in decreased neuronal firing rates and impaired memory formation, making it an attractive AD drug target [55, 56].
Traumatic brain injury (TBI) resulting from road traffic injuries, sports concussions, and other accidents is estimated to affect around 69 million people worldwide each year, making it the biggest contributor to trauma-related disability and death [57]. The response to TBI develops within minutes and may result in neuronal apoptosis, further exacerbated by pro-inflammatory cytokine signaling, which reduces cAMP in microglia. Rolipram treatment of TBI rat models prior to injury has been demonstrated to restore cAMP levels, downregulate pro-inflammatory signaling, and reduce neuronal apoptosis following TBI [58]. SASSI PDE4B2 (in neuronal dendrites) and PDE4D2 (in ipsilateral cortex cells) have been identified as undergoing significant upregulation as early as 30 minutes post-TBI in rat models and maintaining significantly higher levels compared to control animals until 24 hours post-injury [59]. In addition, a study on the effect of pro-inflammatory cytokines on post-TBI microglial cAMP levels has revealed significantly increased protein levels of both PDE4B2 and PDE4A1 within 30 minutes of inflammatory stimulation. Knockdown of PDE4B2 using siRNA has demonstrated its essential role in microglial activation through increased cAMP degradation [60]. Localization studies have indicated that PDE4B2 is expressed in both neurons and infiltrating leukocytes near the contusion site, whereas PDE4D is predominantly found in immune cells but not neurons, suggesting that PDE4B2 may have a role in both inflammation and memory deficits following TBI [61]. Notably, treatment of TBI rat models with a PDE4B-specific inhibitor has been reported to reduce inflammation, neuronal apoptosis, and memory deficits, thus contributing to the growing list of evidence in support of the clinical relevance of PDE4 SASSI in neuroinflammation [62].
Aberrant intracellular signaling underpins carcinogenesis, and, expectedly, altered cAMP levels resulting from changes in PDE4 expression are implicated in malignancy. In vivo studies have demonstrated that PDE4A1 overexpression in murine brain tumor xenografts correlates with lower cAMP levels in tumor cells and significantly decreased cancer cell doubling times. Notably, when combined with first-line therapeutics, rolipram has been shown to improve the survival of mice bearing intracranial tumors [63]. Further work on optic pathway glioma mouse models has affirmed the importance of PDE4A1 in brain carcinogenesis, as cortical PDE4A1 overexpression has been shown to lead to the formation of tumors similar to those observed in patients [64].
In a colorectal carcinoma (CRC) cell line, PDE4B2 has been observed to be upregulated by the mutant Kirsten rat sarcoma (KRAS) oncoprotein, which has been shown to induce acinar structure disruption in three-dimensional (3D) cultures through Ak strain transforming (AKT) phosphorylation and activation. AKT phosphorylation has been reported to be ameliorated by rolipram. Notably, PDE4B2 knockdown has been found to re-establish healthy luminal apoptosis in 3D cultures [65]. These findings have been further corroborated by work on KRAS-mutant mouse models, in which PDE4B2 was demonstrated to enhance tumor growth, while the small molecule PDE4 inhibitor apremilast reduced tumor volume [66]. Altogether, these data strongly suggest that PDE4B2 may be a viable therapeutic target in CRC.
In a study focused on PDE4D oncogenicity, PDE4D shRNA treatment of a melanoma cell line stably expressing exogenous PDE4D2 was reported to exhibit significantly less tumor cell apoptosis compared to control melanoma cells undergoing the same treatment: an indication that PDE4D2 prevents tumor cell death. Notably, upon ectopic PDE4D2 expression, increased proliferation has been observed in both melanoma and gastric cancer cells in support of the hypothesis that PDE4D2 dysregulation is implicated in tumor progression [67].
Chronic obstructive pulmonary disease (COPD) is an irreversible systemic condition affecting more than 200 million people globally and is the third leading cause of death worldwide. Risk factors include genetic predisposition, tobacco smoking, infections, malnutrition, and air pollution. Patients may experience dyspnoea, coughing, and wheezing, which worsen if not managed, as curative treatment is yet unavailable [68].
Progressing COPD is characterized by a significant increase in CD8
The discovery of the dnc gene and its role in D. melanogaster behavior instigated extensive research into rodent and human homologs, leading to the characterization of the PDE4 family of phosphodiesterases. Their essential role in compartmentalized intracellular signaling means that even minute changes in expression or regulation induce abnormal cAMP effector activity and pathology. While full-length PDE4 isoforms have long been in the spotlight as drug targets for the treatment of auto-inflammatory diseases, targeting PDE4 SASSI remains largely uncharted. The present review has summarised the current knowledge of PDE4 SASSI in pathology and has thus highlighted their potential as pharmacological targets in several diseases, for most of which there is no curative treatment available.
Isoform-selective inhibition of PDE4 remains an obstacle, as clinically approved small molecules, including roflumilast, apremilast, and crisaborole, rely on binding the catalytic pocket of cAMP hydrolysis, which is highly conserved across isoforms within each PDE4 subfamily. Non-selective binding to long PDE4 isoforms causes dizziness, nausea, emesis, and gastrointestinal side effects in patients, making PDE4 inhibitors second-line treatment at best [16]. While some selectivity for long isoforms has been achieved with the allosteric inhibitor BPN14770 which has been reported to have higher potency against PDE4D3 and PDE4D7 than against PDE4D2 [73], most molecules in development and in trials either inhibit all PDE4 isoforms or exhibit a higher selectivity for one of the four subfamilies [74]. The major structural difference between long and short isoforms, namely the lack of UCR1 in SASSI and their subsequent inability to form dimers, could be utilized in the development of isoform-selective PDE4 inhibitors. Therapeutics targeting unique domains of PDE4 SASSI have the potential to overcome the long-standing challenge of isoform-selective PDE4 inhibition to ultimately improve patient quality of life.
EK and GSB jointly conceptualized, searched available literature, and wrote the original draft. EK made the figures. GSB edited the final draft. EK made amendments following the review. Both authors read and approved the final manuscript.
Not applicable.
Not applicable.
This research was funded by British Heart Foundation grant number PG/17/26/32881 to GSB.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.