
- Academic Editor
†These authors contributed equally.
Background: Dental follicle cells (DFCs) are promising candidates for tissue engineering. However, the molecular mechanisms that regulate the biological characteristics of DFCs are still unclear. Transient receptor potential melastatin 7 (TRPM7) is a Ca
Dental follicle cells (DFCs) are believed to contain precursor cells for periodontal ligament cells, cementoblasts and osteoblasts. These precursor cells are responsible for periodontal ligament, cementum and alveolar bone formation, respectively, in the process of tooth development [1]. Wise et al. [2] first isolated DFCs from rat molars in 1992. In 2005, Morsczeck et al. [3, 4] successfully isolated DFCs from the human impacted third molars and confirmed they were mesenchymal derived stem cells with multi-differentiation capability in vitro. Over the past few decades, DFCs have received a lot of attention due to their use in regenerative medicine research, as well as their use in basic research for studying molecular processes during in vitro differentiation conditions [5]. The molecular mechanisms that underlie the osteogenic differentiation of DFCs are not yet fully elucidated. However, the Bone Morphogenetic Protein signaling pathway, the canonical Wingless Type Mouse mammary tumor virus (MMTV) Integration Site Family Member (WNT) signaling pathway, the Mitogen-Activated Protein Kinase (MAPK) signaling pathway, and the Notch signaling pathway have all been implicated in the osteogenic differentiation of DFCs [6].
Transient receptor potential melastatin 7 (TRPM7) is a member of the
transient receptor potential
(TRP) family, which is widely
expressed in both electrically excitable and non-excitable cells. TRPM7 is a
bi-functional protein containing a kinase domain fused to an ion channel [7]. As
an ion channel, TRPM7 is a key regulator for Mg
The human sequences encoding TRPM6 or TRPM7 were synthesized and cloned into
pcDNA3.0 vector to generate pcDNA-TRPM6 and pcDNA-TRPM7 plasmids. These were
subcloned into pEGFP-N1 vector with forward primer
(5
Chinese hamster ovary (CHO) cells were cultured with Dulbecco’s Modified Eagle’s
Medium (DMEM; catalog #SH3002302, HyClone, Logan, UT, USA) containing 10% fetal bovine serum (FBS; catalog #FB12999102, Gibco, Carlsbad, CA, USA),
100 units/mL penicillin, and 100 mg/mL streptomycin (catalog #15140148, Gibco,
Carlsbad, CA, USA) in a 5% CO
The ethics committee of Southwest Medical University approved all human cell
culture experiments. Informed consent was obtained from donors who provided teeth
for subsequent research. DFCs were obtained from embedded third molars collected
from 20 individuals (aged 12–20 years). The dental follicle was gently separated
from the tooth and then carefully washed with PBS containing 100 units/mL
penicillin and 100 mg/mL streptomycin. The tissue blocks were minced and digested
in a solution of 1% collagenase and 1% dispase (catalog #17105041, Invitrogen,
Shanghai, China) for 40 minutes at 37 °C. Single cells and digested
tissues were incubated in DMEM supplemented with 10% FBS, 100 units/mL
penicillin and 100 mg/mL streptomycin at 37 °C and with 5% CO
DFCs cultured in 6-well plates (2
Total RNA was isolated from DFCs and CHO cells with ectopic expression of
TRPM6 or TRPM7 using Trizol reagent (catalog
#15596026, Invitrogen, Shanghai, China) according to the manufacturer’s
instructions. The isolated RNA was treated with DNase 1 and reverse transcription
PCR was performed with PrimeScript
Quantitative real-time PCR (qRT-PCR) was used to quantify TRPM7 and osteogenic-related gene expression levels in knockdown cells during osteogenic differentiation. Briefly, total RNA was isolated from DFCs and reverse-transcribed to cDNA using the PrimeScriptTM RT reagent Kit (catalog #RR037B, TaKaRa, Tokyo, Japan). qRT-PCR was performed using the ABI Prism 7300 Real-Time PCR System (Thermo Fisher, MA, USA) with PrimeScript RT-PCR Kit (catalog #RR014B, TaKaRa, Tokyo, Japan). Specific primers used for detection of the target mRNA were (5′-3′): TRPM7 forward CTGGTTCCTCTTCTGGTGCCTTATTC, reverse TGCAACTTGGCTGAGATGGTGTAC; TRPM6: forward AGGGCCTGCAAATCAAAGATGA, reverse: TCTCCAATCAGTCGGCCACA; ALP: forward ACTCTCCGAGATGGTGGTGGTG, reverse CGTGGTCAATTCTGCCTCCTTCC; RUNX2: forward GCAGCAGCAGCAGCAGGAG, reverse: GCACCGAGCACAGGAAGTTGG; BSP: forward TGAAGAAGAAGAGGAGGAAGAGGAAGG, reverse CTCCGCTGCTGCCGTTGC; Human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the internal control to normalize the amount of RNA in each sample. Quantitative data for mRNA expression was calibrated to the results of control cells.
DFCs were cultured in DMEM supplemented with 10% FBS, 10 mM
Cell proliferation of DFCs was determined using Cell
Counting Kit-8 assays (CCK-8, catalog #CK0411, Dojindo, Japan). DFCs were seeded into 96-well plates
(5
Cell migration of DFCs was assayed usingtranswell cell culture chambers (catalog #3384, Corning, NY, USA). The migration of cells was stimulated by DMEM containing 10% FBS in the lower chamber. After 48 hours culture, cells that had migrated through the membrane were stained by 100 ng/mL DAPI for 5 min. To quantify the migrated cells, cells were counted in 5 random fields with a Leica DM2500 optical microscope (Leica Optical, Wetzlar, Germany) and expressed as the average number of cells per field.
Whole cell recordings were performed using an EPC-10 USB amplifier with
PatchMaster software version 2x91 (HEKA Elektronik, Reutlingen,
Germany). Patch pipettes with resistances of 2.0–3.0
M were used. Series resistance was
smaller than 10 M and compensated at least 80% to minimize voltage errors. To
elicit whole cell TRPM7 currents, a standard 2.2 s voltage ramp from –100 mV
to +100 mV was applied. The standard extracellular solution contained (in mM): 140 NaCl, 2 CaCl
DFCs (2
DFCs were washed with PBS and lysed on ice for 30 minutes with Radio
Immunoprecipitation Assay (RIPA, catalog #89900,
Thermo Fisher, MA, USA) buffer containing protease inhibitor (50 mM Tris-HCl,
0.1% Sodium dodecyl sulfate (SDS), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate and 1 mM EGTA).
The lysed cells were centrifuged at 12,000 g at 4 °C for 20 minutes, the
supernatant was collected, and the protein quantified with the BCA protein assay
kit (catalog #23227, Pierce, Rockford, IL, USA). After
normalization, cell lysates were resuspended in SDS sample buffer and denatured
at 95 °C for 5 minutes. Twenty
All data are presented as the mean
The expression of TRPM7 mRNA and protein in DFCs was confirmed using PCR,
western blot and immunofluorescence staining (Fig. 1a–d). Electrophysiological
recordings were then made to determine whether TRPM7 currents were active in
DFCs. In the absence of Mg
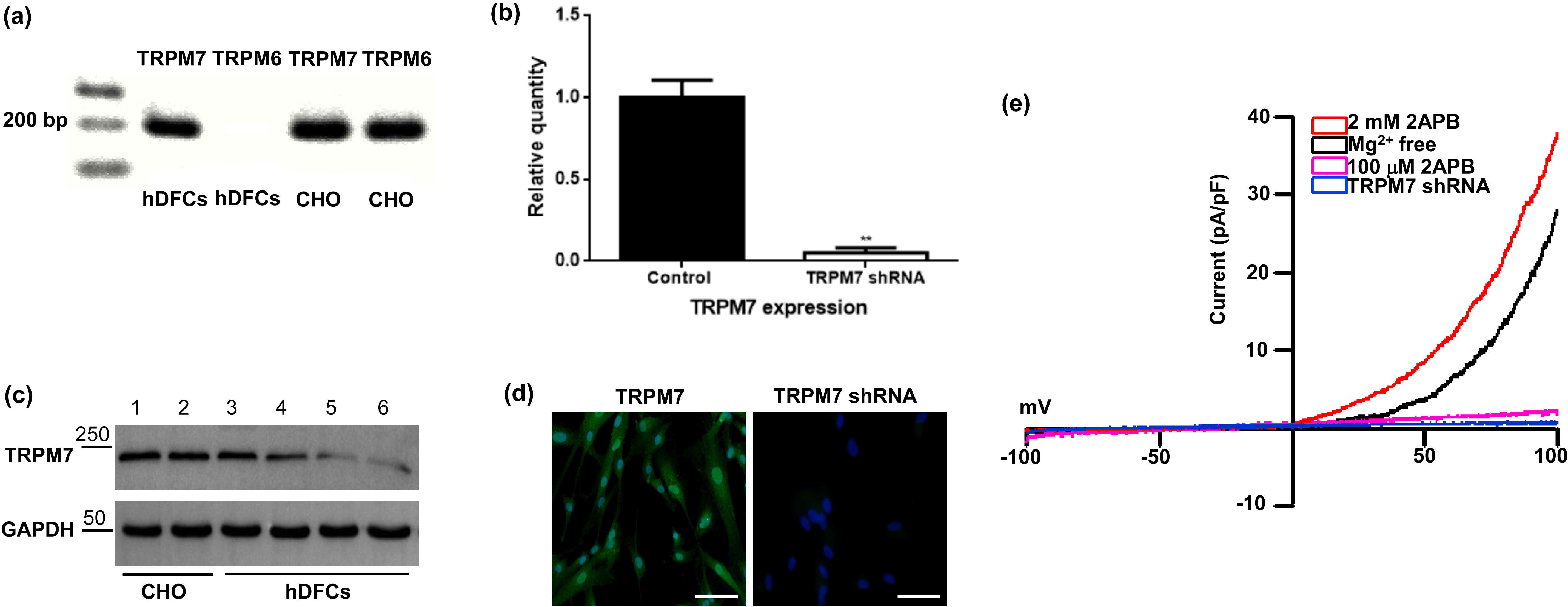 Fig. 1.
Fig. 1.Knockdown of TRPM7 in DFCs using lentiviruses that express
TRPM7-specific shRNA. (a) TRPM7 mRNA, but not TRPM6 mRNA, was detected in DFCs
by reverse transcription PCR. CHO cells with ectopic expression of TRPM6 or TRPM7
were used as positive controls. (b) qRT-PCR showed that TRPM7 gene expression was
significantly reduced in TRPM7 knockdown cells. Quantitative data from three
independent experiments is shown. **, p
We next evaluated whether TRPM7 has a role in the proliferation and migration of DFCs. For this purpose, shRNA-expressing lentiviruses were used to knockdown TRPM7 expression in DFCs. The effectiveness of shRNA was confirmed by performing qRT-PCR, immunofluorescence, western blot and electrophysiological recordings. The results showed significant reductions in TRPM7 mRNA and protein levels, as well as in whole cell TRPM7-like currents in the TRPM7 knockdown cells compared to control cells infected with scrambled shRNA (Fig. 1b–e).
CCK-8 and transwell chamber assays were used to assess the effect of TRPM7 knockdown on the proliferation and migration of DFCs. CCK-8 assay results showed that at all time points (2, 4 and 6 days), the proliferation of TRPM7 knockdown cells was significantly reduced compared to control cells infected with scrambled shRNA (Fig. 2a). Transwell chamber assay results showed that fewer cells with TRPM7 knockdown migrated after 48 hours culture compared to control cells infected with scrambled shRNA (Fig. 2b). Quantitative data showed that TRPM7 knockdown reduced the number of migrated cells by almost half (52.17%) compared to the control cells (Fig. 2c).
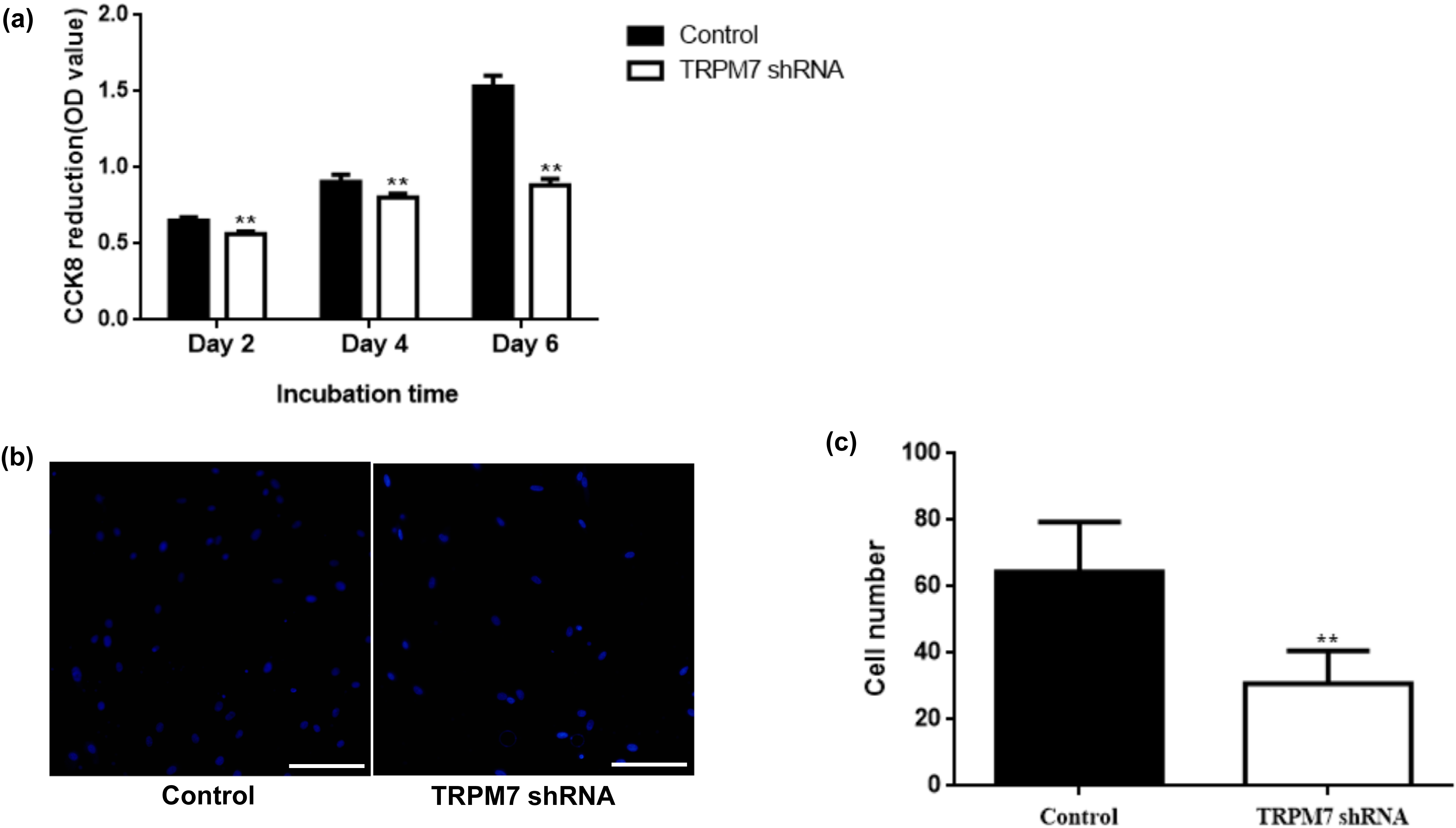 Fig. 2.
Fig. 2.TRPM7 knockdown inhibited the proliferation and migration of
DFCs. (a) CCK-8 assay showed that TRPM7 knockdown inhibited cell proliferation
of DFCs. Quantitative data from five independent experiments are shown. **,
p
To evaluate the effect of TRPM7 on the osteogenic differentiation of DFCs, we cultured these cells in osteogenic differentiation media for 14 days and then determined ALP activity, expression of osteogenic related genes, and formation of mineral deposits. Inhibition of TRPM7 with specific shRNA was found to enhance mineralization (Fig. 3a,b), increase ALP activities (Fig. 3c), and upregulate the expression of ALP, RUNX2 and BSP (Fig. 3d–f) compared to control cells infected with scrambled shRNA.
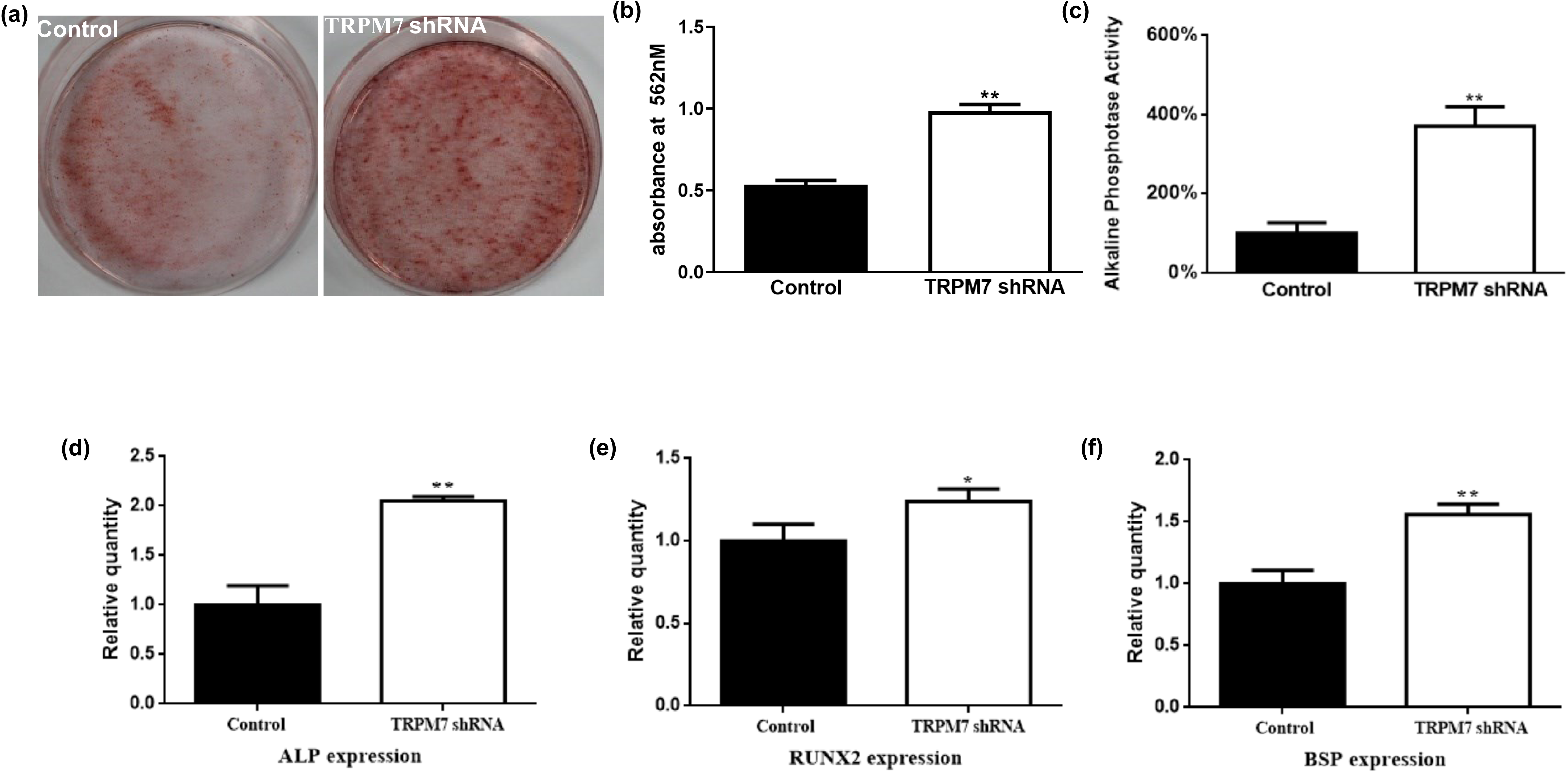 Fig. 3.
Fig. 3.TRPM7 knockdown promoted the osteogenic
differentiation of DFCs. (a,b) Alizarin red staining
showed that TRPM7 knockdown enhanced mineralization.
(c) TRPM7 knockdown increased the ALP activities of
DFCs. Quantitative data from five independent
experiments are shown. **, p
We next examined whether Mg
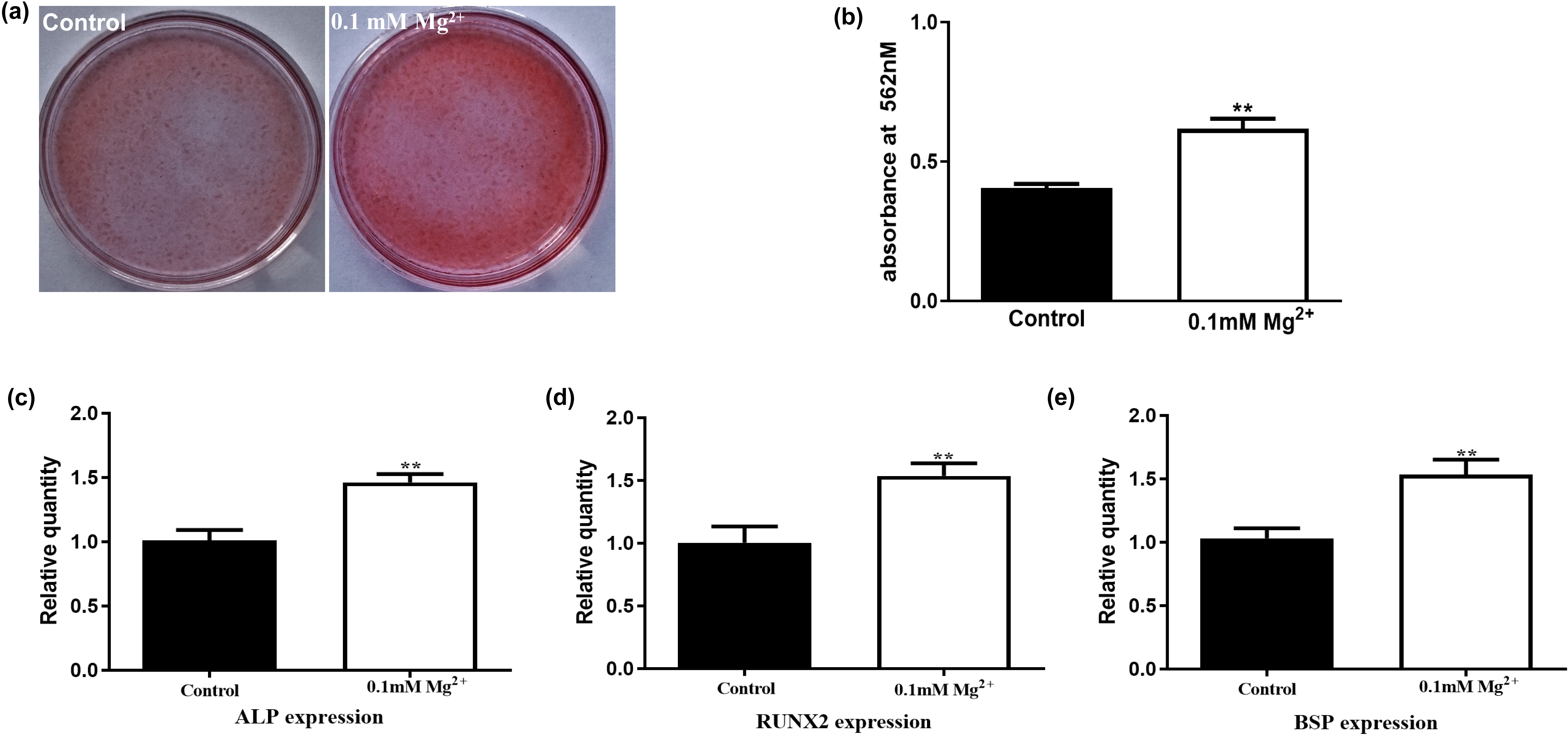 Fig. 4.
Fig. 4.Mg
TRPM7 expression was detected in the ameloblast, odontoblast and osteoblast
cells of dental tissue [19, 20]. Pharmacological block of TRPM7 inhibits
mechanically-evoked Ca
To investigate this, we first confirmed the presence of TRPM7 transcript and
protein in DFCs. TRPM6, a closely related channel with similar function, was not
detected. This agrees with a previous report showing that TRPM7 was more widely
distributed than TRPM6 [22]. In addition, we performed electrophysiological
recordings to determine whether TRPM7 was functionally active in DFCs. In the
absence of intracellular Mg
To study the function of TRPM7 in DFCs, we generated stable knockdown cells with
a lentiviral vector expressing specific TRPM7 shRNA. The knockdown efficiency of
TRPM7 was confirmed by qRT-PCR, western blotting, immunofluorescence and
electrophysiological recordings. Our results showed that knockdown of TRPM7
significantly inhibited the proliferation and migration of DFCs. These results
were expected, since TRPM7 is thought to be the main pathway for Mg
An important observation in the present study was that TRPM7 had an inhibitory
effect on the osteogenic differentiation of DFCs. We observed that ALP
activities, osteogenic-related gene expression, and the number of mineralized
matrix nodules all increased in TRPM7 knockdown cells after osteogenic induction
compared to control cells. Castiglioni et al. [27] previously reported
that silencing of TRPM7 accelerated the osteogenic differentiation of human MSCs.
Interestingly, similar results were obtained when the cells were cultured in
Mg
In conclusion, this study has demonstrated for the first time that TRPM7 channels are functionally expressed in DFCs. Functional studies showed that suppression of TRPM7 inhibited the proliferation and migration of DFCs, while promoting their osteogenic differentiation. These results suggest that TRPM7 has an important role in regulating the biological behavior of DFCs.
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
JZ, JialiL, YH, JiantaoL, SY, LX performed the experiments. JZ and DZ participated in the design of the experiments, analysis of data, and writing of the paper. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The ethics committee of Southwest Medical University approved all human cell culture experiments (20221107004). Informed consent was obtained from donors who provided teeth for subsequent research.
We thank Dr. Weidong Tian, Dr. Weihua Guo and Dr. Li Xie (West China Hospital of Stomatology, Sichuan University, Sichuan, China) for technical assistance.
This work was supported by Sichuan Science and Technology Program (2023NSFSC1522 and 2022YFS0634); Luzhou-Southwest Medical University Joint Project (2020LZXNYDJ05, 2019LZXNYDJ01).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.