








1 Department of Neurosurgery, Affiliated Hangzhou First People's Hospital, Zhejiang University School of Medicine, 310030 Hangzhou, Zhejiang, China
2 The Fourth Clinical Medical College, Zhejiang Chinese Medical University, 310053 Hangzhou, Zhejiang, China
Abstract
Background: Gastrodin (Gas) exhibits anti-inflammatory properties against diseases associated with the central nervous system (CNS). This study aimed to investigate the potential neuroprotective role of Gas in traumatic brain injury (TBI). Methods: A rat TBI model was established in male adult Sprague-Dawley (SD) rats by controlled cortical impingement (CCI), and lipopolysaccharide (LPS) was applied to induce the activation of BV2 microglia and HT22 hippocampal neurons. Neurological deficits, motor function and brain water content were evaluated in TBI rats. TUNEL and Nissl’s staining were applied to measure neuronal degeneration and apoptosis. Microglial activation, the mRNA and protein profiles of pro-inflammatory cytokines were tested by immunohistochemistry (IHC), quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) and enzyme-linked immunosorbent assay (ELISA), respectively. Results: Gas significantly reduced neurological deficits, cerebral edema, and neuronal apoptosis and improved motor function in TBI mice. In addition, Gas inactivated microglia and blocked the production of pro-inflammatory cytokines on the damaged side of the TBI rat brain. In vitro, Gas attenuated BV2 microglia inflammation and reduced HT22 hippocampal neuronal apoptosis. On the other hand, Gas activated the PKA/CREB/BDNF pathway both in vivo and in vitro. Conclusions: Gas blocks microglial activation-mediated inflammation through the PKA/CREB/BDNF pathway, thereby improving neurobehavioral function after TBI, which provides a potential therapeutic benefit for treating TBI.
Keywords
- gastrodin
- neurological protection
- microglial activation
- traumatic brain injury
Traumatic brain injury (TBI) is the primary reason for long-term disability and death among young people, which imposes a significant economic and medical burden on society [1]. TBI can be divided into primary and secondary injuries. The initial damaging force can lead to primary brain injury, resulting in tissue destruction and deformation early after injury. Secondary injuries appear hours, days, or months after the primary injury and include inflammation, brain edema, blood-brain barrier (BBB) rupture, oxidative stress, excitotoxicity, and mitochondrial and metabolic dysfunction [2]. An estimated 10 million people are yearly affected by TBI, and mortality and morbidity are increasing each year, making it a global health challenge.
Inflammation is a critical factor in TBI, leading to extensive cell death and chronic tissue degeneration. Astrocytes, microglia, and central nervous system (CNS) cells are thought to be essential factors in triggering the inflammatory response after injury [3]. Microglia are the main cellular component of the innate immune system of the central nervous system and are the first line of defense in the event of injury or disease. In the injured brain, microglia activate and produce numerous pro-inflammatory and cytotoxic mediators that impede CNS repair and lead to neuronal dysfunction and apoptosis [4]. Additionally, several studies have shown that inactivating microglia in TBI reduces the production of inflammatory cytokines, but the specific mechanism still needs further exploration [5, 6, 7]. Therefore, searching for pharmacological strategies to inhibit microglial status may be an effective way to minimize post-TBI brain damage and improve neurological function.
Gastrodin (Gas), a phenolic glycoside, is the dominating bioactive constituent of Rhizoma Gastrodiae. Since its identification in 1978, Gas has been found to have beneficial effects on the CNS through modulation of neurotransmitters, anti-oxidant and anti-inflammatory effects, and inhibition of microglial activation through the MAPK pathway [8]. It has been confirmed that Gas pretreatment reduces ischemia/reperfusion (I/R) injury-induced neurobehavioral deficits in rats, attenuates the size of cerebral infarction, reverses blood-brain barrier (BBB) injury and alleviates inflammation [9]. Besides, in the rat intracerebral hemorrhage (ICH) model, Gas distinctly improves brain edema and neurological deficits after ICH and represses oxidative stress and neuronal apoptosis in ICH rat brain tissues [10]. More importantly, Yao YY et al. [11] found that Gas abates the proliferation of BV2 microglia stimulated by lipopolysaccharide (LPS) and reduces the expression of inflammatory cytokines in BV2 cells. The same findings are also obtained by Li JJ et al. [12]. Gas inactivates microglia, thereby reducing neuroinflammation. However, research on Gas therapy for TBI is limited. This paper aims to investigate the effect of Gas on inflammation after TBI and its related mechanisms and to provide a basis for the clinical practice of Gas in treating TBI.
BV2 microglia and HT22 hippocampal neurons were obtained from the Chinese Academy of Sciences (Shanghai, China). All cell lines were tested for mycoplasma and no mycoplasma contamination was observed. All cell lines were detected by Short Tandem Repeat (STR) test and all were detected correctly. Cells were maintained in DMEM with 10%
FBS and 1% penicillin/streptomycin. The cell viability was examined
periodically, and the medium was replaced every three days. The logarithmic
growth phase cells were selected. BV2 microglia (5
The animal test was authorized by the Experimental Animal Ethics Committee of
Zhejiang University School of Medicine and implemented under strict supervision (EAECZUSM20210013).
Forty adult male Sprague-Dawley (SD) rats (220–270 g) were purchased from the
Institute of Experimental Animals of Wuhan University. Rats were placed in a
temperature (23
Rats were divided into sham, Gas, TBI, and TBI+Gas groups (10 rats in each group). The stereotactic impacter Impact One™ (Leica Biosystems, Deer Park, IL, USA) was applied to cause controlled cortical impingement (CCI) damage in rats [13]. The rats were immobilized in a stereotactic instrument and anesthetized with isoflurane through a nasal mask. The induction rate was 3%, and the maintenance rate was 2%. A midline incision was made in the skull. Unilateral craniectomy was carried out between Bregma and Lambda with a hand drill (5 mm in diameter). The rats were hit with a 4.0 mm stainless steel flat impacter tip. The conditions were as follows: stereotactic coordinates (AP: –2.26 mm and ML: +2.0 mm), depth (0.65 mm), speed (4.0 m/s), residence time (200 ms), and angle (0.4°). After the injury, the bleeding was cleaned up, the incision was sutured, and the anesthesia was immediately removed. After the surgery, the rats were placed in cages on heating pads. The recovery of each rat was observed until they were standing on their four paws. The sham-operated rats underwent the same process as those with CCI injury, without craniotomy and later hits. Rats in the Gas group and TBI+Gas group were intraperitoneally injected with Gas (100 mg/kg; Sigma, USA) one hour before CCI, once daily for seven days. In contrast, the sham and TBI groups were not treated.
As previously stated [14], the rats were decapitated after the nerve injury was
assessed. Their brains were harvested immediately at 3 and 7 days after the
surgery. For craniotomy, a portion of the cerebral cortex was isolated (about 2
mm; 200
The Rotarod test was utilized to test motor coordination and balance [15, 16]. Before CCI induction, train rats to hold on to the rotarod for 120 s at 10 rpm (adaptation phase). After the rat was accustomed to running on the stick, the speed was accelerated to 40 rpm for 300 s. The mean time to fall of the last three trials was recorded as the baseline latency. After CCI, rats were examined three times on days 1, 3, 7, 14 and 21, and the mean fall latency was recorded.
TUNEL staining and Nissl staining were performed with the method of Wang H [17]. First, antigen-retrieved brain slices were incubated with a TUNEL reaction mix (Beyotime, China) for one hour at room temperature. After washing with phosphate buffered saline (PBS) for 20 min, the slides were incubated with DAPI for 5 min. Then, the sections were rinsed twice and observed with an inverted fluorescence microscope. ImageJ software was used to count TUNEL-positive cells. The number of apoptotic cells relative to DAPI was considered the apoptotic index. For Nissl staining, sections were stained in Nissl staining solution (Beyotime, China) for 5 min, washed with double distilled water, and mounted with Permount. This photo was taken under an optical microscope. Neurons with visible nuclei and relatively intact cell morphology were counted by ImageJ (version 2, LOCI, University of Wisconsin, Madison, WI, USA) and Cellcounter software 1.0 (Ningbo Shunyu Technology, China).
Rats were sacrificed under chloral hydrate anesthesia. The injured side brain
tissue was taken, stored in 100 g/L paraformaldehyde solution, and fixed at room
temperature for 24 hours. Cut the brain tissue into 3–4
Total RNA in tissues was extracted using the TRIzol reagent (Invitrogen,
Waltham, MA, USA) according to the manufacturer’s instructions. The RNA
concentration and purity were determined with Nanodrop-spectrophotometer
(Invitrogen, Waltham, MA, USA). Complementary DNA (cDNA) was synthesized from 1
| Target | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| IL-1 |
GGATGATGACGACCTGC | CTTGTTGGCTTATGTTCTG |
| IL-6 | TTTCACCAGGCAAGTCTCCT | AGACAGCCACTCACCTCTTC |
| TNF- |
CTTATCTACTCCCAGGTTCTCTTCAA | GAGACTCCTCCCAGGTACATGG |
| GAPDH | GGGA GCCAAAAGGGTCAT | GAGT CCTTCCACGATACCAA |
WB was carried out using the methods of predecessors [18].
The membranes were incubated with the primary antibodies (1:1000; Abcam, Waltham, MA, USA) overnight at 4 °C as follows: Bad (ab32445, Abcam, Waltham, MA,
USA), Bax (ab32503, Abcam, Waltham, MA, USA), Bcl-2 (ab32124, Abcam, Waltham, MA,
USA), iNOS (ab178945, Abcam, Waltham, MA, USA), TLR4 (ab13556, Abcam, Waltham,
MA, USA), NF-
The interleukin (IL)-1
Cell viability was determined by CCK-8 assay. The cells were plated into 96-well
plates at a density of 1
The cells were trypsinized and harvested by centrifugation at 1500 rpm for 3
min. The cell apoptosis was determined by the apoptosis detection kit
(Shanghai Aladdin biological technology Co., Ltd). Briefly, the cells were washed
with PBS twice and then incubated with 400
Statistical analysis was performed by SPSS17.0 statistical software (IBM SPSS,
Chicago, IL, USA). One-way analysis of variance (ANOVA) was used to compare
between groups. The measurement data were presented as mean
To understand the effect of Gas (100 mg/kg) on the neurological function of TBI
rats, we implemented a modified Neurological Severity Score (mNSS), rotarod test
and the wet/dry method to test the neurological function, motor function and
brain water content of TBI rats 1, 3, 7, 14 and 21 days after TBI. The findings
demonstrated that mNSS in the sham and Gas group did not change at the
corresponding time points (with a score of 1–3). Nevertheless, neurological
function was seriously damaged by TBI. After the treatment of Gas, the mNSS of
rats in the TBI+Gas group was significantly lower than that of the TBI group
(p
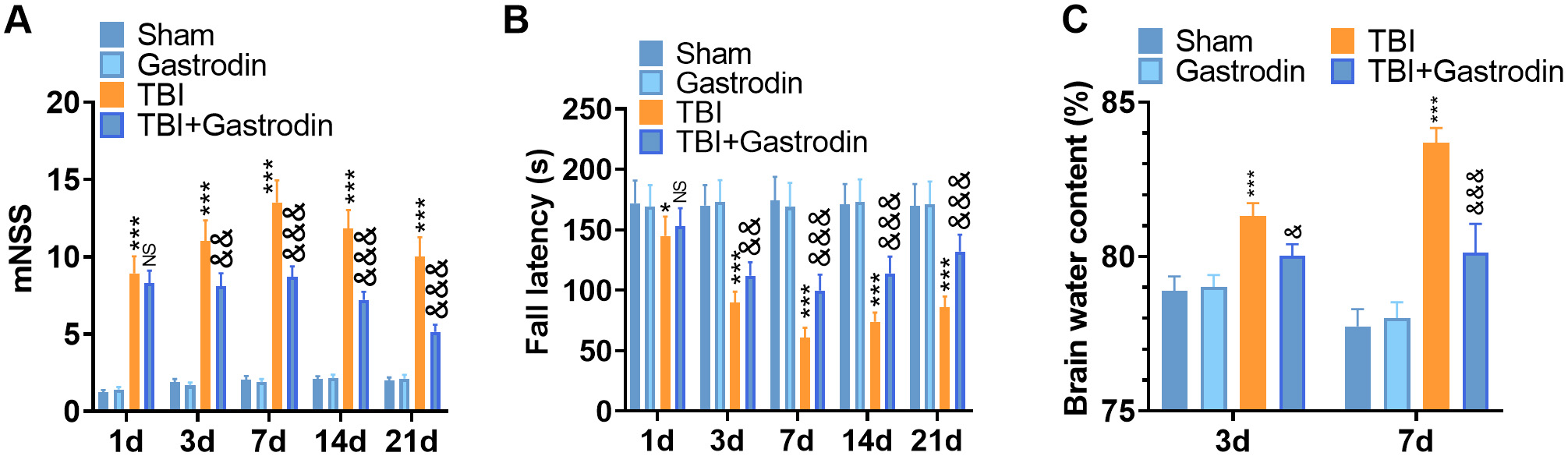 Fig. 1.
Fig. 1.Gas reduced the neurological deficits and brain water content of
TBI rats. (A,B) The recovery of the nervous system was analyzed by mNSS (A), and
Rota-rod (B) tests at 1, 3, 5, 7, 14 and 21 days after TBI. (C) The water content
in the brain of TBI rats was assessed by the dry and wet method at 3, 7 and 21
days after TBI. *p
Nissl and TUNEL staining were performed to verify neuronal apoptosis in the
damaged side of the cerebral cortex of TBI rats. The results illustrated that the
neurons in the sham group were clear and complete. In contrast, damaged neurons
in the TBI group exhibited irregular cell bodies, enhanced crinkling and nuclear
pigmentation, and increased TUNEL-positive cells. After the treatment of Gas, the
neuronal loss and TUNEL-positive neurons decreased in the TBI group (p
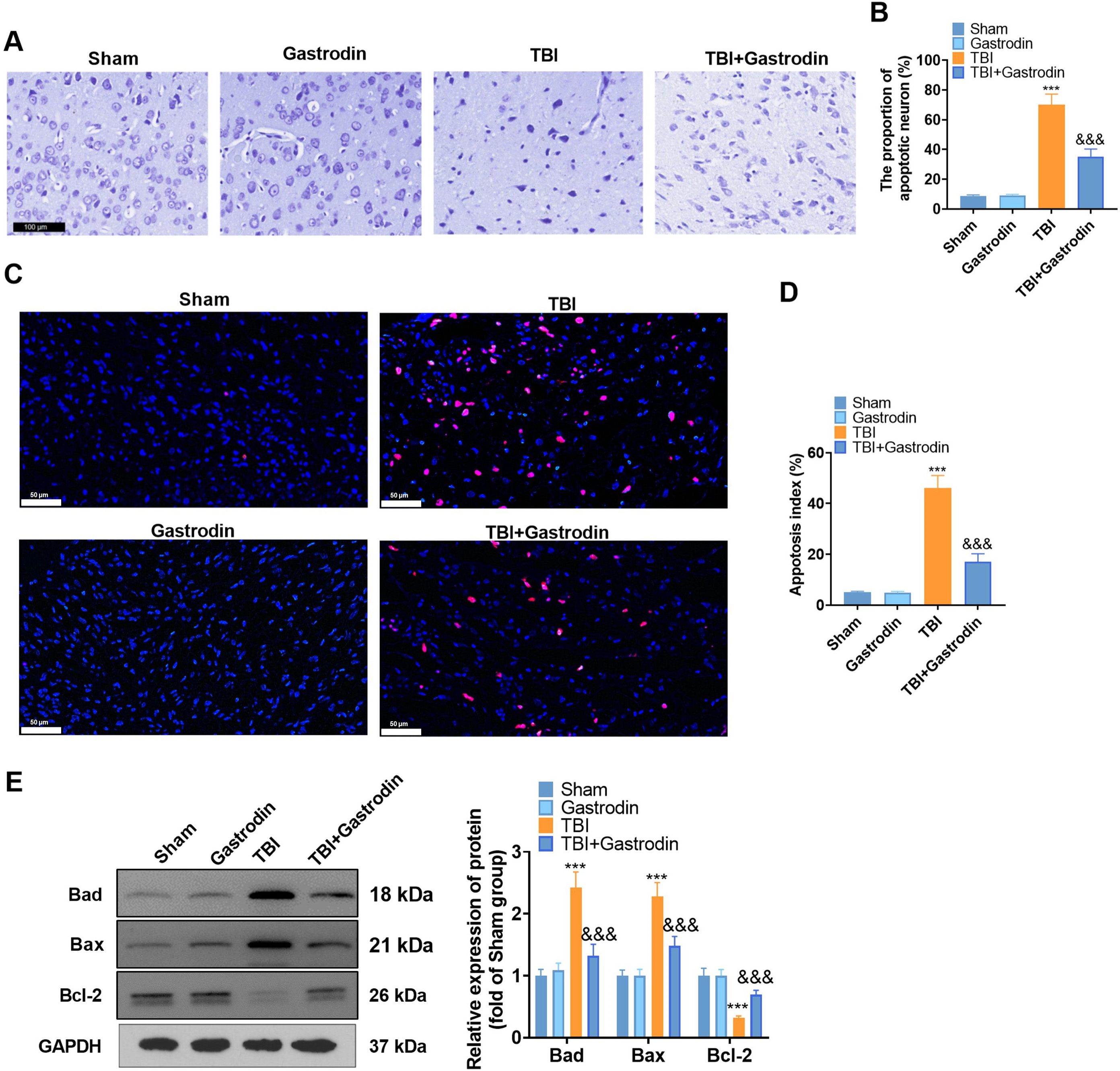 Fig. 2.
Fig. 2.Gas inhibited neuronal degeneration and apoptosis after brain
injury in rats. (A,B) Representative photomicrograph (A) (scale = 100
First, IBA1-labeled microglia in the brain tissues of TBI rats were determined
by IHC. The results indicated that IBA1 microglia were significantly increased in
the TBI group compared with the sham group. However, the IBA1 microglia were
reduced after Gas intervention (p
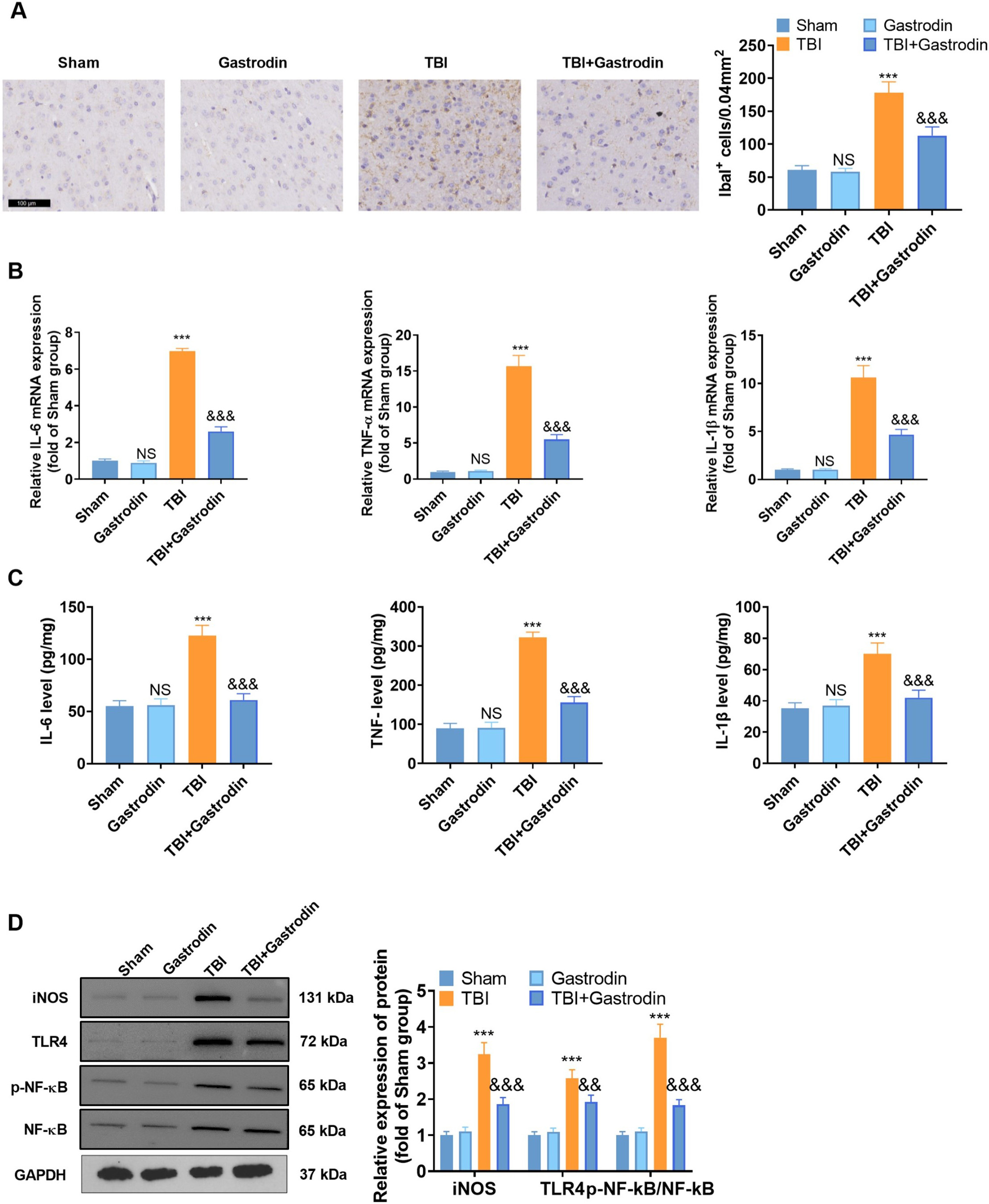 Fig. 3.
Fig. 3.Gas repressed microglia-mediated inflammations in TBI rats. (A)
IHC was utilized to check the representative images and quantification of
IBA1-positive cells in the damaged side of the cortex of TBI rats on day 7, with
a scale of 100
To explore the molecular mechanism of Gas in vivo, the effects of Gas
on the PKA/CREB/BDNF pathway were determined. Our results indicated that the PKA
level was decreased, the phosphorylation of CREB was inhibited, and Bcl2 was
significantly downregulated in the TBI group compared with the sham group.
However, Gas intervention yielded opposite results (p
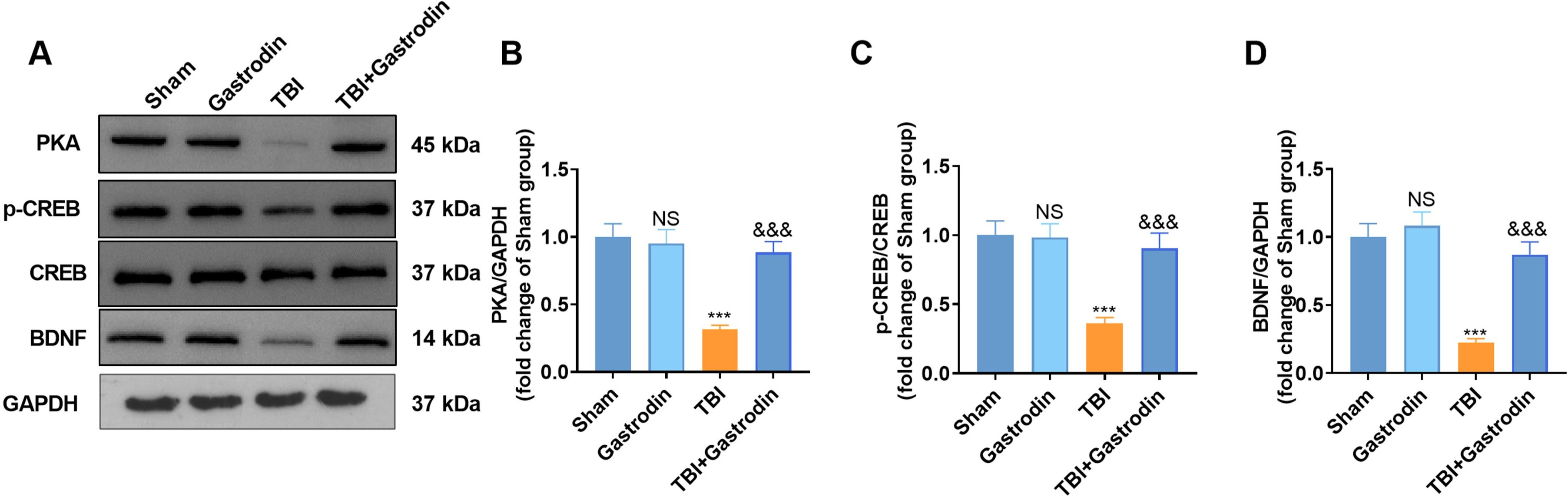 Fig. 4.
Fig. 4.Gas activated the expression of PKA/CREB1/Bcl2. (A–D) WB was utilized to test the PKA expression, the CREB phosphorylation, and the BDNF profile in the damaged side of the cortex of TBI rats on day 7. NS p
Inflammatory responses are the main pathological mechanism of brain injury.
Therefore, the influence of Gas (40
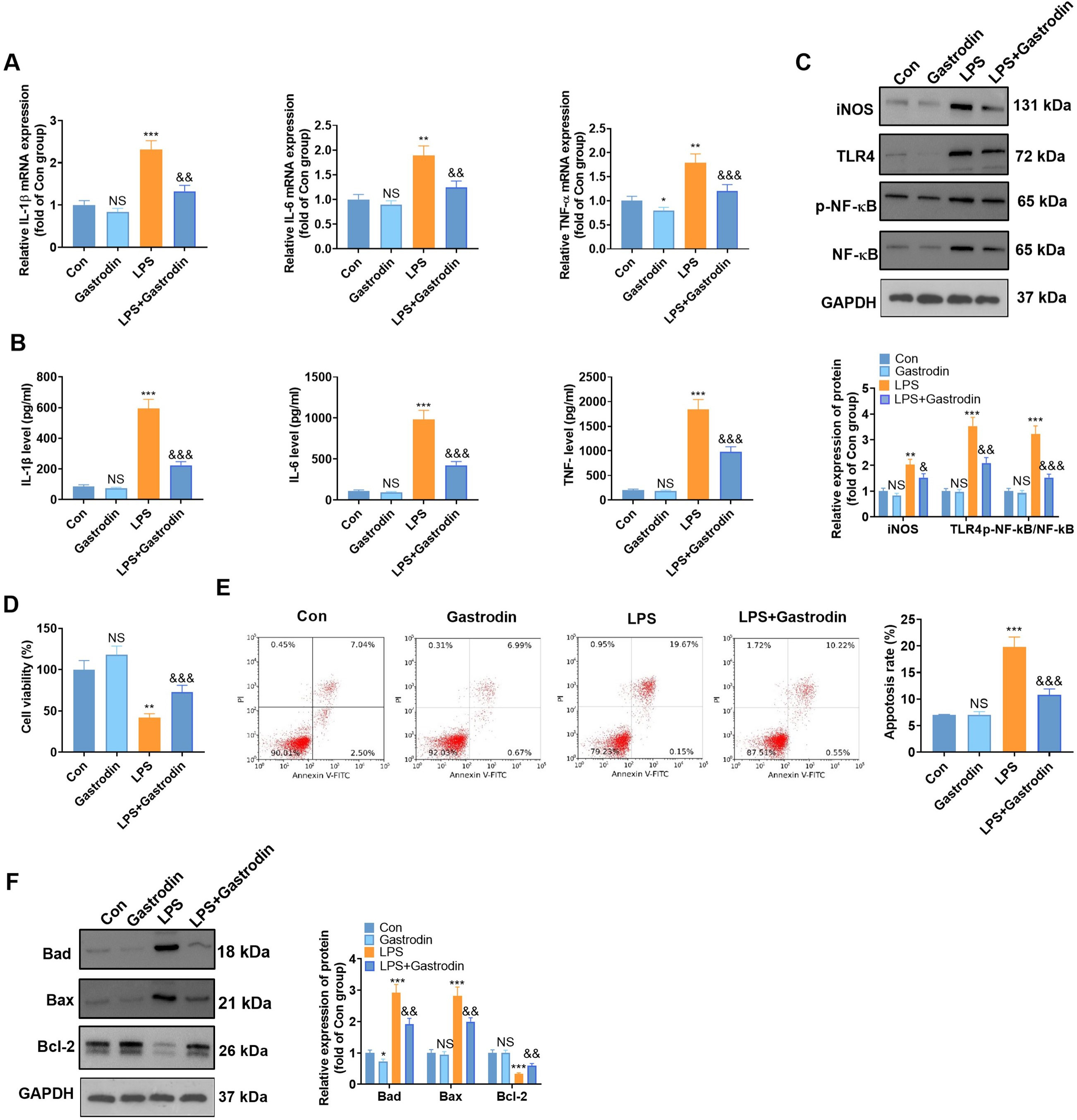 Fig. 5.
Fig. 5.Gas attenuated LPS-mediated inflammatory responses and neuronal
apoptosis in BV2 microglia (suggestion: in row E, it would be better to use a
bold or larger font to make the labels easy to read). BV2 microglia and HT22
hippocampal neurons were treated with LPS (1 g/mL) for 24 hours with or without
Gas (40
The effects of Gas in vitro have been determined. First, the
PKA/CREB/BDNF pathway expression in LPS-treated BV2 microglia and HT22
hippocampal neurons was evaluated. The results confirmed that PKA was
downregulated, the phosphorylation of CREB was reduced, and the expression of
BDNF was significantly attenuated in the LPS group compared with the control
group (p
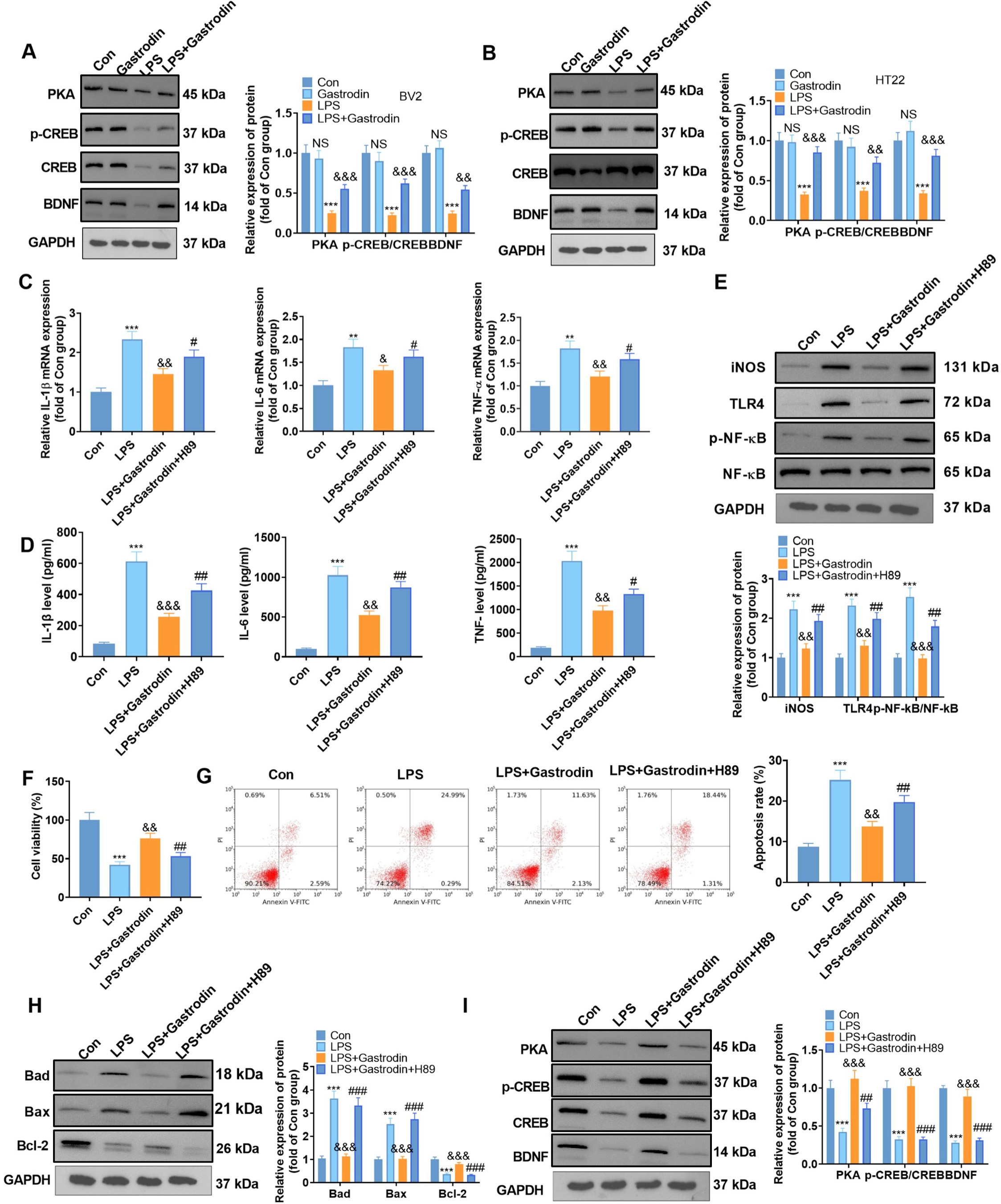 Fig. 6.
Fig. 6.Inhibiting the PKA/CREB/BDNF pathway weakened the
anti-inflammatory and anti-apoptotic effects of Gas on cells (suggestion: for row
G, please refer to the suggestion for Fig. 5 row E). (A,B) WB was employed to
measure the PKA expression, CREB phosphorylation, and the BDNF profile in BV2
microglia and HT22 hippocampal neurons. (C,D) qRT-PCR (C) and ELISA (D) were
applied to define the expression of inflammatory cytokines in BV2 microglia
treated with H89 (10
TBI is the cause of various neurological and cognitive deficits as well as psychosocial dysfunction. The incidence of TBI is in the millions every year and is increasing globally [20]. Although many promising neuroprotective therapeutic options have emerged from preclinical research primarily targeting lesions, they can still not be applied clinically [21]. Therefore, the development of effective drugs for the treatment of TBI is crucial. In this study, we explored the neuroprotective effects of Gas in a rat model of TBI and assessed the potential function of the PKA/CREB/BDNF pathway. The findings suggest that Gas exerts neuroprotective effects by activating the PKA/CREB/BDNF pathway.
Gastrodin (Gas; 4-hydroxybenzyl alcohol
4-O-
Cyclic adenosine monophosphate (cAMP), the first and second messenger identified, contributes to cell signaling and regulates various physiological and pathological processes. cAMP mainly regulates the transcription of multiple target genes through protein kinase A (PKA) and its downstream effectors, such as cAMP response element binding protein (CREB) [30]. Previous studies have shown that drugs can reduce LPS-induced inflammatory cytokine production by modulating the cAMP-PKA pathway in BV2 cells [31]. For example, Morin suppressed LPS-stimulated microglia inflammation by upregulating the PKA/CREB pathway [32]. BHDPC induces phosphorylation of PKA and CREB to suppress neuroinflammation in LPS-activated BV2 microglia and promote M2 polarization of microglia [33]. Furthermore, after TBI, the cAMP/PKA/CREB pathway is downregulated, while phosphodiesterase 4 activates CREB to improve cognitive impairment [34]. In this study, we found that Gas activates the PKA/CREB pathway in a rat model of TBI. Moreover, inhibition of the PKA/CREB pathway reversed Gas-mediated anti-inflammatory and anti-apoptotic effects. These results suggest that Gas protects against TBI-mediated nerve injury by activating the PKA/CREB pathway.
The protein family of B-cell lymphoma 2 (Bcl-2) is an important factor in the apoptotic pathway. The Bcl-2 family members mainly regulate apoptosis, and abnormal expression of Bcl-2 is associated with multiple diseases, including cancer, neurodegenerative diseases, local ischemia and autoimmune diseases [35]. Bcl2 is the target of CREB. A previous study demonstrated that inhibiting equilibrative nucleoside transporter 1 suppressed neuronal apoptosis and ameliorated neurological impairment through activation of cAMP/PKA/CREB pathway [36]. In ischemic brain injury, propofol increases the contents of cleaved caspase-3 and Bax in rat hippocampal neurons by suppressing the activity of cAMP-dependent proteins and Bcl-2 [37]. Moreover, BDNF, as a neurotrophin, has been confirmed as a vital cytokine involved in synaptic transmission and long-term potentiation (LTP) in the hippocampus and other brain regions [38], and it also protects neurons from apoptosis in acute brain injury [39, 40, 41]. Therefore, we investigated the expression and role of the PKA/CREB/BDNF axis in TBI. The results demonstrated that Bcl2 and BDNF were downregulated in the brain tissues of TBI rats, LPS-induced BV2 microglia and HT22 hippocampal neurons. In contrast, the Gas enhanced the expression of Bcl2 and BDNF, while inhibition of the PKA/CREB pathway by H89 weakened Bcl2 and BDNF levels. These results confirmed that Gas eases TBI by activating PKA/CREB/BDNF pathway.
This study contained limitations, and our results must be further validated in
clinical applications. Currently, there is only the optimal model of TBI
in vivo, and there is no excellent stimulation method to completely
simulate the occurrence of TBI in vitro. This study employed
LPS-mediated chemical brain injury to mimic this process. When TBI occurs, the
human environment changes dramatically, and microglia polarize into one of two
distinct phenotypes: M1 and M2. M1 microglia play a role in promoting the
development of inflammation. Within injured tissue, microglia exist in various
states of activation and retain the ability to alter their functional phenotype
during inflammatory responses. When an injury occurs, microglia polarize towards
a pro-inflammatory (M1) phenotype, produce pro-inflammatory cytokines such as
TNF-
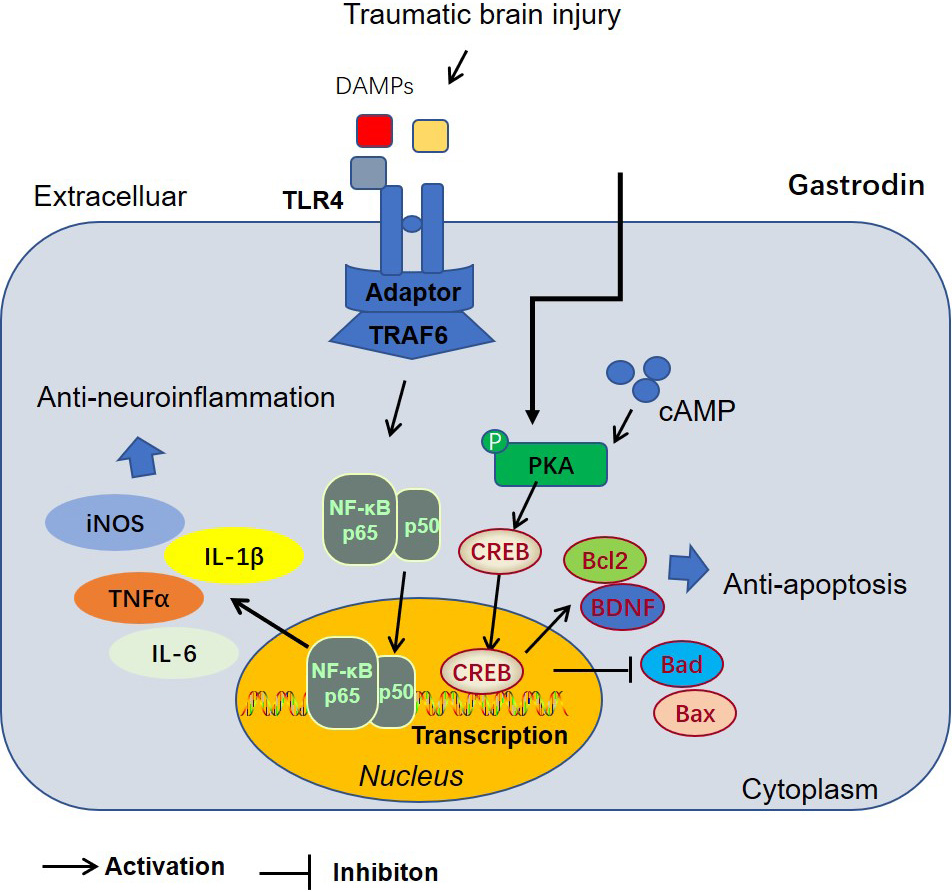 Fig. 7.
Fig. 7.The mechanism’s diagram of Gas-mediated anti-apoptosis and anti-neuroinflammation via inhibiting the PKA/CREB1/Bcl2 pathway.
In conclusion, our results indicated that Gas improves neural function and reduces neuronal apoptosis in TBI rats by targeting PKA/CREB/BDNF, suggesting that Gas is a new potential therapeutic approach for TBI treatment. To our knowledge, this is the first report on the role of Gas in TBI.
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
CS, WZ and QD designed the research study. CS and WZ performed the research. LW analyzed the data. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The animal test was authorized by the Experimental Animal Ethics Committee of Zhejiang University School of Medicine and implemented under strict supervision (EAECZUSM20210013).
Not applicable.
This research was supported by the Medical Health Science and Technology Project of the Zhejiang Provincial Health Commission (2021KY229).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.