


1 Department of Medicine, Uniformed Services University of the Health Sciences, Bethesda, MD 20814, USA
Abstract
Maintaining a balance between the supply and demand of oxygen is vital for
proper organ function. Most types of acute kidney injury (AKI) are characterized
by hypoxia, a state where the supply of oxygen cannot match the demand for normal
cellular activities. Hypoxia results from hypo perfusion and impaired
microcirculation in the kidney. It inhibits mitochondrial oxidative
phosphorylation, resulting in a decrease in production of adenosine triphosphate (ATP), which is essential
to power tubular transport activities, especially reabsorption of Na
Keywords
- mitochondria
- oxygenation
- hypoxia
- Na+-H+ exchanger 3
- Na+-dependent glucose transporter 2
- Na+ reabsorption
Acute kidney injury (AKI) is defined by a sudden decrease in glomerular
filtration rate (GFR), occurring within hours to weeks, along with the retention
of nitrogenous waste products and renal parenchymal damage. Examples of AKI
include acute tubular necrosis, acute interstitial nephritis, and
glomerulonephritis. Based on the Kidney Disease: Improving Global Outcomes
criteria, AKI is diagnosed by an absolute increase in serum creatinine levels of
at least 0.3 mg/dL (26.5
Acute kidney injury is associated with poor outcomes. Acute kidney injury is
independently associated with both short and long-term morbidity and mortality
[6, 10, 11, 12, 13]. For example, a recent study of 1286 COVID-19 patients in Belgium
demonstrated that all stages of AKI were associated with increased ICU mortality
rates with 9.3% at stage 1, 40.1% at stage 2 and 47.0% at stage 3 compared to
3.6% with no AKI [14]. Furthermore, the incidence of AKI is steadily rising due
to an aging population, increased prevalence of CKD, and improved recognition by
physicians [15]. In addition to the high human cost, AKI imposes a heavy
financial burden on society. In the United States, the in-hospital costs for AKI
ranged from
In order to achieve clearance, the human kidney filters approximately 180 liters
of blood each day. This filtrate is composed of both harmful metabolites and
essential elements for life, such as NaCl, amino acids, and glucose.
Approximately 99% of water and ions, and 100% of amino acids and glucose are
reabsorbed by the renal tubules through the transcellular and paracellular
pathways. The Na
Despite diverse etiologies, the majority of AKI sub types share common pathophysiologic mechanisms, including microvascular dysfunction and inflammation [3]. Microvascular dysfunction leads to hypoxia. Hypoxia is a state where oxygen supply cannot meet oxygen demand, which has a profound impact on kidney function (Fig. 1). Artificially induced hypoxia by increasing oxygen consumption with triiodothyronine without other compounding factors induces kidney injury in rats [25]. Similarly, treating rats with normobaric hyperoxia per se helps renal recovery from warm ischemia-induced injury [26]. Hypoxia compromises mitochondrial aerobic metabolism by diminishing nicotinamide adenine dinucleotide hydrogen (NADH) supply and electron transport [27]. Hypoxia leads to the accumulation of non-esterified fatty acids, which contributes to activation of pro-inflammatory pathways and produces reactive oxygen species, resulting in apoptosis and necrosis [28, 29]. Furthermore, inflammation directly inhibits mitochondrial respiration. As a result, the ability of mitochondria to generate ATP is compromised and the cellular ATP levels are reduced in AKI [30]. Therefore, AKI has been broadly characterized as a state of tubular ATP depletion [31].
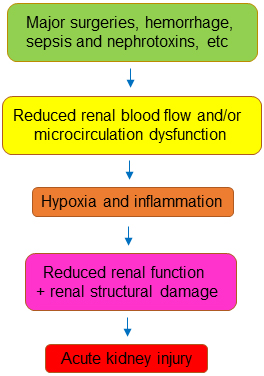 Fig. 1.
Fig. 1.A common mechanism for acute kidney injury.
To make matters worse, the efficiency of the kidney in using oxygen to reabsorb
Na
To restore the balance between oxygen supply and demand, many studies have
examined increasing oxygen supply. Examples include fluid resuscitation,
vasoconstrictors and removing the underlying cause of AKI (e.g., antibiotics and
nephrotoxic medications). These approaches have been reviewed previously [41, 42, 43].
While these approaches unarguably improve patient outcomes, they are clearly
insufficient. In most tissues, increasing blood flow will increase tissue
oxygenation, however, the kidney is an exception to this rule. The variation in
plasma Na
Reducing oxygen demand is another way to maintain the supply-demand balance of
oxygen when supply is low. Western painted turtles (Chrysemys picta) can
survive under water with little oxygen for as long as 30 hours. They tolerate
anoxia in part by reducing Na
How can reducing tubular cell injury and death help preserve and/or delay
deterioration of GFR? The renal tubular cells are the primary site of injury in
AKI. When the tubular cells undergo necrosis or apoptosis, they detach from the
supporting basement membrane and obstruct the tubular lumen, causing back leakage
of fluid with resultant decreased clearance. Even a sub-lethal injury can disrupt
tight junctions, causing a loss of epithelial integrity and back leakage of
fluid. The fluid back leakage further diminishes already impaired glomerular
filtration induced by hypoperfusion of the kidney, eventually contributing to the
filtration failure [58, 59, 60, 61]. Therefore, reducing oxygen demand through inhibition
of Na
The present review summarizes progress in balancing supply and demand to prevent or treat AKI in patients (Table 1, Ref. [62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87]) and animal models. Acute kidney injury is also associated with oxidative stress, which in turn increases oxygen consumption resulting in kidney tissue hypoxia [88, 89]. While various antioxidants have been shown to improve renal oxygenation and ameliorate AKI, reviewing the effect of antioxidants is beyond the scope of this article [90, 91, 92, 93].
| Medications | Mechanisms | Inducers of AKI | Timing therapy | Patient # | Results | References | ||
|---|---|---|---|---|---|---|---|---|
| Prevention | Treatment | Control | Medications | |||||
| A. Proximal tubules | ||||||||
| 1. Inhibition of NHE3 | ||||||||
| Acetazolamide | Reducing production of H |
Contrast | X | 46 received HCO |
50 received acetazolamide | Scr decreased | [62] | |
| Contrast | X | 96 received HCO |
96 received acetazolamide | Scr decreased | [64] | |||
| eGFR increased | ||||||||
| Cisplatin | X | 20 received mannitol | 15 received acetazolamide | AKI risk decreased from 30% to 8.9% | [63] | |||
| Coronary artery bypass surgery | X | 65 received placebo | 65 received acetazolamide | ineffective | [65] | |||
| ICU setting | X | 868 | AKI 26.8% vs vancomycin 16% and gentamicin 20% | [66] | ||||
| Dopamine | Inhibiting NHE3 translation and surface expression and promoting NHE3 degradation | Operation for cardiac diseases | X | 40 received furosemide, bumetanide or ethacrynic acid | 60 received furosemide, mannitol and dopamine | Dialysis decreased from 90% to 6.7% | [82] | |
| Cardiac surgery | X | 40 received saline | 42 received dopamine | ineffective | [84] | |||
| Fenoldopam | Dopamine receptor agonist | Post operation | X | 87 received furosemide | 39 received furosemide and fenoldopam | Tend to increase urine output (p = 0.06) | [68] | |
| Fenoldopam | Dopamine receptor agonist | X | Total patient # | [67] | ||||
| Post operation | 958 | Reduced onset and incidence of AKI | ||||||
| 917 | ineffective | |||||||
| Contrast-induced | 501 | Reduced onset and incidence of AKI | ||||||
| 714 | ineffective | |||||||
| ICU setting | 417 | Reduced onset and incidence of AKI | ||||||
| 155 | ineffective | |||||||
| Human atrial natriuretic peptide (hANP) | Possibly modulating dopamine system | ICU setting | X | 30 received placebo | 29 received hANP | Improved creatinine clearance | [83] | |
| hANP | ischemic or nephrotoxic insults | X | 114 received placebo | 108 received hANP | Increased morbidity and mortality | [85] | ||
| ARBs and ACEIs | Inhibiting the stimulatory effect of angiotensin II on NHE3 | Major operation | X | 18,871 received no ARBs or ACEIs | 268 received ARBs and 1137 received ACEIs | Reduced incidence of AKI | [72] | |
| ARBs and ACEIs | ICU setting | X | 3179 received no ARBs or ACEIs | 3179 received ARBs or ACEIs | Reduced all-cause mortality | [73] | ||
| ARBs and ACEIs | Multiple inducers | X | 40,000 received no ARBs or ACEIs | 30,801 received ARBs or ACEIs | Reduced all-cause mortality and recurrent AKI | [74] | ||
| ARBs and ACEIs | COVID-19 | X | X | 279 received no ARBs or ACEIs | 164 received ARBs or ACEIs | Reduced all-cause mortality | [69] | |
| ARBs and ACEIs | COVID-19 | X | 220 received no ARBs or ACEIs | 80 received ARBs or ACEIs | Ineffective | [70] | ||
| ARBs and ACEIs | COVID-19 | X | 100 received no ARBs or ACEIs | 30 received ARBs or ACEIs | Increased mortality and risk of AKI | [71] | ||
| Spironolactone | Inhibiting aldosterone-dependent Na |
Cardiac surgery | X | 118 received placebo | 115 received spironolactone | Ineffective | [75] | |
| Clonidine | Inhibiting sympathetic nerve-mediated activation of NHE3 | Non-cardiac surgery | X | 3452 received placebo | 3453 received clonidine | Ineffective | [76] | |
| Dexmedetomidine | Inhibiting sympathetic nerve-mediated activation of NHE3 | Aortic surgery | X | 54 received placebo | 54 received dexmedetomidine | Reduced incidence of AKI | [78] | |
| Dexmedetomidine | Laparoscopic prostatic surgery | X | 44 received placebo | 45 received dexmedetomidine | Ineffective | [77] | ||
| Dexmedetomidine | Sepsis | X | 719 received placebo | 719 received dexmedetomidine | Improved renal recovery rate and decreased in-hospital mortality | [79] | ||
| 2. Sglt2 inhibitors | ||||||||
| Empagliflozin | Inhibiting Sglt2 | Decompensated heart failure with and without diabetes | X | 9 received placebo | 10 received empagliflozin | Reduced tubular injury | [80] | |
| Empagliflozin | Decompensated heart failure with and without diabetes | X | 30 received standard medical care | 10 received standard medical care plus empagliflozin | Increased urine output | [86] | ||
| Dapagliflozin | Inhibiting Sglt2 | COVID-19 | X | 606 received placebo | 613 received dapagliflozin | Ineffective | [81] | |
| B. Thick ascending limb of the Loop of Henle | ||||||||
| Furosemide | Inhibiting NKCC2 | Cardiac surgery | X | 283 received placebo | 283 received furosemide | Ineffective | [87] | |
Scr, serum creatinine concentrations; NHE3, Na
Inhibition of NHE3 with the NHE3 inhibitor #4167 protects against acute
rejection of renal grafts in a rat model, an effect associated with preservation
of the renal ATP levels [94]. Inhibitors of NHE3, 5-(N-ethyl-N-isopropyl)
amiloride and S3226 protect against ischemia/reperfusion-induced AKI in rodents
[95, 96]. Hypoxia induces intracellular acidosis. Activation of Na
Acetazolamide is an inhibitor of carbonic anhydrase. It is used clinically to
treat glaucoma, epilepsy, high altitude sickness, and congestive heart failure
[100]. It is also a diuretic, because inhibition of carbonic anhydrase indirectly
leads to inhibition of NHE3 due to reduced production of H
Dopamine inhibits NHE3 activity by inhibiting NHE3 translation [112], promoting ubiquitin-dependent NHE3 degradation [113], decreasing NHE3 exocytosis and cell membrane recycling [114, 115]. Dopamine has been shown to increase renal oxygenation in post-cardiac surgery patients. This effect was associated with profound pre- and post-glomerular vasodilation [116]. Fenoldopam is a dopamine receptor agonist, which has been used clinically to treat hypertension in part because of its inhibition of NHE3 [117]. Fenoldopam was effective in reducing the onset of postoperative AKI in adult patients, when used prophylactically (Table 1) [67, 68, 118]. However, other studies have failed to demonstrate a beneficial effect for fenoldopam (Table 1). Furthermore, its potential benefit to prevent AKI after cardiac surgery in pediatrics remains uncertain [119].
Atrial natriuretic peptide is a peptide hormone that is produced by the walls of the heart in response to an increase in blood volume and pressure. It reduces blood volume and pressure in part through inhibition of NHE3 in the proximal tubules [120, 121]. Its analogs brain-type and c-type natriuretic peptides have similar functions [122]. How ANP inhibits NHE3 remains incompletely understood, but evidence indicates that it may be mediated in part by modulating the renal dopaminergic system [123]. It appears that while infusion of ANP raises oxygen supply by increasing renal blood flow, it also increases GFR and the subsequent burden for tubular Na reabsorption [44]. However, one small study in high risk surgery patients demonstrated a potential protective effect for ANP on kidney function (Table 1) [122]. Atrial natriuretic peptide is degraded by the endopeptidase neprilysin. Strategies that inhibit neprilysin offer an alternative means by which to raise the ANP levels. The challenge of this approach is that neprilysin also degrades endothelin, bradykinin, vasopressin and angiotensin II, which are critical for many physiologic activities. To circumvent this potential complication, Novartis Pharmaceuticals manufactures a drug named LCZ696, a mixture of the neprilysin inhibitor sacubitril and the angiotensin II receptor 1 blocker valsartan and markets it as Entresto®. Entresto protected the kidney from ischemic AKI in a porcine model of partial nephrectomy [124]. Multiple clinical trials have shown that Entresto does not increase the risk of AKI in patients with heart failure [122, 125]. However, whether Entresto has protective and/or therapeutic benefits against AKI in patients remains unknown.
It has been well described that angiotensin II stimulates NHE3, which contributes to angiotensin II-induced hypertension [126, 127, 128]. Angiotensin II increases the levels of NHE3 mRNA and protein [129], stimulates cellular trafficking and exocytotic membrane insertion [130, 131], counteracts inhibitory phosphorylation by cAMP/PKA [132], and increases transport activity [133]. Not surprisingly, angiotensin II increases oxygen consumption by renal mitochondria [134]. While angiotensin receptor blockers (ARBs) have repeatedly shown benefits in animal models of AKI [135, 136, 137, 138] and CKD in humans [139, 140, 141], the results from clinical trials of AKI with ARBs and angiotensin converting enzyme inhibitors (ACEIs) have had mixed results (Table 1). One potential adverse impact of ACEIs and ARBs in the setting of AKI is that they could worsen renal hypoperfusion. In the cases where ACEIs and ARBs have shown potential benefit, namely hypertensive COVID-19 and sepsis, it remains unclear whether the effect was mainly due to control of blood pressure or reducing oxygen consumption in the proximal tubules [69, 142, 143]. Conversely, other studies have shown either no difference or increased risk of AKI and mortality in the hospitalized COVID-19 patients treated with ACEIs or ARBs [70, 71, 144, 145].
The results from trials examining patients undergoing major surgeries are also mixed. One study showed that ARBs and ACEIs were associated with a reduced risk of AKI after major surgery [72]. However, Zhou et al. [146] reported no significant association between perioperative use of renin-angiotensin system inhibitors and postoperative AKI in patients undergoing cardiac surgery. Other studies generally agree that ACEIs or ARBs increase survival, but also increase acute kidney disease, which is defined as persistent AKI from 7–90 days [73, 74, 147].
Aldosterone binds to the mineralocorticoid receptors to increase sodium
reabsorption and potassium secretion in the distal nephron. The expression of
mineralocorticoid receptors has been detected in other tissues as well, where its
activation may be pathophysiologic. Angiotensin II stimulates the biosynthesis
and release of aldosterone from the adrenal cortex zona glomerulosa [148, 149].
Aldosterone stimulates NHE3 and Na
Angiotensin 1-7 is generated mainly by angiotensin-converting enzyme 2 and exerts its actions via activation of its receptor Mas. Its functions frequently oppose angiotensin II [157]. Angiotensin 1-7 has been shown to inhibit NHE3 activity in the proximal tubules of normotensive and hypertensive rats [158, 159]. Angiotensin 1-7 also inhibits NHE3 activity by modulating the renal dopamine system [160]. Angiotensin 1-7 decreases oxygen consumption in the thick ascending limb of the loop of Henle and presumably in the proximal tubules as well [161]. Angiotensin 1-7 alleviates AKI in a variety of animal models [162, 163, 164, 165]. However, whether it has a similar effect in humans remains unknown.
Adenosine is present at low concentrations in the extracellular space, but its
levels are greatly increased in conditions of metabolic stress, such as hypoxia
as a result of enzymatic cleavage of the nucleotide adenosine
5
The kidney is densely innervated by sympathetic nerves. Activation of these
nerves increases renal vascular resistance and reduces renal blood flow by
releasing norepinephrine and renin [170, 171]. Moreover, activation of these
nerves increases NHE3 and Na
It is important to emphasize that the renoprotective effects of fenoldopam, ANP, activation of adenosine receptor A1, inhibition of angiotensin II system and sympatholytics are not entirely mediated by inhibition of NHE3. The well-known hemodynamic effects of these approaches also contribute significantly to their reno-protection [67, 118, 122, 157, 176, 177].
Na
Na
Na
Chen et al. [211] recently reported the development of the third
generation of a synchronization modulation electric field device. This device
consists of three phases: synchronization, modulation and maintenance. In the
synchronization phase, the device uses electric field to synchronize the
Na
Endothelin-1 is an endogenous 21 amino acid peptide that has powerful vasoactive
properties. In the kidney, endothelin-1 is produced by endothelial, epithelial
and mesangial cells. Endothelin-1 acts through binding to its type A and type B
endothelin receptors to modulate renal blood flow, GFR, reabsorption of sodium
and water, and acid-base balance [212, 213, 214]. Ischemia/reperfusion of the kidney
increases endothelin concentration in the renal cortex and decreases renal blood
flow [215, 216]. Endothelin receptor antagonists such as ambrisentan and bosentan
attenuated ischemia/reperfusion-induced experimental AKI [217, 218]. However, the
reno-protective effect is due to an increase in oxygen supply from increased
renal blood flow rather than a reduction of oxygen consumption [216, 217, 218, 219], because
endothelin-1 actually inhibits Na
The pathophysiology of AKI is complex, involving hypoperfusion, impaired microcirculation, hypoxia, oxidative stress, abnormal coagulation, and inflammation. There is no single approach that can address all aspects of this complicated pathophysiology. Further, addressing one aspect may exacerbate other aspects, even within the concept of reducing oxygen demand. For example, although furosemide reduces oxygen consumption by inhibiting NKCC2 in the thick ascending limb of the Loop of Henle, it also activates the renin-angiotensin-aldosterone system. Combining different treatments that increase oxygen supply and decrease oxygen consumption or different approaches to reduce oxygen consumption may be a better approach. A good example is the synergistic effect observed with using fenoldopam in combination with furosemide in treatment of AKI in critically ill surgical patients [68].
ACEIs, angiotensin converting enzyme inhibitors; AKI, acute kidney injury; ANP,
atrial natriuretic peptide; ARBs, angiotensin receptor blockers; CKD, chronic
kidney disease; NHE3, Na
The content and views expressed in this article are the sole responsibility of the author and do not necessarily reflect the views or policies of the Uniformed Services University or Department of Defense of USA. Mention of trade names, commercial products, or organizations does not imply endorsement by the Uniformed Services University or Department of Defense of USA.
XZ conceived ideas, searched for references, drafted, edited and approved the manuscript.
Not applicable.
The author would like to thank Lieutenant Colonel Ian J. Stewart for his help with editing this manuscript.
This work was supported in part by the United States of America Department of Defense grants W81XWH-22-2-0068 and MED-83-12715.
The author declares no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.