
- Academic Editor
Background: Neonatal acute respiratory distress syndrome (ARDS) is a clinical disorder characterized by excessive acute inflammatory response in lung parenchyma and has high morbidity and mortality. However, the therapeutic treatments are still lacking. The aim of this study is to evaluate the role of unfractionated heparin in neonatal ARDS and explore the underlying mechanism of its effects. Methods: To conduct the ARDS model, the mouse pups were treated by intraperitoneal injection of lipopolysaccharide (LPS) (10 mg/kg). For unfractionated heparin intervention group, C57BL/6 mouse pups received a single subcutaneous injection of unfractionated heparin (400 IU/kg) 30 minutes prior to LPS. The survival rate was recorded for each group. Histological analysis was used to evaluate lung injury. MPO (myeloperoxidase) concentration level in lung tissues and extracellular histones in serum were detected by enzyme linked immunosorbent assay (ELISA). A commercially available kit was used to detect inflammatory cytokine levels in serum. Real time quantitative polymerase chain reaction (qPCR) and western blot were used to detect the mRNA and protein in the JAK2/STAT3 signaling pathway, respectively. Results: Intervention of unfractionated heparin significantly increased the survival rate of mouse pups with ARDS, restored lung architecture, inhibited neutrophil infiltration as evidenced by reduced MPO concentration, and attenuated the LPS-induced inflammatory responses, characterized by the down-regulation of proinflammatoy factors and up-regulation of anti-inflammatory factor when compared with the ARDS group. In addition, the concentration of extracellular histones, which have been proven to be mediated in the pathogenesis of ARDS, was diminished by unfractionated heparin. Moreover, the protein expressions of p-JAK2 (Y1007/1008) and p-STAT3 (Y705) in the ARDS group were remarkably up-regulated, which were reversed by unfractionated heparin. Conclusions: Unfractionated heparin protects LPS-induced ARDS via inhibiting JAK2/STAT3 pathway in neonatal mice, which might present a novel therapeutic target for ARDS of neonates.
Firstly described in 1967 by Ashbaugh et al. [1], acute respiratory distress syndrome (ARDS) is characterized by acute diffuse, bilateral pulmonary injury resulting in increased alveolocapillary permeability, interstitial and alveolar haemorrhage, and the development of nonhydrostatic pulmonary edema in patients. In 2017, De Luca et al. [2] proposed the Monteux definition for neonatal ARDS. Like ARDS in children and adults, neonatal ARDS has its own peculiarities, including infectious triggers, incidence, and mortality. Although great improvement has been made in ARDS of neonates these years, it remains a life-threatening problem with high morbidity and mortality [3]. A multi-center trial of China reported that the mortality rate of newborns with severe ARDS and gestational age more than 36 weeks was 25.2% [4]. One of common pathological factors of ARDS is the systemic inflammation accompanied by accumulation of inflammatory cells and release of inflammatory cytokines [5, 6]. However, the pathogenesis of neonatal ARDS remains unclear. Treatments for neonatal ARDS including ventilation strategies (e.g., high-frequency oscillatory ventilation (HFOV)), supportive cares, and pharmacotherapies (e.g., surfactant, inhaled nitric oxide, and neutrophil elastase inhibitor). However, the therapeutic effects of these pharmacotherapies for ARDS were inconsistent in most of the clinical trials. And the drugs that target a variety of biological pathways, including inflammation, endothelial injury, and epithelial injury, have been verified to be inconsistently effective [7].
Therapeutic doses of heparin has been applied in patients with intravascular
coagulation and sepsis for more than 20 years [8]. Some studies have showed that
heparin has both anticoagulant effects and anti-inflammatory effects in acute
lung injury models [9, 10]. Recently, more and more studies focused on heparin
therapy on ARDS in experimental models or in patients. Wang et al. [11] noted
that unfractionated heparin ameliorated the endotoxemia-induced lung injury in
rats. Dixon et al. [12] reported that nebulised heparin limited
progression of lung injury in patients with ARDS. In addition, heparin was
reported to exert anti-inflammatory effect in acute lung injury (ALI) of rats via
inhibiting transforming growth factor-
The Janus kinase/signal transducer and activator of the transcription (JAK/STAT) signaling pathway is mediated in a variety of cellular processes such as inflammation, apoptosis, and development [16], and JAK2/STAT3 is a proinflammation and apoptosis pathway [17]. JAK2 is a nonreceptor tyrosine kinase, and the phosphorylation of the tyrosine 1007 (Y1007) and tyrosine 1008 (Y1008) residues plays a regulatory role in the function of JAK2 [18, 19]. Tyrosine 705 (Y705) is a major phosphorylation site of STAT3, and activation of STAT3 on Y705 is mediated by JAK upon stimulation of the heterodimeric glycoprotein 130 (gp130)/cytokine-specific receptor [20]. JAK2/STAT3 pathway has been reported to be inhibited in ALI of mice [21, 22, 23]. Since JAK2/STAT3 pathway is involved in the pathogenesis of ARDS and regulating the coagulation activity and inflammatory response in lungs [24], and heparin exerts both anti-coagulant and anti-inflammatory properties, we hypothesized that unfractionated heparin might attenuate LPS-induced neonatal ARDS through inhibition of JAK2/STAT3 signaling pathway. The present study was designed to verify the hypothesis that unfractionated heparin protects neonatal mice against LPS-induced lung injury by suppressing the JAK2/STAT3 pathway activation.
This study was approved by the institutional guidelines of the Animal Care and Use Committee of Children’s Hospital of Chongqing Medical University, China and was conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Adult and neonatal C57/BL6 mice were housed in a SPF environment with a 12-h light-dark cycle, and were allowed access to feed and tap water ad libitum. Within 12 hours of birth, pups from multiple litters were pooled and randomly distributed to experimental groups.
The neonatal ARDS model of mouse was conducted according to the previous experience, with some adjustments [25]. C57BL/6 mouse pups (day of life 6) were randomly divided into three groups: the control group, the LPS-induced ARDS group, and the ARDS+heparin group. Pups of the LPS-induced ARDS group received intraperitoneal injection of 10 mg/kg of LPS Escherichia coli O111:B4; Sigma, St. Louis, MO, USA) diluted in sterile saline to a final volume of 20 µL; For the control group, pups were given an equal volume of sterile saline via intraperitoneal injection; and pups of the ARDS+heparin group were treated with unfractionated heparin (Macklin Biochemical Co., Ltd , Shanghai, China) via subcutaneous injection at 400 IU/kg per pup 30 minutes prior to LPS, based on the previous studies [26, 27]. The mice were allowed to recover overnight. All mice were euthanized by decapitation at 24 h post-LPS administration, and the lungs and blood samples were harvested for histological and biochemical assessments.
A total of 30 mouse pups were grouped and treated as above (n = 10). The number of dead pups in each group was observed and recorded every six hours for a total of 24 hours. The mortality during the observation period was calculated.
Fresh postnatal lungs on postnatal day (PND) 6 were harvested and fixed with 4% paraformaldehyde and paraffin embedded for histology. The lungs were then processed to obtain 5 µm thick paraffin sections, followed by staining with hematoxylin and eosin (H&E). Histological characteristic was evaluated by a pathologist blinded to the experiment. The lung inflammatory score was categorized according to the sum of the score for neutrophil infiltration, alveolar congestion, hemorrhage, and thickness of alveolar wall/hyaline membrane formation [28].
Commonly used to evaluate the degree of neutrophil accumulation in lungs [29, 30], MPO concentration was detected in our study. Briefly, lung tissues were excised after 24 h of LPS-administration, and were homogenized and dissolved in extraction buffer for the analysis of MPO concentration using a commercially available mouse MPO ELISA kit (Solarbio life sciences, Beijing, China). Absorbance was determined at 450 nm.
We first measured the extracellular histones of serum using a histone H4
detection kit (USCN, Wuhan, China). We then measured a panel of multiple
cytokines of serum using a commercially available multiplex immunoassay
(MILLIPLEX MAP Mouse Cytokine/Chemokine Magnetic Bead Panel, Millipore,
Burlington, MA, USA), which can simultaneously quantify the following five
cytokines: IFN-
Extraction of total RNA from the lung tissue was conducted using
SteadyPure Quick RNA Extraction Kit (Accurate Biotechnology, Hunan,
China). The extracted RNA was synthesized into cDNA using reverse transcription
PCR (Accurate Biotechnology, Changsha, Hunan, China). The SYBR Green kit (SYBR Green
Premix Pro Taq HS qPCR Kit (Accurate Biotechnology, Hunan, China)) with specific
primers for JAK2 and STAT3 (Accurate Biotechnology, Hunan, China) was used to
perform real time quantitative PCR. Glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) was used to normalize the input mRNA levels (JAK2, STAT3) as an
endogenous housekeeping gene. Primers used for quantitative PCR include
JAK2f: 5′-CAACCTCAGCGGGACTAAGA-3′ and JAK2r:
5′-GGGGCAGCATTTGGTAAACT-3′. STAT3f: 5′-ATGCTTGTCGGTTGGAGGTG-3′ and
STAT3r: 5′-GGGAGGGAGTAGGGTGATGA-3′. The results are shown as
the mean 2
Extraction of total protein from the lung tissue was conducted using RIPA lysis
buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100,
1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate (SDS) with extra added
phenylmethylsulfonyl fluoride (PMSF) and a phosphatase inhibitor (Beyotime Biotechnology, Beijing, China).
Protein quantification was detected using a BCA Protein Assay Kit (Beyotime
Biotechnology, Beijing, China). Next, protein was separated on 7.5% SDS-PAGE
gels and transferred onto PVDF membranes (Millipore, Billerica, MA, USA). After
incubating with primary antibodies against JAK2 (abcam, Cambridge, CA, USA), STAT3 (abcam,
Cambridge, CA, USA), phospho-JAK2 (Y1007/1008) (abcam, Cambridge, CA, USA), phospho-STAT3 (Y705)
(abcam, Cambridge, CA, USA), and
GraphPad prism 8.3.0 (GraphPad Software Inc., La Jolla, CA, USA) was used to
analyze the data. All data were expressed as the mean
The survival of mouse pups were observed for 24 consecutive hours, and the 24-hour survival rate was calculated. Results in Fig. 1 showed that mice survival rate in the ARDS group (30%) was significantly reduced than the control group (100%), while the decrease in survival rate was significantly increased after pharmacological inhibition with unfractionated heparin.
 Fig. 1.
Fig. 1.Unfractionated heparin improves the survival rate of neonatal
ARDS mice. The survival rate was analyzed using the log-rank test. **p
To examine the protective effect of unfractionated heparin against LPS-induced neonatal ARDS, pathological analysis was performed for each mouse and the lung injury score was calculated. As shown in Fig. 2a, multifocal alveolar hemorrhage, disruption of alveolar wall, and massive infiltration of inflammatory cells were observed in mice of the ARDS group, whereas injected with unfractionated heparin alleviated LPS induced damages. In addition, administration of LPS dramatically increased lung injury score (Fig. 2b), which was significantly reduced after pharmacological inhibition with unfractionated heparin.
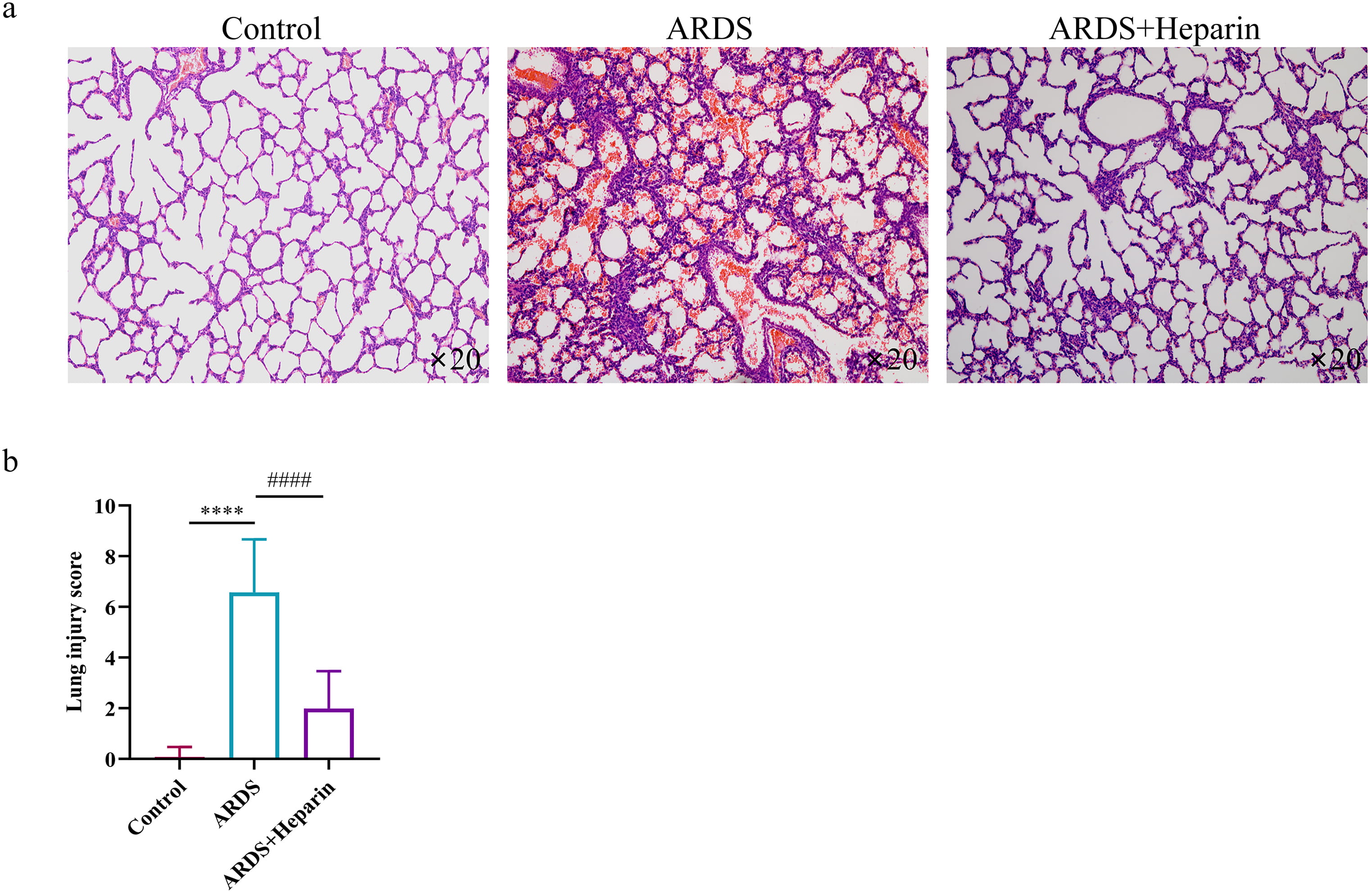 Fig. 2.
Fig. 2.Unfractionated heparin ameliorates the morphological
characteristics of LPS-induced ARDS. Heparin (400 IU/kg) was administered 30
minutes before LPS administration. (a) Representative pictures of H-E stained
lung tissues, magnification (
To investigate the infiltration and accumulation of neutrophils in the lung tissues, analysis of MPO concentration was detected according to the manufacturer’s instruction. As shown in Fig. 3, significant increase of the lung MPO concentration was observed in the ARDS group. While the increase in MPO concentration was significantly decreased after pharmacological inhibition with unfractionated heparin. These data suggested that unfractionated heparin can relieve lung inflammation in neonatal ARDS mice.
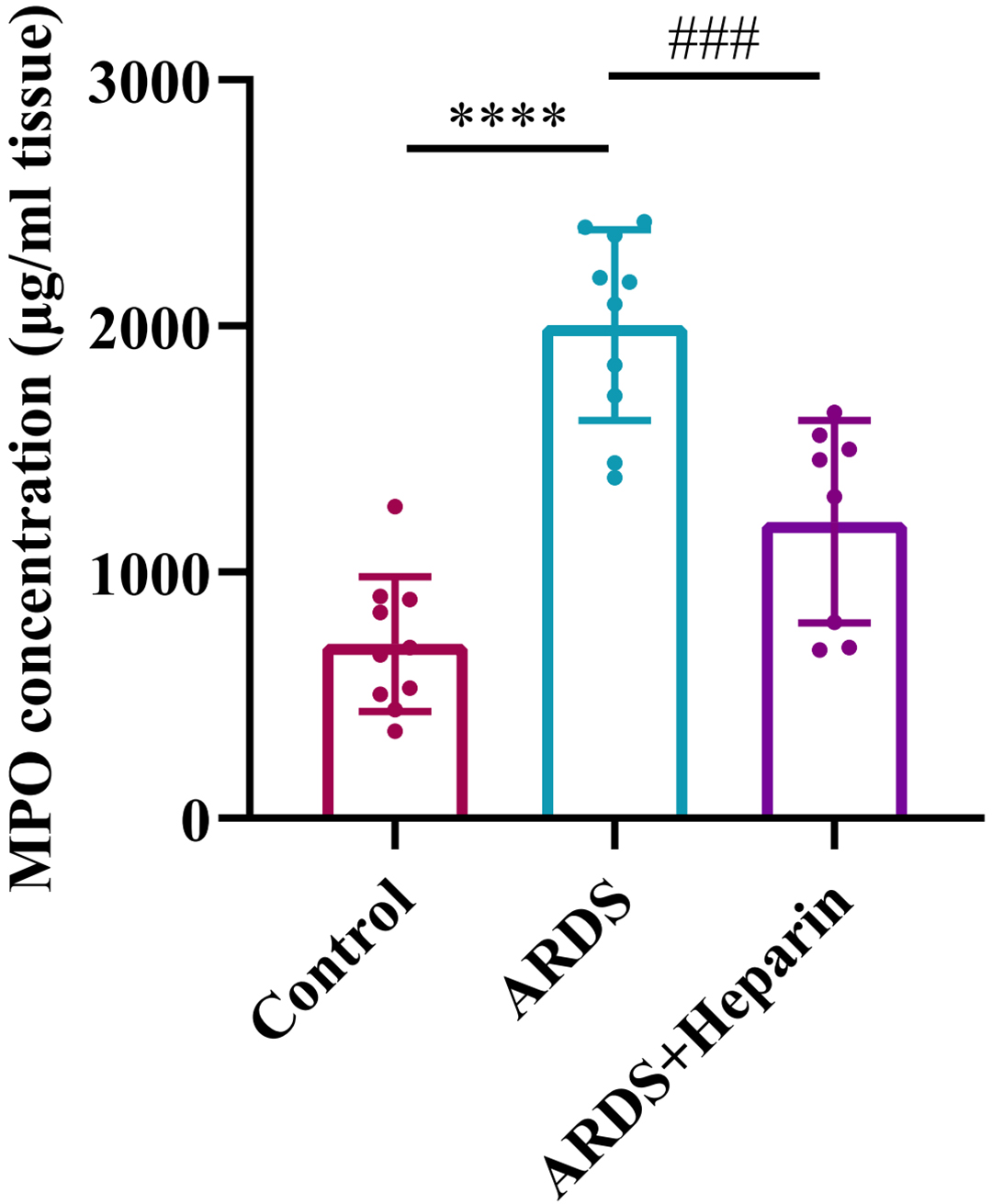 Fig. 3.
Fig. 3.Unfractionated heparin reduced the expression of MPO in neonatal
ARDS mice. Lung tissues were collected at 24 h after LPS or normal saline
administration. Data are means
Studies have shown that extracellular histones are inflammatory mediators involved in the pathogenesis of ARDS [8, 9, 31]. To determine whether unfractionated heparin could inhibit the release of extracellular histones, the histone H4 level in serum was detected by ELISA. We found that after lung injury, the level of histone H4 was significantly increased in neonatal ARDS mice, and unfractionated heparin significantly reduced the levels of histone H4 in serum (Fig. 4).
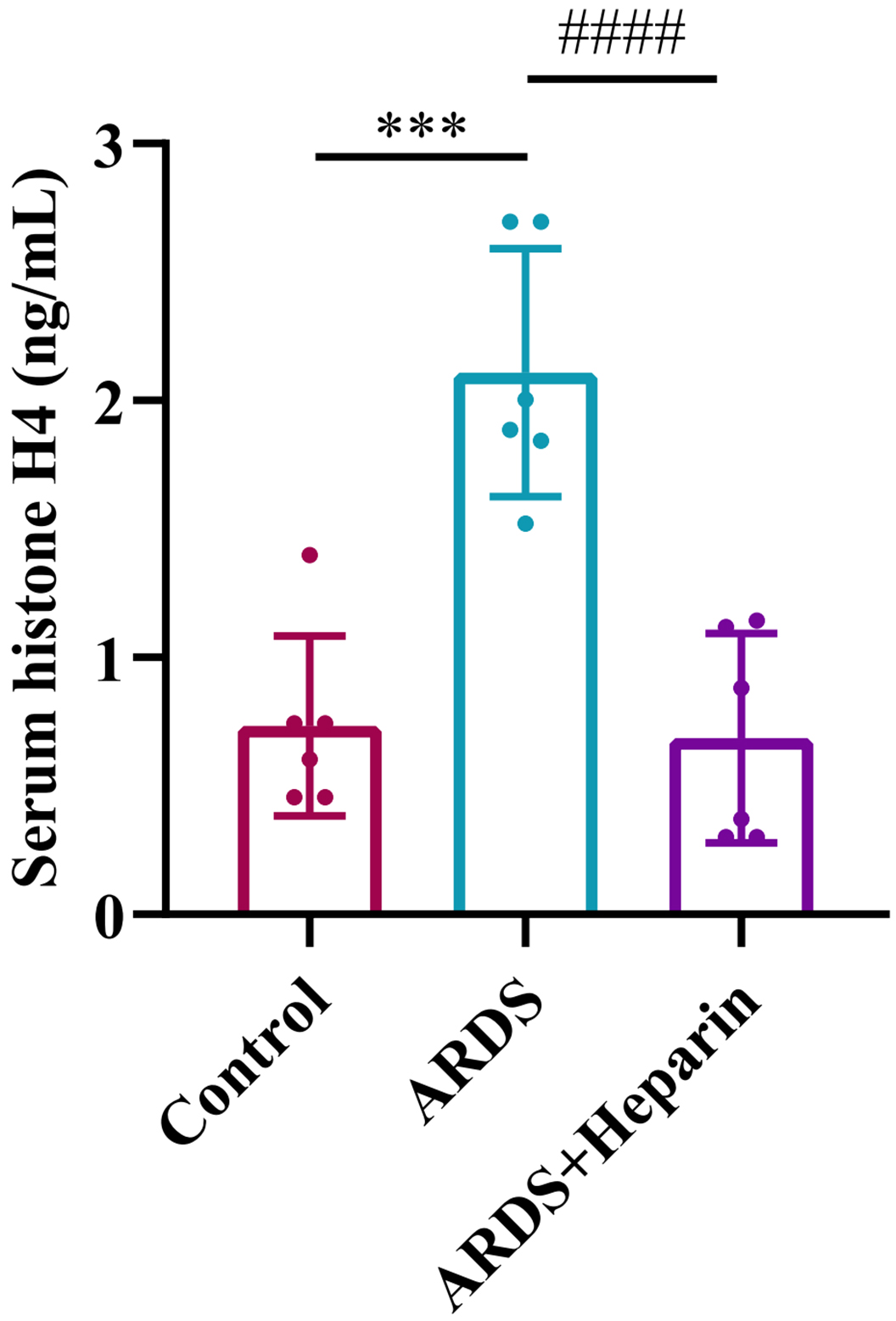 Fig. 4.
Fig. 4.Unfractionated heparin reduced the expression of extracellular
histones in serum of neonatal ARDS mice. Blood samples were collected at 24 h
after LPS or normal saline administration. Data are means
To confirm the role of unfractionated heparin in the regulation of inflammatory
response in neonatal ARDS mice, we measured the related inflammatory cytokines of
IFN-
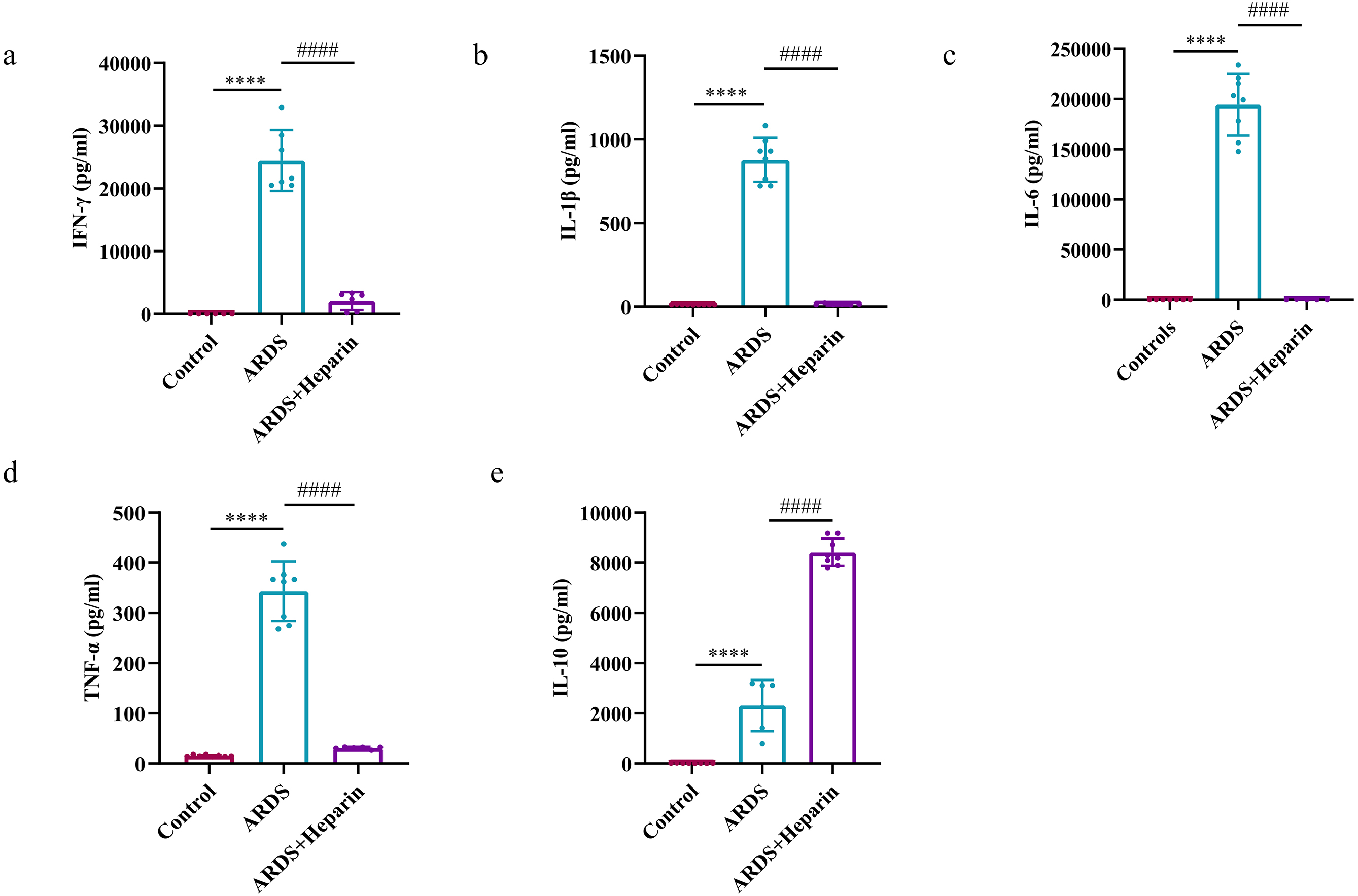 Fig. 5.
Fig. 5.Unfractionated heparin diminished the expression of
pro-inflammatory cytokines and augmented the expression of anti-inflammatory
cytokine in the serum of neonatal ARDS mice. Blood samples were collected at 24
h after LPS or normal saline administration. (a) IFN-
As shown in Fig. 6a,b, the mRNA expressions of JAK2 and STAT3 in the ARDS group was significantly up-regulated compared with that in the control group, but were reduced in presence of unfractionated heparin, respectively. Western blot analysis also showed that the protein expressions of phosphorylated JAK2 (p-JAK2) (Y1007/1008) and p-STAT3 (Y705) in the ARDS group were both significantly increased as compared to that in the control group (Fig. 7a). When unfractionated heparin was applied, the protein expression of p-JAK2 (Y1007/1008) was down-regulated (Fig. 7b), but no change on the expression of JAK2 (Fig. 7c). Similarly, the protein expression of p-STAT3 (Y705) (Fig. 7d) but no STAT3 (Fig. 7e) was significantly up-regulated, while the increased expression of p-STAT3 (Y705) was significantly decreased after administration of unfractionated heparin (Fig. 7d), but no change on the expression of STAT3 (Fig. 7e). These results demonstrated that unfractionated heparin may relieve the inflammatory response during ARDS via inactivation of the JAK2/STAT3 signaling pathway.
 Fig. 6.
Fig. 6.Expression levels of JAK2/STAT3 signaling pathway in lung
tissues of neonatal ARDS mice by qRT-PCR (a,b). Lung tissues were collected at
24 h after LPS or normal saline administration. Data are means
 Fig. 7.
Fig. 7.Expression levels of JAK2/STAT3 signaling pathway in lung
tissues of neonatal ARDS mice by western blot (a–e). Lung tissues were
collected at 24 h after LPS or normal saline administration. Data are means
Plenty of studies have shown that LPS is a perfect component to establish animal models of ARDS [32]. In the current study, we found that intraperitoneal injection of LPS to the six-day-old neonatal C57BL/6 mice resulted in the destruction of alveolar structure, which was in accordance with the previous research [24].
Possessing high affinity for antithrombin and a large number of heparin-binding proteins such as coagulation and fibrinolytic factors, chemokines/cytokines, histone-like protein, extracellular matrix proteins, and immune response mediators, heparin exits both anticoagulant and anti-inflammatory properties [33, 34]. However, the mechanisms by which heparin exerts its therapeutic effects on ARDS have remained poorly understood. In this study, we found that unfractionated heparin improved the survival rate, attenuated LPS-induced physiological and hematological changes, as evidenced by attenuated pulmonary pathologic damages, reduced levels of extracellular histones and pro-inflammatory cytokines, increased level of anti-inflammatory cytokine, and dramatically reduction of neutrophil infiltration in the lungs as assessed by MPO in LPS-induced ARDS in neonatal mice. These data indicate that unfractionated heparin may protect lungs from the inflammatory effects of LPS-induced neonatal ARDS by altering the inflammatory profile.
In animal models and patients of ARDS, extracellular histones have been proven to be associated with damages [35, 36]. Study showed that heparin/low molecular weight heparin exerted a positive effect on mortality in COVID-19 patients by neutralizing the extracellular histones [37]. And multiple studies reported that heparin may neutralize extracellular histones, thus reducing the toxicity of extracellular histones [38, 39]. Contrast to heparin that has high negative charges, histones is rich in positive charges resulting in strong affinity toward heparin [38]. By forming a non-toxic complex through interaction of heparin and histones to prevent histone interactions with endothelial cells and platelets, which could protect against thrombocytopenia, tissue damage, and death induced by histones [40, 41]. In the present study, a marked increase of extracellular histones in the serum of neonatal ARDS mice was observed than the controls, while unfractionated heparin significantly reduced the level of extracellular histones, suggesting that extracellular histones mediate in the systematic inflammatory changes in neonatal ARDS, and unfractionated heparin might exert its anti-inflammatory property by binding to histones.
It is well demonstrated that inflammatory response plays profound roles in ARDS
[5]. Heparin has been reported to be able to bind to the vast majority of
chemokines and cytokines [42]. In animal models, heparin could decrease the
production of pro-inflammatory factors [43]. IL-6 is a pleiotropic cytokine
considered to play a critical role in inflammation [44]. By binding to IL-6
receptor on the cell surface, IL-6 associates with the signal transducing
Growing evidence suggests that the JAK2/STAT3 signaling involves in lung injury and other pulmonary pathophysiology [60, 61, 62]. The phosphorylation of JAK2 subsequently leads to the phosphorylation and dimerization of STAT3 [63]. Activation of STAT3 requires phosphorylation of the Y705 residue, which is constitutively hyperphosphorylated and then translocated into nucleus where STAT3 binds to specific DNA response elements and regulates target gene transcription [64]. Piao et al. [21] showed inflammation and oxidative stress in mice with lung injury were significantly attenuated by inhibiting the JAK2/STAT3 pathway. Zhao et al. [65] demonstrated that bleomycin-induced acute lung injury was also alleviated by inhibiting the JAK2/STAT3 signaling pathway. In the present study, the results showed that unfractionated heparin downregulated the levels of mRNA expression of JAK2 and STAT3 and the protein expression of p-JAK2 (Y1007/1008) and p-STAT3 (Y705) induced by LPS. The JAK2/STAT3 signaling pathway can modulate gene expression in many biological pathways such as cell survival, proliferation, and inflammation [66]. And the suppression of JAK2/STAT3 signaling pathway by reducing the mRNA and phosphorylation expression of JAK2 and STAT3 with no changed total expressions has been reported to attenuate the inflammatory response in lung disease [21, 62], which is consistent with our results. Possible reason is that the transcription activation might be regulated by interaction of JAK2/STAT3 with other regulatory factors owing to phosphorylation. However, the reason of no changed total expressions of JAK2 and STAT3 among the three groups is not clear because remarkable decrease of p-JAK2 (Y1007/1008) and p-STAT3 (Y705) in the heparin intervention group were observed, which should be explored in the further research.
There are some limitations in our study. Firstly, ARDS is a complex disorder that is difficult to reflect in a simplified model such as the LPS model. Secondly, we only identified the role of unfractionated heparin in LPS-induced neonatal ARDS, whereas its role in other neonatal ARDS models (such as aspiration-induced ARDS) requires further study. If the beneficial efficacy is consistently determined, these preclinical results should be transferred to clinical trials to further assess the effects of unfractionated heparin on ARDS of neonates.
In conclusion, our experimental results demonstrated that unfractionated heparin significantly improved the survival rate, attenuated the lung architecture damage, reduced the concentration of MPO and extracellular histones, diminished the levels of proinflammatory cytokines, and increased the anti-inflammatory cytokine of IL-10 as compared to the ARDS group. Consistently, PCR data and western blot results indicated that the gene and protein expression levels of phosphorylated JAK2/STAT3 also increased in the ARDS group and decreased in the heparin intervention group. These results suggested that unfractionated heparin may alleviate LPS-induced neonatal ARDS by reducing inflammatory response and inhibition of JAK2/STAT3 pathways. Which provides a new perspective for the study of its molecular mechanism.
The raw data supporting the conclusions of this article will be made available by the corresponding author YS, without undue reservation.
YS and JX designed the study. JX performed the experiment. QA and LB analyzed and interpreted the data. JX drafted the manuscript. All authors reviewed and approved the manuscript.
This study was approved by the Review and Ethics committee of the Children’s Hospital of Chongqing Medical University (Ethic Approval Number: CHCMU-IACUC20220429009).
Dr Xiong thank Mr. Bo Wang for his help and support during the experiment.
This study was supported by grants of NCRCCHD-2020-GP-03 from the National Clinical Research Center for Child Health and Disorders (Republic of China, Chongqing) and CSTC2021jscx-gksb-N0015 from the Ministry of Science and Technology (Republic of China, Chongqing).
The authors declare no conflict of interest. YS is serving as Guest Editor of this journal. We declare that YS had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to GP.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.