

 , Donge Tang 1,*, Yong Dai 1,*
, Donge Tang 1,*, Yong Dai 1,*1 Clinical Medical Research Center, The Second Clinical Medical College of Jinan University (Shenzhen People's Hospital), 518020 Shenzhen, Guangdong, China
2 Institute of Nephrology and Blood Purification, The First Affiliated Hospital, Jinan University, 510632 Guangzhou, Guangdong, China
3 Central Laboratory, Guangxi Health Commission Key Laboratory of Glucose and Lipid Metabolism Disorders, The Second Affiliated Hospital of Guilin Medical University, 541001 Guilin, Guangxi, China
4 Clinical Lab of Shenzhen Pingshan People's Hospital, Shenzhen Pingshan People's Hospital, 518118 Shenzhen, Guangdong, China
†These authors contributed equally.
Abstract
Background: Osteopetrosis represents a rare genetic disease with a wide range of clinical and genetic heterogeneity, which results from osteoclast failure. Although up to 10 genes have been identified to be related with osteopetrosis, the pathogenesis of osteopetrosis remains foggy. Disease-specific induced pluripotent stem cells (iPSCs) and gene-corrected disease specific iPSCs provide a platform to generate attractive in vitro disease cell models and isogenic control cellular models respectively. The purpose of this study is to rescue the disease causative mutation in osteopetrosis specific induced pluripotent stem cells and provide isogenic control cellular models. Methods: Based on our previously established osteopetrosis-specific iPSCs (ADO2-iPSCs), we repaired the point mutation R286W of the CLCN7 gene in ADO2-iPSCs by the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) mediated homologous recombination. Results: The obtained gene corrected ADO2-iPSCs (GC-ADO2-iPSCs) were characterized in terms of hESC-like morphology, a normal karyotype, expression of pluripotency markers, homozygous repaired sequence of CLCN7 gene, and the ability to differentiate into cells of three germ layers. Conclusions: We successfully corrected the point mutation R286W of the CLCN7 gene in ADO2-iPSCs. This isogenic iPSC line is an ideal control cell model for deciphering the pathogenesis of osteopetrosis in future studies.
Keywords
- CRISPR/Cas9
- gene correction
- osteopetrosis
- iPSCs
- CLCN7
Osteopetrosis represents a rare genetic disease which results from osteoclast failure [1, 2]. The clinical manifestations are heterogeneous and range from asymptomatic to severe [3]. The typical feature of osteopetrosis is an increased skeletal mass [2]. In the intermediate/severe forms, patients also present with osteomyelitis, anemia with extramedullary hematopoiesis, cranial nerve involvement, short stature, malformations and brittle bones [4]. Based on genetic pattern, osteopetrosis is classified into autosomal recessive osteopetrosis (ARO), autosomal dominant osteopetrosis (ADO) and X-linked osteopetrosis, in which, ADO is the most frequent form of osteopetrosis with a global incidence of 5/100,000 [5, 6]. To date, the genes identified to affect osteopetrotic patients are all involved in the function of the osteoclast, including LRP5, CLCN7, TCIRG1, TNFS11, CAII, OSTM1, PLEKHM1, TNFRS11A, SNX10, NEMO [7]. Although some palliative treatments are utilized for osteopetrosis treatment [4], such as pain infections could be treated using analgesics and antibiotics, multiple fractures could be treated by surgery, and hematopoiesis cloud be improved by erythropoietin and glucocorticoids [4], there is no cure for osteopetrosis. Hematopoietic stem cell transplantation (HSCT) provides severe patients an effective therapy to improve the quality of their life [4]. However, as it currently stands, HSCT has many limitations and its lethal complications have hampered its applicability for ADO patients [8, 9]. Therefore, advanced basic research which is focused on the pathogenesis of osteopetrosis should be performed to provide innovative orientations for therapeutic approaches to osteopetrosis.
Induced pluripotent stem cells (iPSCs), which possess infinite proliferative activity, exhibit multipotent differentiation potential and harbor the genetic information from their parental cells, have provided a previously unattainable platform to generate attractive in vitro cell models that could accurately mirror diseases phenotypic manifestations [10]. Due to the promises of iPSCs, an increasing number of disease-specific iPSCs or control iPSCs have been derived from somatic cell samples from patients or control individuals respectively [11]. Differentiated targeted cells of interest that can manifest disease features have been employed to discover pathophysiological conditions. Gene corrected disease-specific iPSCs on causative mutation(s) exhibit rescued phenotypes, which have unlocked new possibilities to delve disease mechanisms as a resource of isogenic control cellular models [12, 13]. Among the reported genome engineering technologies, clustered regularly interspaced short palindromic repeats-associated 9 (CRISPR/Cas9) technology possesses the capacity to precisely modify genes [14, 15]. The reported research using CRISPR/Cas9 to correct the mutations in patient-specific iPSCs revealed that the combination of patient-specific iPSCs with CRISPR/Cas9 holds a substantial promise to elucidate the role of targeted genes in the pathophysiology of diseases [16, 17, 18, 19].
Previously, we have reported an osteopetrosis family carrying a heterozygous
mutation, c.856C
Animal studies were approved by Guilin Medical University Laboratory Animal Ethics Committee (Approval No. GLMC 202003175). Four weeks-old SCID Beige mice were purchased from the Beijing Vital River Laboratory Animal Technology Co., Beijing, China and kept in the central laboratory of the Second Affiliated Hospital of Guilin Medical University. Mice were sacrificed by cervical dislocation. The criteria for euthanizing animals prior to the planned end of the experiment: (1) Animals are on the verge of death or unable to move, (2) Dyspnea, (3) Diarrhea or incontinence, (4) The body weight of animals decreased by 20% of that before the experiment, (5) Inability to eat or drink, (6) Experimental animals show obvious anxiety, restlessness, or tumor weight exceeding 10% of the animal’s own body weight, (7) Experimental animals show paralysis, persistent epilepsy or stereotyped behavior, (8) The skin damage area of animals accounts for more than 30% of the whole body, or there is infection and purulence. All mice need to be sacrificed at the conclusion of the experiment. The cell line ADO2-iPSC used in this study was reported in our previous work [20] and stored in the central laboratory of the Second Affiliated Hospital of Guilin Medical University. The ADO2-iPSCs cell line is negative for mycoplasma. The identical STR profiles were confirmed between ADO2-iPSCs and the proband [20]. The construction of ADO2-iPSCs was approved by the Medical Ethics Committee of Shenzhen Peoples Hospital (the Second Clinical Medical College of Jinan University) and informed consent was obtained from the participant donor [20].
CRISPR/Cas9-mediated homology-directed repair (HDR) technology was employed for
single base correction at the CLCN7 locus in ADO2-iPSC line. The
single-guide RNA (sgRNA) sequences were designed using the online tool
(http://www.deephf.com/index/#/cas9) to target CLCN7 mutant sequence
(c.856C
A polymerase chain reaction (PCR) method was carried out for sgRNA fragment
amplification. The total volume of amplification reaction was 30 µL
consisting of 18 µL of ddH
| Step | Temperature | Ramp Rate | Time |
| Initial Denaturation | 95 °C | –2.4 °C/s | 10 min |
| Annealing | 95 °C–85 °C | –0.3 °C/s | |
| 85 °C | 1 min | ||
| 85 °C–75 °C | –0.3 °C/s | ||
| 75 °C | 1 min | ||
| 75 °C–65 °C | –0.3 °C/s | ||
| 65 °C | 1 min | ||
| 65 °C–55 °C | –0.3 °C/s | ||
| 55 °C | 1 min | ||
| 55 °C–45 °C | –0.3 °C/s | ||
| 45 °C | 1 min | ||
| 45 °C–35 °C | –0.3 °C/s | ||
| 35 °C | 1 min | ||
| 35 °C–25 °C | –0.3 °C/s | ||
| 25 °C | 1 min | ||
| Hold | 4 °C | Hold |
The subclones of iPSCs were collected and total DNA was isolated using the QuickExtract™ DNA Extraction Solution 1.0 (no. QE09050, Epicentre, Madison, WI, USA) according to the manufacturer’s instructions. Briefly 500–1000 cells were collected and transferred into 50 µL of rapid extraction buffer and mixed by vortexing for 15 seconds. The mixture was incubated at 64 ℃ for 6 minutes and 98 ℃ for 2 minutes. The DNA was stored at –20 ℃ for up to 1 week, or at –70 ℃ for longer periods.
A nested PCR method was carried out in this study. The total volume of
amplification reaction was 50 µL consisting of 15 µL of ddH
G-banding karyotype analysis was performed for karyotyping iPSC lines [20]. The iPSCs were harvested at 80% confluency and treated with 50 ng/mL colchicine for 2 h at 37 ℃. The single-cell suspension of iPSCs was swollen by exposure to KCl hypotonic solution (0.075 M) and fixed in 3:1 methanol to acetic acid. A minimum of 30 metaphase spreads were analyzed for each sample. The samples were examined and photographed using Lieca GSL120 (Leica Biosystems, Richmond, IL, USA).
Total proteins were extracted using RIPA lysis buffer (no. KGP702-100, KeyGEN
BioTECH, Nanjing, Jiangsu, China), and the protein concentration was determined
using BCA assay. The same amount of protein (20 µg) from each sample was
loaded to SDS-PAGE and transferred to nitrocellulose membranes. Following
blocking with 5% skimmed milk for 2 h at room temperature, the membranes were
first incubated with primary antibodies against CLCN7 (no. DF3932, Affinity
Biosciences, Melbourne, Victoria, Australia, dilution, 1:1000) or
Flow cytometric analysis was used for pluripotency validation [20]. GC-ADO2-iPSCs were dissociated with Solase dissociation buffer (no. RP01021, Nuwacell Biotechnologies Co., Ltd., Hefei, Anhui, China) and incubated with the antibodies for SSEA-4 (no. 560128, BD Pharmingen, San Diego, CA, USA) or TRA-1-81 (no. 560161, BD Pharmingen, San Diego, CA, USA) at 4 ℃ for 20 min in dark. Samples were then analyzed using a FACScan flow cytometry (BD Biosciences, San Diego, CA, USA), and data were analyzed using the FlowJo Software (version 7.6.5, FlowJo LLC, Ashland, OR, USA).
Teratoma assay was performed to evaluate the pluripotency of GC-ADO2-iPSCs in vivo [20]. SCID Beige mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. GC-ADO2-iPSCs were resuspended in 50% Matrigel (no. 354,277, Corning Inc., Corning, NY, USA) with PBS and injected into the posterior thigh muscles of three 4-weeks-old SCID Beige mice. After 2 months, there is a little protrusion at the injection site and the mice were sacrificed and the tumors were harvested and processed for hematoxylin and eosin (H&E) staining. The other hind leg muscle of the identical mice received an injection of Matrigel in the same volume as a control. Ear tags were used to identify mice.
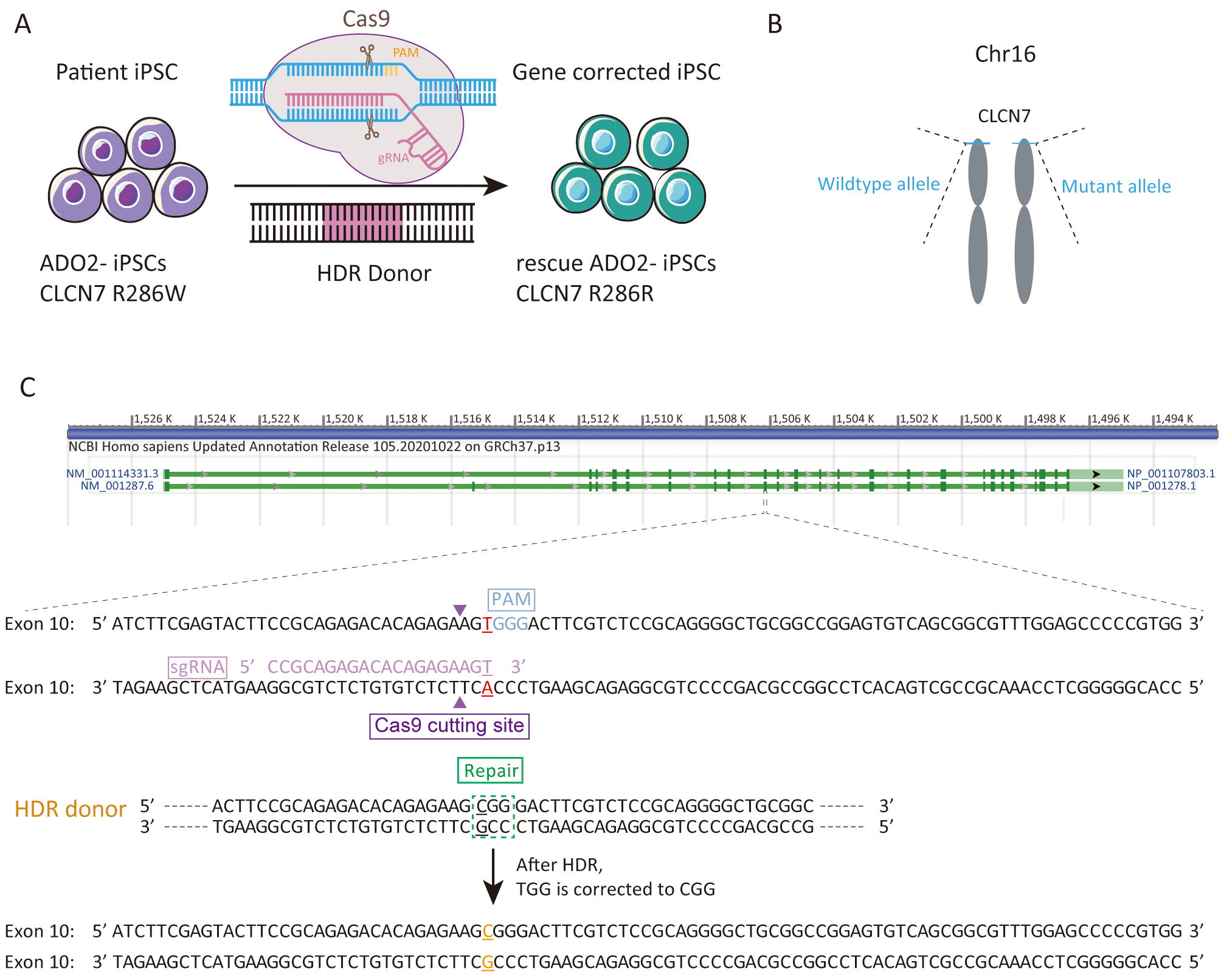
We sought to repair the disease causative mutation in osteopetrosis patient-derived iPSCs (ADO2-iPSCs) [20] through in situ gene targeting using CRISPR/Cas9 mediated HDR system (Fig. 1A). The CLCN7 gene is located at 16p13.3 (Fig. 1B) that encompasses ~30 kb of genomic sequence [22, 23]. The mutant exon 10 of CLCN7 (ATCTTCGAGTACTTCCGCAGAGACACAGAGAAGTGGGACTTCGTCTCCGCAGGGGCTGCGGCCGGAGTGTCAGCGGCGTTTGGAGCCCCCGTGG) was the target for sgRNA design using the design tool (Fig. 1C). The top 5 sgRNAs and their efficiencies were listed in Table 2. One sgRNA (sgRNA5) which overlapped the mutation site of CLCN7 with the efficiency of 0.646979 was selected as the sgRNA for inducing double-strand breaks (DSBs) near the mutation site. The sgRNA fragment was PCR amplified. The plasmid bearing both Cas9 and sgRNA scaffold backbone (pSpCas9n(BB)-2A-GFP) was used in our study because it could facilitate the fluorescence-based clones screening or selection [21]. The oligo pair encoding the sgRNA sequence was annealed and ligated into the plasmid (pSpCas9n(BB)-2A-GFP) (Supplementary Fig. 1A). The donor vector was constructed by cloning the homology arms in the pUC57 plasmid backbone (Supplementary Fig. 1B). Upon lipofection with Cas9/sgRNA plasmids and donor vector, iPSCs were cultured under feeder-free conditions.
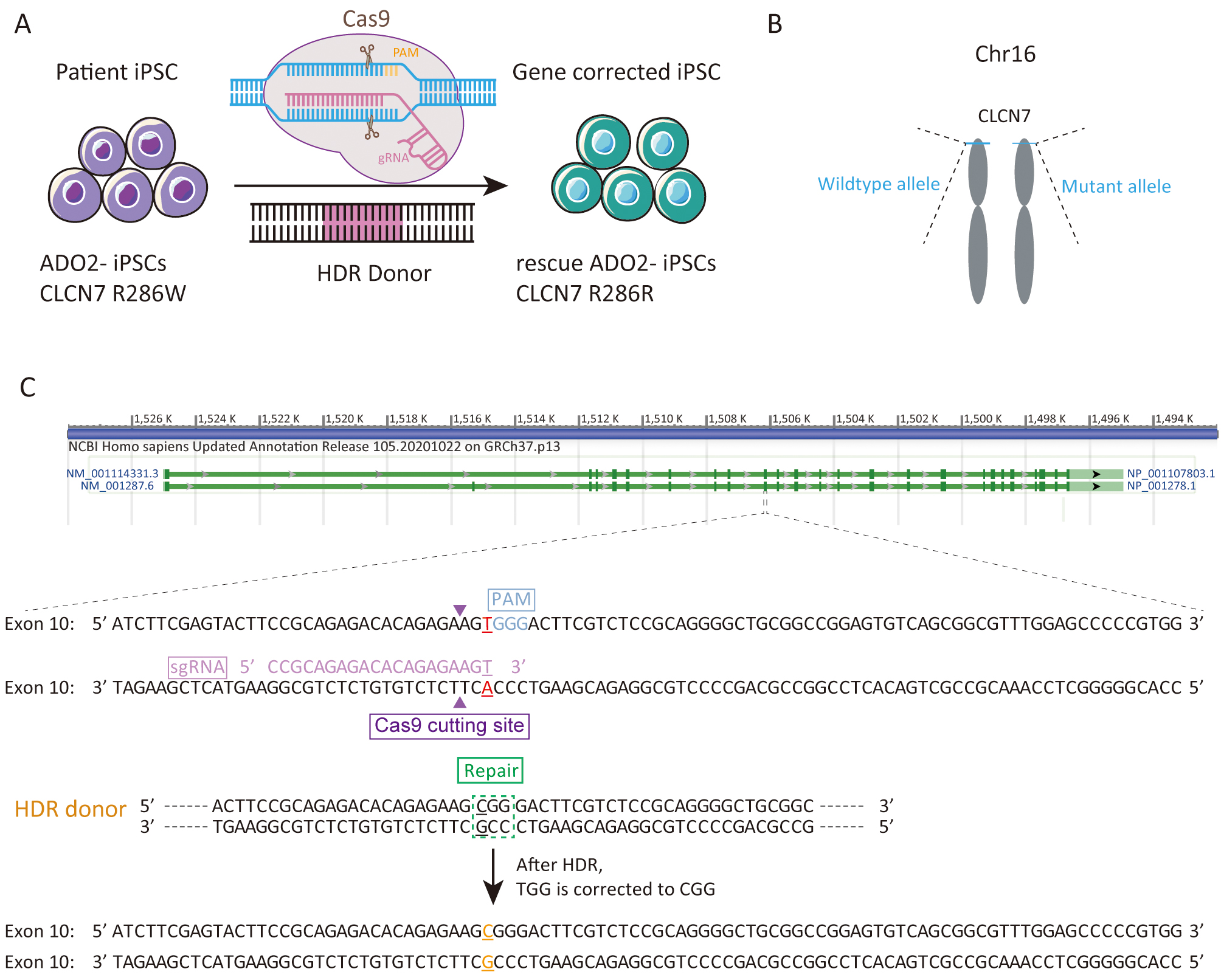 Fig. 1.
Fig. 1.Experimental design for the correction the R286W mutation of CLCN7 in ADO2-iPSCs. (A) Schematic representation of gene correction in ADO2-iPSCs. (B) The mutant allele position on the chromosome. (C) Strategy for reversion of the CLCN7 R286W mutation. The sgRNA sequence was highlighted in pink. PAM was highlighted in blue.
| gRNA ID | Strand | Cut position | gRNA sequence | PAM | Efficiency |
| sgRNA1 | + | 30 | TCCGCAGAGACACAGAGAAG | TGG | 0.681511 |
| sgRNA2 | + | 49 | GTGGGACTTCGTCTCCGCAG | GGG | 0.663927 |
| sgRNA3 | + | 48 | AGTGGGACTTCGTCTCCGCA | GGG | 0.652291 |
| sgRNA4 | + | 70 | GGCTGCGGCCGGAGTGTCAG | CGG | 0.648056 |
| sgRNA5 | + | 31 | CCGCAGAGACACAGAGAAGT | GGG | 0.646979 |
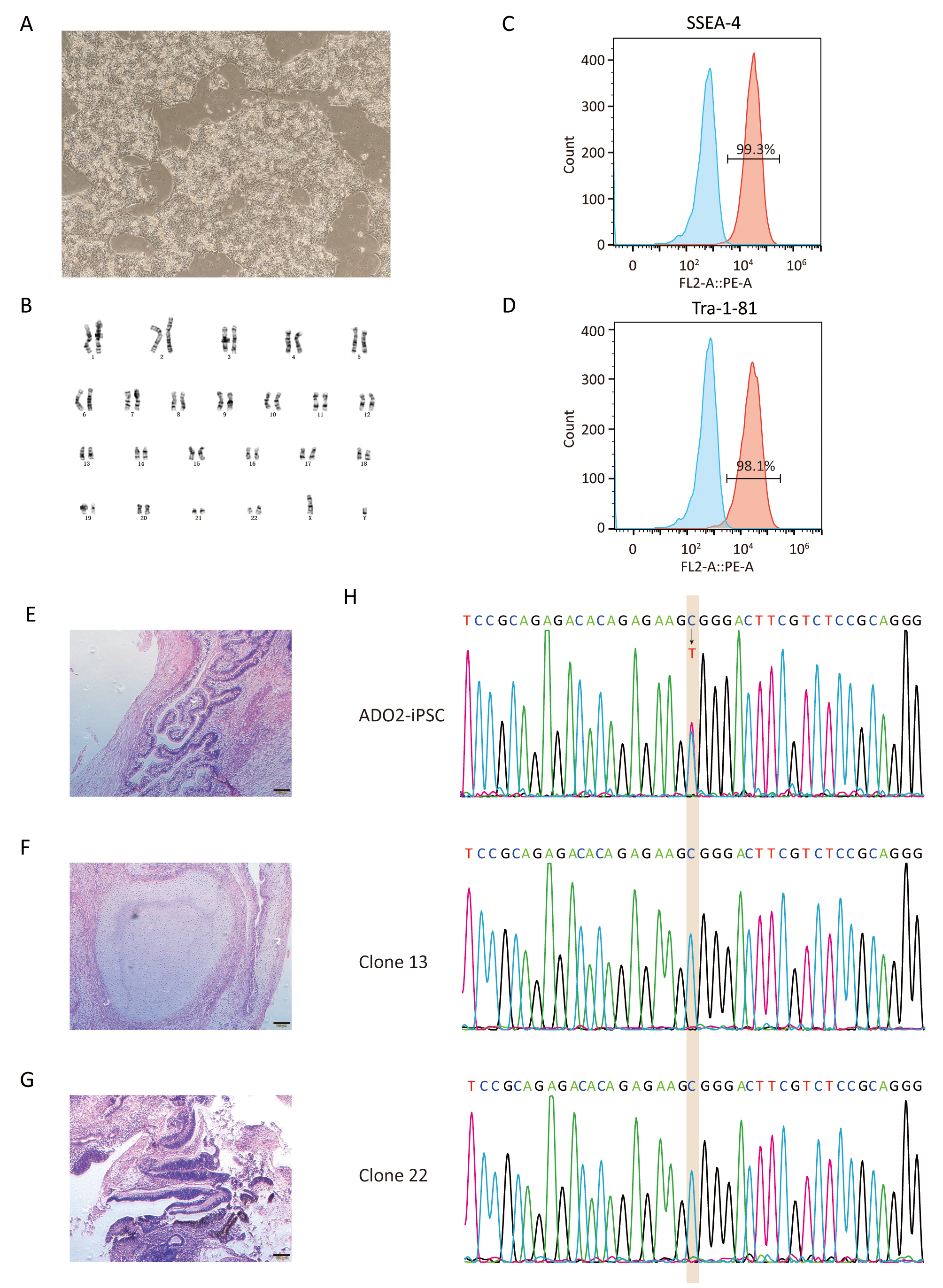
After antibiotic selection using Blasticidin, 11 resistant clones were manually picked and expanded. Their DNAs were extracted and amplified via nested PCR. The resulting amplification products were firstly assessed by agarose gel electrophoresis. As shown in Fig. 2, 10 clones were positive for the targeted 724 bp of amplification product. Clone 15 showing multiple bands was excluded from further sequencing. Two positive clones with site-specific correction of CLCN7 gene were further confirmed by genomic sequencing (Fig. 3H).
 Fig. 2.
Fig. 2.Representative images showing the PCR assay for CLCN7 detection in different GC-ADO2-iPSC clones and ADO2-iPSCs. Clone 13-clone23 represent GC-ADO2-iPSC clone 13- GC-ADO2-iPSC clone23.
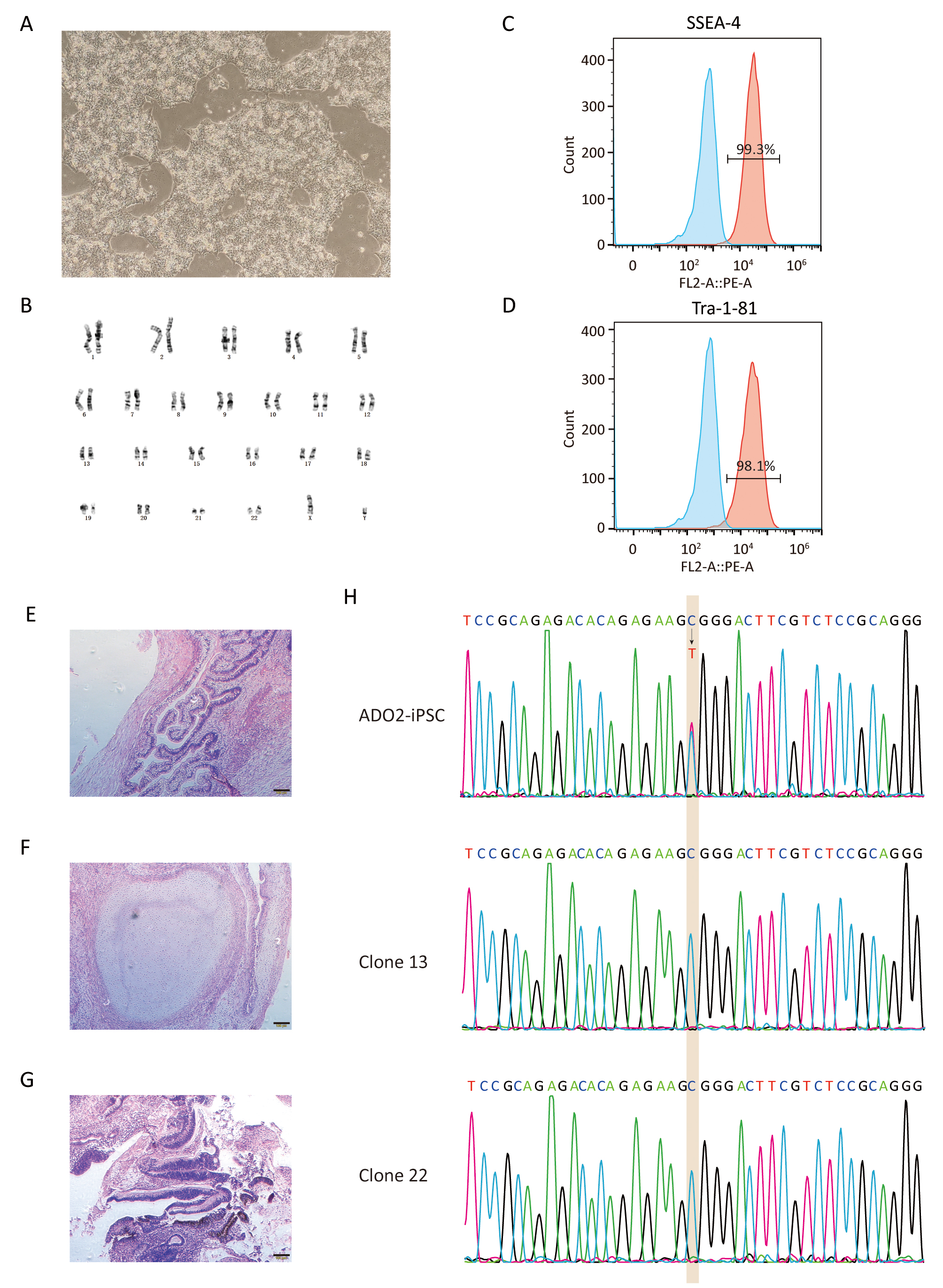 Fig. 3.
Fig. 3.Characterization of GC-ADO2-iPSCs. (A) Images of GC-ADO2-iPSCs colonies cultured on matrigel. (B) GC-ADO2-iPSCs displayed a normal karyotype. (C,D) Flow cytometric analysis of pluripotency markers SSEA-4 and Tra-1–81 of GC-ADO2-iPSCs. (E,F,G) H&E staining of teratomas derived from immunodeficient mice injected with GC-ADO2-iPSCs shows tissues representing all 3 embryonic germ layers: endoderm (intestinal epithelial cells), mesoderm (cartilage) and ectoderm (neural rosettes). Scale 100 µm. (H) The Sanger sequencing results for clone 13 and clone 22 of GC-ADO2-iPSCs.
After verified by genomic sequencing, gene-corrected ADO2-iPSCs (GC-ADO2-iPSCs) were expanded on Matrigel-coated plates with a passage every 3–4 days. As shown in Fig. 3A, GC-ADO2-iPSCs resembled a representative hESC morphology including refractive clone edges and high nuclear/cytoplasmic ratio [24]. Chromosome GTG band analysis revealed the chromosome integrity and stability of GC-ADO2-iPSCs with a normal diploid 46, XY karyotype (Fig. 3B), which is consistent with the karyotype analysis result of ADO2-iPSCs [20]. FACS analyses revealed that 99.3% and 98.1% of the cell population of GC-ADO2-iPSCs expressed pluripotency cell surface markers SSEA-4 and TRA1-81 respectively (Fig. 3C,D). In order to check the in vivo pluripotency of GC-ADO2-iPSCs, a teratoma formation assay was performed. GC-ADO2-iPSCs were transplanted into SCID Beige mice and the formation of teratomas was observed 8 weeks following the injection. Hematoxylin and eosin staining confirmed that the teratomas had derivatives of all three germ layers, such as the intestinal epithelial cells developed from the endoderm, the cartilage differentiated from the mesoderm, and the neural rosettes formed from the ectoderm (Fig. 3E,F,G). These results revealed the establishment, the pluripotency and multilineage differentiation of GC-ADO2-iPSCs.
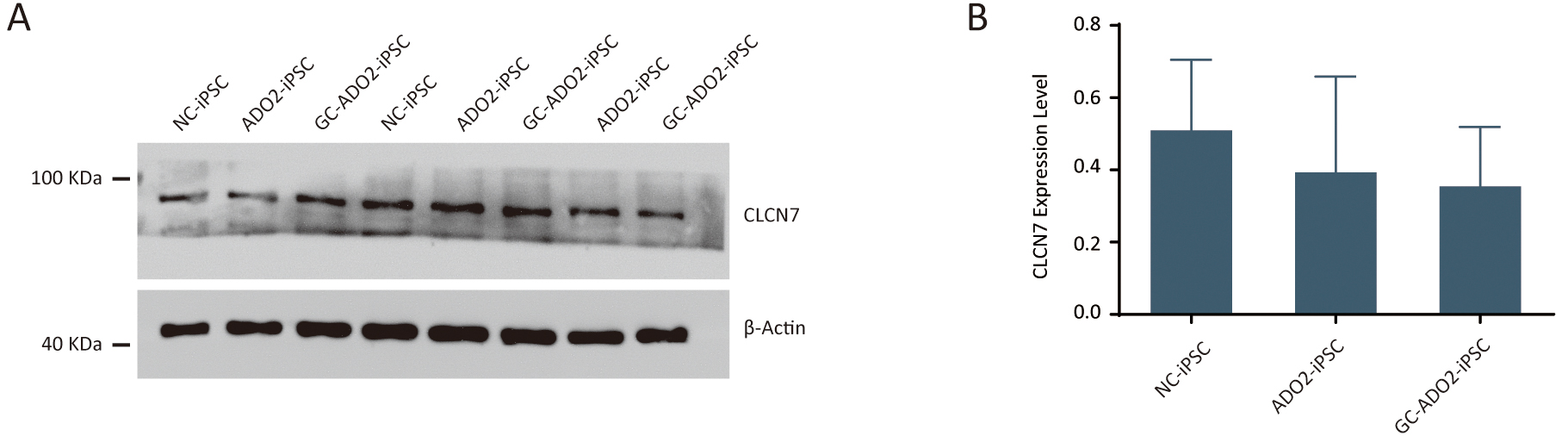
To assess the expression of CLCN7, we extracted proteins from the wild-type
iPSCs, ADO2-iPSCs, and GC-ADO2-iPSCs. Western blot analysis was performed to
determine the relative CLCN7 expression. As shown in Fig. 4A,B, the expression
levels of CLCN7 in the wild-type iPSCs, ADO2-iPSCs, and GC-ADO2-iPSCs showed no
significant differences, which indicated that c.856C
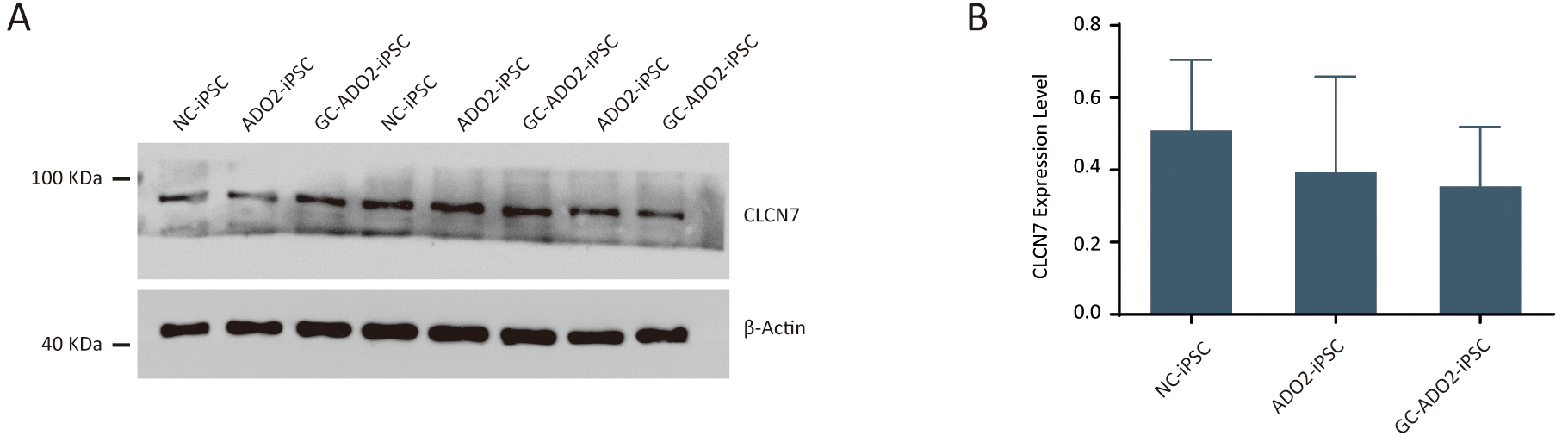 Fig. 4.
Fig. 4.The expression analysis of CLCN7 in ADO2-iPSC and GC-ADO2-iPSC.
(A) Western blotting analysis imaging of CLCN7 in ADO2-iPSC and GC-ADO2-iPSC.
(B) Quantification of relative levels of CLCN7 in ADO2-iPSC and GC-ADO2-iPSC.
CLCN7 levels were normalized by
The gene CLCN7 is located at chromosome 16p13.3 in humans having a
genomic size of approximate 30 kb [23]. The translation product of this gene,
voltage-gated chloride channel 7 (CLCN7), belongs to the CLC chloride channel
family of proteins [25]. For mammals, CLCN7 is broadly expressed and mainly distributed in the ruffled membrane of osteoclasts and the membranes of late endosomes and lysosomes [26]. By regulating the balance of Cl
iPSCs carry the genetic information of their parental cells, possess indefinite propagation ability, have trilineage differentiation potential. Thus, iPSCs provide an ideal platform to investigate the mechanism of various disease, especially of genetic diseases. Up to now, genetic diseases specific iPSCs have become an indispensable research tool for genetic diseases, considerable amounts of which have been successfully established and utilized in mechanistic research and drug estimation. Besides, with the speedy development of genetic engineering techniques, gene corrected patient specific iPSCs, which carry the exact genetic information of their donor cells except disease causative mutation(s), are easy to construct. Benefited from that, they are more appealing as control samples compared with allogeneic healthy samples. Vast quantities of promising results have been reported focusing on genetic corrections of disease-causing genes for elucidating the disease pathology or developing cell or gene therapies.
Previously, rescued bone resorbing activity of the osteoclasts differentiated from gene corrected iPSCs derived from osteopetrotic mice were demonstrated [34]. Another study revealed that the cell function of iPSCs-derived osteoclasts could be rescued by transgenic expression of the functional TCIRG1 gene to osteopetrotic fibroblasts [35]. A similar study showed that infantile malignant osteopetrosis-specific iPSCs exhibited partially corrected phenotype after transfected a lentiviral vector expressing wild type TCIRG1 [36]. Inspired by these encouraging works, we performed gene correction on ADO2-iPSCs by CRISPR/Cas9 mediated HDR. The assessment performed in our study showed the successful correction of the mutation on CLCN7 gene in ADO2-iPSCs. The long-term goal of this study is to generate rescued osteoclasts from GC-ADO2-iPSCs and further study CLCN7 mutations related osteopetrosis pathobiology.
In clinic, HSCT is the only curative for autosomal recessive osteopetrosis [4],
which can prevent or reverse most bone-related abnormalities, but has no obvious
effect on progressive neurodegeneration [37]. Mesenchymal stem cell
transplantation (MSCT) is a considerable strategy to replace the osteoblast
precursor population [38]. However, graft versus host disease (GVHD) caused by
allogeneic cell transplantation can result in lethal complications. The
transplantation of autologous gene-repaired MSC, which derived from gene-repaired
patient-derived iPSC, has the advantage of low immunological rejection, which
could help to increase the curative rate and reduce costs. The GC-ADO2-iPSCs is a
promising seeding cell for autologously treating osteopetrosis. The efficacy of
MSCT or HSCT has been evaluated based a number of mouse models of osteopetrosis
[39, 40]. Thus, it is a recommended study strategy using GC-ADO2-iPSCs derived MSC
to treat the mouse models of osteopetrosis. We would like to point out that there
is still a long way to applying this method to clinical use, because the
tumorigenicity, immunogenicity, and heterogeneity of iPSCs are the most
concerning issues [18, 41]. Small interfering RNA (siRNA) therapy also showed
massive potential for treating osteopetrosis. Anna Teti and her group
demonstrated that Clcn7
Our results demonstrated that iPSCs derived from ADO2 patient could be repaired at the genome level. However, our research presents some limitations. Firstly, the characterization in our study is based on a single gene repaired iPSC line. In the future, experimental designs may consider multiple gene repaired iPSC lines to provide more source cells. Secondly, in this study, the function evaluation of GC-ADO2-iPSCs is at on iPSCs stage. Future studies are needed to examine the derived osteoclast from GC-ADO2-iPSCs, since osteoclasts are the most affected cells in osteopetrosis patients.
In summary, we successfully corrected the point mutation R286W of the CLCN7 gene in osteopetrosis iPSCs (ADO2-iPSCs) by CRISPR/Cas9 mediated homologous recombination and obtained the gene corrected cell line GC-ADO2-iPSCs. GC-ADO2-iPSCs had a normal karyotype and expressed pluripotency markers. The wild type sequence of CLCN7 in GC-ADO2-iPSCs was confirmed by sequencing analysis. GC-ADO2-iPSCs have great potential to offer an attractive resource of isogenic control cellular models for osteopetrosis research.
ARO, autosomal recessive osteopetrosis; ADO, autosomal dominant osteopetrosis; HSCT, hematopoietic stem cell transplantation; iPSCs, induced pluripotent stem cells; CRISPR/Cas9, clustered regularly interspaced short palindromic repeats-associated 9; ADO2-iPSCs, osteopetrosis-specific iPSCs; GC-ADO2-iPSCs, the gene corrected ADO2-iPSCs; HDR, homology-directed repair; sgRNA, single-guide RNA; IARO, intermediate autosomal recessive osteopetrosis; ADO2, autosomal dominant osteopetrosis type II; CLCN7, chloride channel 7.
The data and materials generated during the current study are available from the corresponding author.
DL—conception and design, data collection, data analysis, manuscript writing, review and editing; MO—conception and design, data collection, financial support, review and editing; WZ—conception, design and review and editing; QL—data analysis, review and editing; WC—data collection, review and editing; CM—Validation, review and editing; WL—Validation, review and editing; GD—data collection, financial support, review and editing; LY—conception and design, supervision, review and editing; PZ—conception and design, data collection, review and editing; DT—conception and design, data collection, review and editing; YD—conception and design, data collection, supervision, financial support, review and editing. All authors have reviewed it critically for important intellectual and approved final manuscript.
Animal studies were approved by Guilin Medical University Laboratory Animal Ethics Committee (Approval No. GLMC 202003175).
We thank the online tool for the single-guide RNA (sgRNA) sequences design (http://www.deephf.com/index/#/cas9).
This work was supported by the National Natural Science Foundation of China (82060393), the Science and Technology Plan of Shenzhen (JCYJ20180305163846927), the Natural Science Foundation of Guangxi (2020GXNSFAA159124) and the Scientific Research Project of Health System in Pingshan District of Shenzhen (202139). And all the fundings did not involve in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.