1. Introduction
Mitochondria are multifunctional organelles crucial for cellular energy
production and the formation of reactive oxygen species (ROS) [1]. In addition,
mitochondria are involved in many other processes critical to cell function and
dysfunction, including maintaining metabolic and ionic homeostasis, calcium
signaling, cell differentiation and growth, cell cycle control, and cell death.
Mitochondria generate a significant amount of energy in the form of the
adenosine 5′-triphosphate (ATP) molecule needed to power the cell’s
biochemical reactions [2]. The tricarboxylic acid cycle (the TCA cycle) and
oxidative phosphorylation are the mitochondrial stages of cellular respiration
leading to the oxidation of reduced carbon compounds. The four electron transport
complexes of the respiratory chain (complexes I–IV) and ATP synthase (complex
V), which directly catalyzes the formation of ATP, form the oxidative
phosphorylation system located in the inner mitochondrial membrane (Fig. 1). The
mitochondrial coenzyme Q (mtQ)-reducing pathway of the respiratory chain consists
of complex I (NADH:mtQ oxidoreductase) and complex II (succinate
oxidoreductase:mtQ), which transfer electrons from reduced nucleotides,
nicotinamide adenine dinucleotide (NADH) and dinucleotide adenine (FADH),
respectively. Complex III (mtQH:cytochrome c oxidoreductase,
bc complex), cytochrome c, and finally complex IV
constitute the mtQH-oxidizing pathway in the respiratory chain. Electron
transport in the respiratory chain and ATP production are coupled by a proton
electrochemical gradient formed by proton pumps (complexes I, III and IV) across
the inner mitochondrial membrane. In addition to ATP synthesis by ADP
phosphorylation, the proton electrochemical gradient can drive proton leak
pathways, which do not generate ATP but may protect mitochondria from oxidative
damage by decreasing mitochondrial ROS (mtROS) formation [3]. Moreover, the
protonmotive force is coupled directly to the transport of several metabolites
and calcium ions across the inner mitochondrial membrane and to the nicotinamide
nucleotide transhydrogenase that regulates mitochondrial NADPH levels and
mitochondrial redox balance.
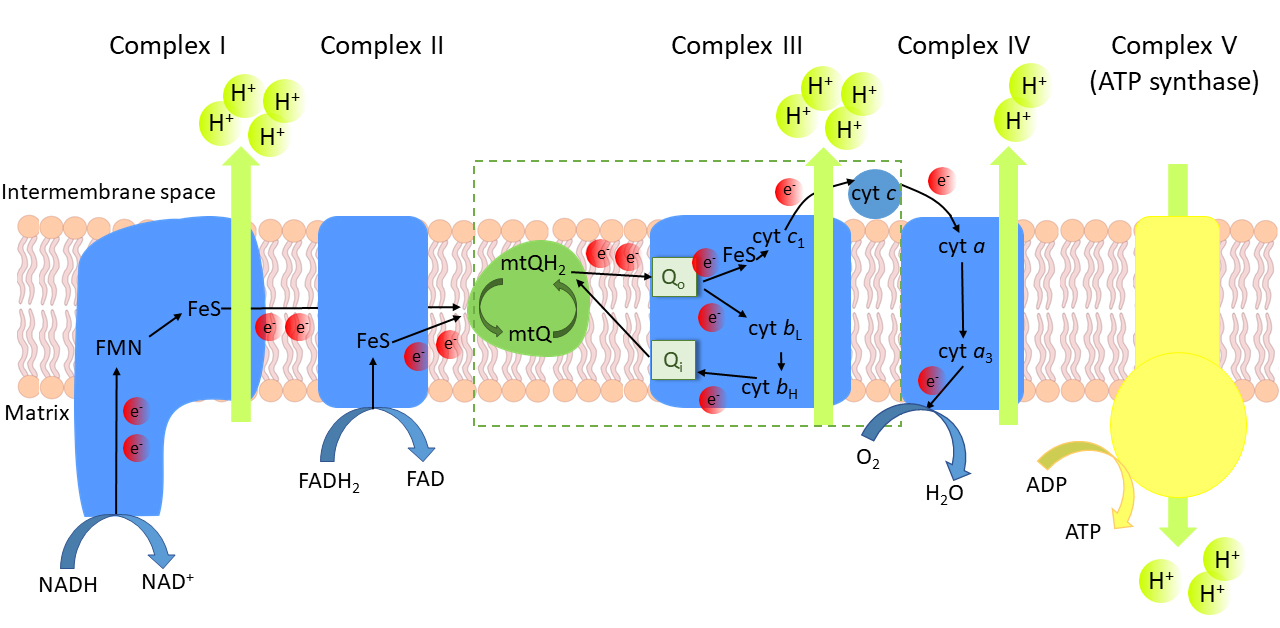 Fig. 1.
Fig. 1.
The oxidative phosphorylation system in the inner
mitochondrial membrane. Electron (e) transfer cofactors:
FMN, flavin mononucleotide; FeS, iron-sulfur cluster; heme b and
heme b of cytochrome b; heme c of
cytochrome c; heme c of cytochrome c; mtQ,
oxidized mitochondrial coenzyme Q; mtQH, reduced mtQ, ubiquinol. Q,
mtQH binding site of complex III; Q, mtQ binding site of complex III.
The mtQ cycle is marked in the box.
1.1 Structure and Redox States of Q
Coenzyme Q (Q, ubiquinone or
2,3-dimethoxy-5-methyl-6-polyprenyl-1,4-benzoquinone) is a ubiquitous fat-soluble
vitamin-like compound distributed in all cell membrane systems, including the
plasma membrane and all intracellular membranes, primarily the inner
mitochondrial membrane [4, 5, 6]. Q consists of a redox active benzoquinone ring
with two methoxy groups, one methyl group, and an isoprenoid side chain with a
species-specific length (6 to 10 units) (Fig. 2). For example, ten isoprenoid
units (Q10) have been described as the most commonly found Q in humans and nine
units (Q9) in rodents (Q6) [7]. Some species have more than one Q isoform, e.g.,
rodent tissues contain different proportions of Q9 and Q10. In cells, Q is
present in three different redox states, i.e., fully oxidized (ubiquinone, Q),
intermediate semioxidized (semiubiquinone radical, QH), and
fully reduced (ubiquinol, QH) (Fig. 2) [8, 9, 10]. Thus, Q has the ability to
accept and transfer one or two electrons, which is crucial for its role as a
one-electron carrier in the mitochondrial electron transport chain because of the
iron-sulfur (FeS) clusters, which can only accept one electron at a time [11],
and for its role as a powerful antioxidant scavenging free radicals [12, 13].
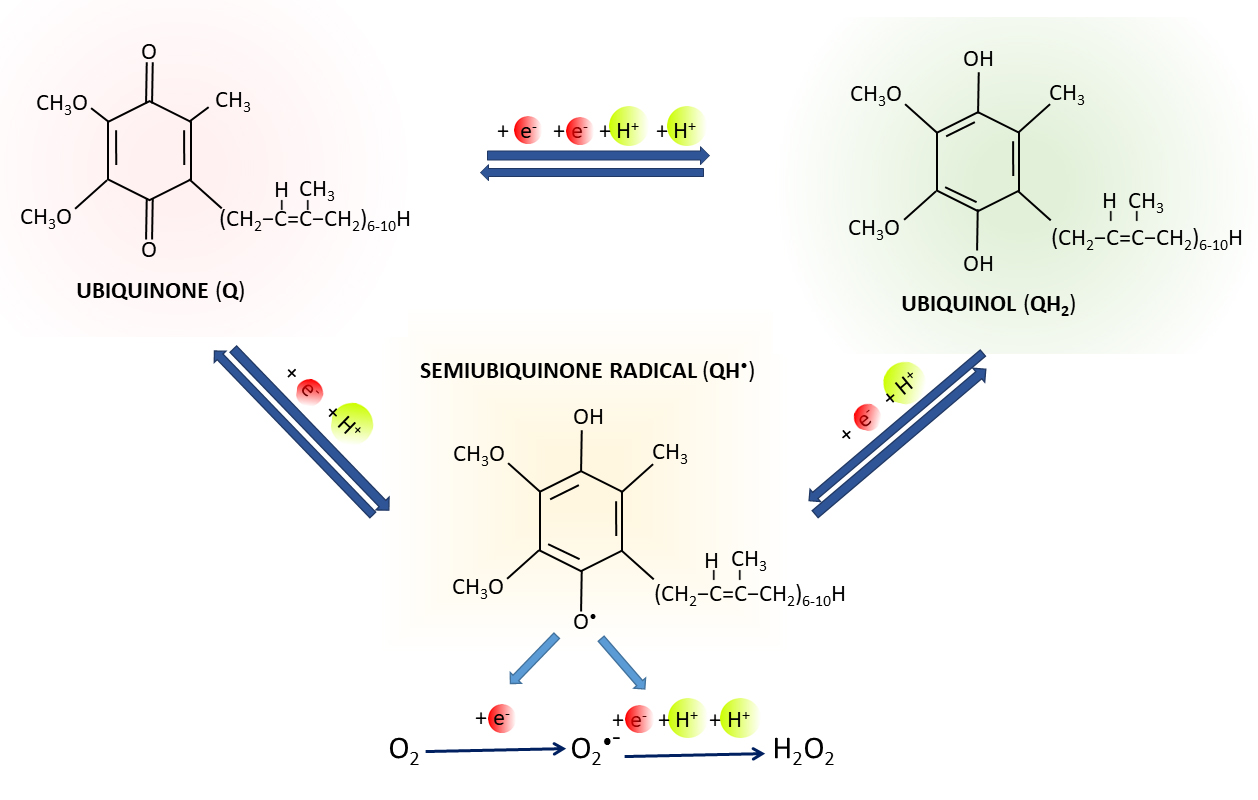 Fig. 2.
Fig. 2.
Q redox states. Oxidized form of Q (ubiquinone) can be reduced
to ubiquinol (QH) by two one-electron reactions through semiubiquinone
(QH) intermediate, or by one reaction of two electrons.
Superoxide (O) and hydrogen peroxide
(HO) can be produced by the leakage of electrons from the redox group
of QH with unpaired electron.
1.2 Q Biosynthesis
Unlike other lipophilic antioxidants such as vitamin A, carotenoids, and vitamin
E, which are usually obtained from dietary sources, Q can also be produced
endogenously [14]. The synthesis of Q takes place in two main steps, one of which
provides the quinone ring, while the other produces the isoprenoid tail (for
reviews see [15, 16, 17, 18, 19, 20]). The 4-benzoquinone ring is synthesized by converting
tyrosine (or phenylalanine) to 4-hydroxybenzoate, while the polyprenyl tail is
provided via the mevalonate pathway, which also synthesizes cholesterol and
dolichols, i.e., large isoprenoid chains comprised of multiple isoprene units.
The synthesis of Q precursors (4-hydroxybenzoate and polyprenylpyro-phosphate)
takes place in the cytosol, and the last stages of Q biosynthesis take place on
the matrix side of the inner mitochondrial membrane [21, 22]. The quinone head is
attached to a polyprenylated tail by a prenyl transferase (COQ2) which, together
with other enzymes, forms a multiprotein Q biosynthetic complex on the matrix
side of the inner mitochondrial membrane [7, 21]. After multiple modifications of
the aromatic ring, including methylation, decarboxylation, hydroxylation and
deamination (catalyzed by COQ3, COQ5, COQ6, and COQ7, respectively), the final Q
molecule is formed. Defects in Q synthesis, including the highly regulated
mitochondrial biosynthetic complex, can lead to a reduction in mtQ levels and
thus to disturbance of mtQ redox homeostasis and oxidative stress [23].
Despite the vast number of publications on both cellular and mitochondrial Q,
its antioxidant and bioenergetic functions, role in health and disease, the
literature is surprisingly limited regarding the relationship between Q redox
state and ROS production in mitochondria under physiological and pathological
conditions. In addition, the role of the mtQ redox state (mQH/mtQ ratio) is
poorly understood because determining its value is technically difficult. In this
review, we focus on factors affecting mtQ redox homeostasis and its relationship
to mtROS production. We first describe the mtQ-reducing and mtQ-oxidizing
pathways of the mitochondrial electron transport chain and the factors
influencing the redox state or reduction level of the mtQ pool. We then discuss
the relationship between mtQ and mtROS formation, with an emphasis on the
mtQ-biding sites of mtROS production, the dependence of mtROS formation on mtQ
reduction level, and the relationship between mtQ content and mtROS production.
Finally, we provided an introduction to the role of reduced mtQ as an
antioxidant. In conclusions, we describe the technical progress in determining
the redox state of Q in cells, tissues and isolated mitochondria.
2. mtQ as an Electron Carrier in the Mitochondrial Respiratory Chain
mtQ forms the mtQ pool in the lipid phase of the inner mitochondrial membrane.
mtQ plays a central role as an electron carrier in the mitochondrial respiratory
chain [24, 25, 26]. Dehydrogenases that oxidize respiratory substrates reduce mtQ
to mtQH, and mQH-oxidizing pathway(s) convert mtQH to mtQ (Fig. 1). mtQ regulates the flow of electrons in the respiratory chain and the building
of a proton electrochemical gradient (including the mitochondrial membrane
potential, mt) across the inner mitochondrial membrane [3, 18, 27, 28]. Therefore, mtQ is the only mobile nonprotein electron carrier in the
respiratory chain involved in the formation of the protonmotive force and,
consequently, the oxidative phosphorylation process leading to ATP synthesis.
2.1 mtQ-Reducing Pathways
mtQ is reduced to mtQH by several dehydrogenases that oxidize reducing
equivalents derived from various food fuels, i.e., glucose, fats and amino acids
[29]. Thus, mtQ is a common reservoir of electrons that contribute to oxidative
oxidative phosphorylation, regardless of source.
Complex I and complex II of the mitochondrial respiratory chain reduce mtQ pool
with electrons from the reduced nucleotides (NADH and FADH, respectively)
derived mainly from the TCA cycle and thus from the oxidation of major food
molecules. In the rotenone-sensitive complex I, two electrons are transferred
from NADH to the flavin mononucleotide (FMN) and then, through the iron-sulfur
centers to mtQ, which is reduced to mtQH [30, 31, 32]. The malonate-sensitive
complex II is an enzyme involved in the TCA cycle and oxidizes succinate to
fumarate. Complex II transfers two electrons from FADH through the
iron-sulfur centers to mtQ, thereby also introducing two electrons into the
mitochondrial respiratory chain [33, 34, 35].
In addition to the basic dehydrogenase complexes of the respiratory chain, i.e.,
complex I and complex II, mtQ receives electrons from a number of electron
transporting flavoproteins that supply electrons to the mtQ pool [36, 37, 38]. mtQ is
reduced by electron transfer flavoprotein (ETF):mtQ oxidoreductase (ETF:mtQ
oxidoreductase) that oxidizes FADH derived from -oxidation of
fatty acids in the mitochondrial matrix [39, 40]. Electrons are also delivered to
the mtQ pool from the oxidation of dihydroorotate to orotate by dihydroorotate
dehydrogenase, a key mitochondrial enzyme in pyrimidine nucleotide biosynthesis
[41, 42]. Another mtQ-reducing dehydrogenase located on the outer surface of the
inner mitochondrial membrane is FAD-linked glycerol-3-phosphate dehydrogenase,
which catalyzes the conversion of glycerol-3-phosphate to dihydroxyacetone [43, 44]. The oxidation of glycerol-3-phosphate by mitochondrial glycerol 3-phosphate
dehydrogenase is a major pathway for the transfer of cytosolic reducing
equivalents from lipid and carbohydrate metabolism to the mitochondrial electron
transport chain. In addition, mtQ accepts electrons from FAD-linked proline
dehydrogenase, which is involved in the metabolism of amino acids [45, 46]. In
the branched mitochondrial respiratory chain of plants, fungi, and some protists,
mtQ receives electrons from alternative rotenone-resistant NAD(P)H dehydrogenases
located on the outer and inner surfaces of the inner mitochondrial membrane [47, 48]. Among the mtQ-reducing dehydrogenases, only complex I of mitochondrial
respiratory chain pumps protons and therefore participates in the generation of
the proton electrochemical gradient.
2.2 mtQH-Oxidizing Pathway(s)
mtQ metabolism depends on mtQH reoxidation by the mitochondrial
mtQH-oxidizing pathway(s). In mammalian mitochondria, the cytochrome
pathway (complex III, cytochrome c, and finally complex IV) is the only
mtQH-oxidizing pathway in the respiratory chain [2, 27]. The antimycin A-
and myxothiazol-sensitive complex III and the cyanide-sensitive complex IV pump
protons through the inner mitochondrial membrane to the intermembrane space and
therefore participates in the generation of the protonmotive force. In the
branched mitochondrial respiratory chain of plants, fungi, and some protists,
there is an additional antimycin A- and cyanide-resistant alternative quinol
oxidase (AOX) that oxidizes mtQ, bypassing the cytochrome pathway [49, 50].
Transfer of electrons in AOX is not accompanied by proton pumping and thus
mitochondrial ATP production.
Complex III (mtQH:cytochrome c oxidoreductase, cytochrome
bc complex) of the cytochrome pathway oxidizes the mtQH pool
directly, catalyzing a series of electron transfer reactions from mtQH to
cytochrome c, called the mtQ cycle (Fig. 1, marked box) [51, 52, 53]. Thus,
complex III connects two mobile electron carriers of the respiratory chain, mtQ
and cytochrome c. The reducing force of the mtQH pool and the
oxidizing force of the cytochrome c pool influence the redox state of
the four metal redox centers in complex III: hemes b and
b in cytochrome b, and heme c and the
Rieske FeS center in cytochrome c. mtQ reduction and mtQH
oxidation reactions take place at two separate catalytic centers. The
myxothiazol-sensitive Q site is the site of binding and oxidation of
QH located toward the intermembrane space, while the antimycin A-sensitive
Q site is the site of binding and reduction of mtQ located on the matrix
side.
The mtQ cycle of complex III involves all three forms of Q (fully oxidized,
semioxidized, and fully reduced) (Fig. 3) and participates in the generation of
the protonmotive force as during mtQH oxidation protons are released into
intermembrane space [51, 52, 53, 54]. The mtQ cycle involves a series of one-electron
redox reactions as a result of bifurcation of electrons at the Q site (Fig. 1, marked box). At this site, one electron from mtQH is transferred to the
Rieske protein, then to cytochrome c, and finally to cytochrome
c; the second electron is translocated through heme b and
heme b to the catalytic site Q, where mtQ is reduced to a
semiubiquinone radical (QH) [51, 52, 53, 54, 55]. The oxidation of two
mtQH molecules involves the reduction of two cytochrome c
molecules and completes the reduction of semiubiquinone to mtQH at the
Q site. Thus, a complete enzymatic cycle of complex III gives the oxidation
of one mtQH with the reduction of two molecules of cytochrome c and the return of half of the electrons within the mtQ pool.
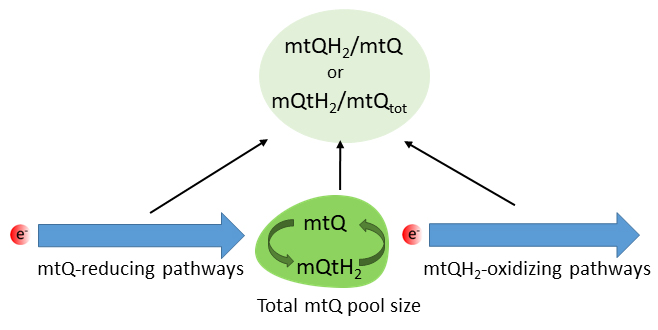 Fig. 3.
Fig. 3.
Factors influencing the mtQ reduction level. mtQ,
oxidized mitochondrial coenzyme Q; mtQH, reduced mtQ, quinol; mtQ,
total mtQ (mtQ + mtQH) in the inner mitochondrial membrane.
Although one of the main functions of the mtQ-reducing and mtQH-oxidizing
pathways is to maintain the mtQ redox homeostasis, there are few studies
describing a direct relationship between their activity and the level of mtQ
reduction (or mtQ redox state) and mtROS production under physiological and
pathological conditions.
3. Redox State or Reduction Level of the mtQ Pool
mtQ levels, its redox state and its interaction with mtQ-dependent enzymes may
play a key role in modulating mitochondrial homeostasis. One of the main
properties of the mtQ pool in the inner membrane is its redox state or mtQ
reduction level, defined as the mtQH/Q ratio or the ratio of mtQH to
the total mtQ content in the membrane (mtQH/mtQ ratio),
respectively. The redox state of mtQ depends on the several factors, including
the activity of mtQ-reducing pathways (respiratory substrate-oxidizing
dehydrogenases), the activity of mtQH-oxidizing pathway(s) (the cytochrome
pathway and AOX), and the size of the mtQ pool of the inner mitochondrial
membrane (Fig. 3). The mtQH/mtQ ratio is sensitive to an imbalance between
the supply of electrons from reducing equivalents and the need for ATP. It
changes rapidly and reversibly in response to changes in the function of
mitochondria. Interestingly, it has recently been proposed that succinate evolved
as a signaling modality because its concentration reflects the mtQ redox state
[56]. By balancing with the mtQ pool, succinate enables the transfer of the mtQ
redox state from the mitochondria to the rest of the cell, to the circulation and
other cells.
The redox state of the mtQ pool reflects the bioenergetic mitochondrial activity
of cells and is considered to be one of the markers of oxidative stress [29, 52, 57, 58, 59, 60, 61]. It represents the level of mtROS formation, and thus mitochondrial
oxidative stress, and the need for protective antioxidants in mitochondria. It
should be noted that mitochondrial reactive nitrogen species are also key
contributors to oxidative stress [62, 63]. Nitric oxide (NO) can react with
ubiquinol (QH) to form nitric oxide radical (NO), which
can react with O to form peroxynitrite
(ONOO), and semiubiquinone
(QH), which can react with O to form superoxide
(O) [64].
The size of the mtQ pool together with cellular Q may be modified by various
factors, including endurance training [65, 66, 67], statin-induced mtQ synthesis
impairment [68], cell age and oxygen level [69, 70, 71] or certain diseases,
including cardiovascular and neurodegenerative disease or cancer [22, 60, 72, 73]. However, the size of the mtQ pool is rarely determined. Similarly,
alteration in the redox state of the mtQ pool has so far been determined for only
a few physiological and pathophysiological conditions, including endurance
training [65, 66, 67] and statin treatment [68]. Interestingly, the size of the
reduced nonoxidizable pools of mtQ may reflect the cell’s need for Q as an
antioxidant [74]. Namely, we have recently shown that in the tissues of rats with
different energy requirements (heart, brain, liver and lungs), the production of
ROS at the tissue and mitochondrial levels was strongly related to the available
reduced nonoxidizable Q and mtQ pools, i.e., the cellular QH and mtQH
content measured in the absence or depletion of oxidative metabolism substrates
[74].
The mtQ reduction level adapts dynamically to variable metabolic conditions in
the cell. Therefore, another possible function of the mtQ reduction level is
related to the action of mitochondrial uncoupling proteins (UCPs), which mediate
regulated proton leakage across the inner mitochondrial membrane, leading to the
dissipation of energy stored in the proton electrochemical gradient [75, 76].
First, it was observed that mtQ is an obligatory cofactor for UCP function [77],
and the regulation of uncoupling activity by mtQ depends on its reduction level
[78]. Moreover, we have reported that mtQH but not mtQ acts as a negative
modulator for the UCP inhibition by purine nucleotides [79].
4. mtQ and mtROS Formation
Mitochondria are an important source of ROS (mtROS) in eukaryotic cells.
Superoxide (O) and hydrogen peroxide
(HO) are produced by the leakage of electrons from the redox centers
of the mitochondrial respiratory chain and substrate oxidation-associated
enzymes, leading to a one-electron or two-electron reduction of oxygen [36, 80, 81]. mtQ participates in the generation of
O from semiubiquinone radicals by the
leakage of electrons from its redox group with unpaired electron (Fig. 2).
mtROS are formed as a by-product of normal aerobic metabolism or under
conditions of metabolic stress leading to oxidative stress. Under physiological
conditions, mtROS can act as beneficial signaling molecules sent from the
mitochondria to other parts of the cell that is essential for cellular adaptive
responses [36, 82, 83, 84]. Dysfunction of the mitochondrial respiratory chain leads
to decreased energy production and mtROS overproduction. Under oxidative stress
conditions, excessive mtROS production can lead to a range of oxidative damage,
including oxidative modification of cellular macromolecules such as lipids, DNA
and proteins, which is the cause of aging and many diseases. In addition, damaged
mitochondria release apoptotic factors that act as signals inducing cell death.
In mammalian mitochondria, at least eleven sites can generate mtROS
(O and/or HO) in the
electron transport chain (seven sites) and related enzymes (four sites) involved
in the oxidation of the reduced substrates, including the TCA cycle and
-fatty acid oxidation enzymes (Fig. 4) [80, 85]. The sites of
O/HO production are present
in respiratory chain complexes (complexes I, II and III) and dehydrogenases
(mitochondrial glycerol-3-phosphate dehydrogenase, dihydroorotate dehydrogenase,
and ETF:mtQ oxidoreductase), the redox cofactor of which is mtQ. mtQ is directly
involved in the formation of
O/HO at four mtQ-binding
sites, i.e., the mtQ-reducing sites of complex I (I) [86], mitochondrial
glycerol-3-phosphate dehydrogenase (G) [87], dihydroorotate dehydrogenase
(D) [88], and the mtQH-oxidizing site of complex III (III)
[89]. Other dehydrogenases linked to mtQ, such as proline dehydrogenase, feed
electrons into the mtQ pool and then reduce other sites that produce
O/HO [90]. However, it is
possible that these dehydrogenases also produce
O/HO at mtQ-reducing sites,
but this production is too small to be detected [80]. Changes in the bioenergetic
states of mitochondria, as a consequence of alterations of mitochondrial
substrate availability and the reduction states of mitochondrial redox centers,
including mtQ, affect the rate of
O/HO production in various
mitochondrial sites [80, 85]. There are seven characterized
O/HO production sites that
are not directly related to mtQ reduction but contain redox-active flavin (FMN or
FAD): I of complex I [86], II of complex II [91], E of
ETF/ETF:mtQ system [92], and O, B, A, and P sites of
2-oxoacid dehydrogenase complexes, i.e., 2-oxoglutarate dehydrogenase (the TCA
cycle enzyme), branched-chain 2-oxoacid dehydrogenase, 2-oxoadipate
dehydrogenase, and pyruvate dehydrogenase complexes, respectively [93]. The
2-oxoacid dehydrogenase complexes contain a dihydrolipoamide dehydrogenase
subunit with redox-active FAD, which acts as an
O/HO production site. They
feed electrons to NAD. Other NAD-linked dehydrogenases, such as malate
dehydrogenase, isocitrate dehydrogenase (TCA cycle enzymes), and glutamate
dehydrogenase reduce other sites that produce
O/HO [80]. Electrons from
NADH formed in all NAD-linked dehydrogenases are transferred to the flavin (FMN)
in the NADH-oxidizing site of complex I (I) and then to the mtQ-reducing
site of complex I (I).
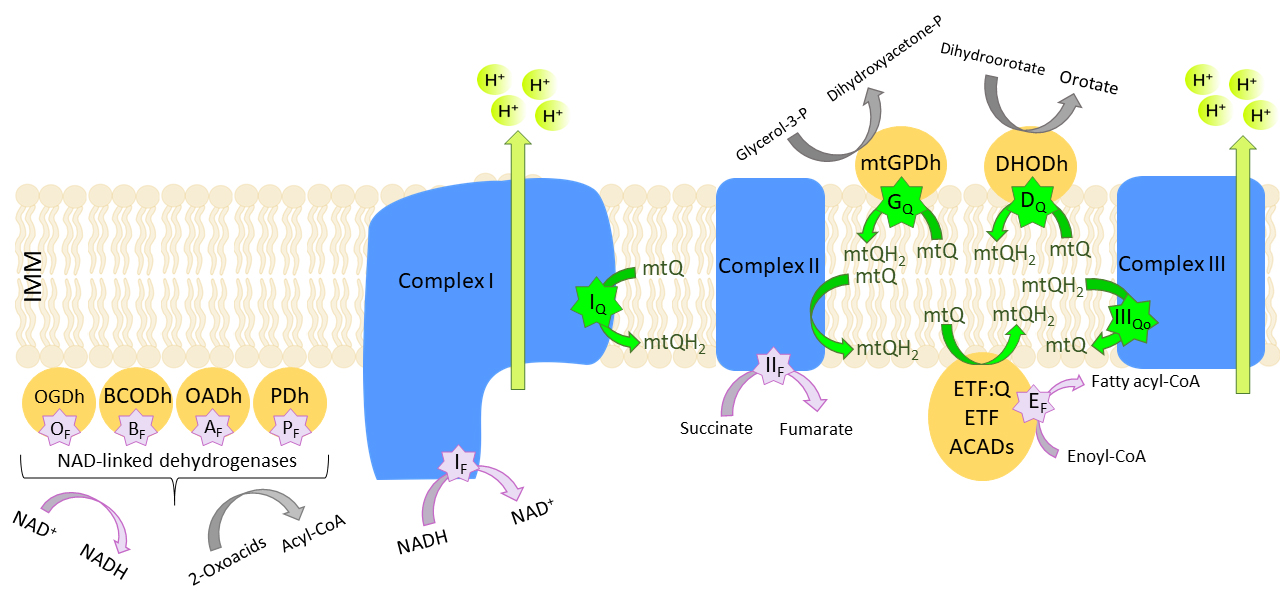 Fig. 4.
Fig. 4.
Eleven mitochondrial sites of
O/HO formation. mtQ,
ubiquinone; mtQH, ubiquinol; ETF, electron transfer flavoprotein; ETF:Q,
ETF:Q oxidoreductase; ACADS, acetyl-CoA dehydrogenase; IMM, inner mitochondrial
membrane. Flavin-dependent (F, purple stars) or mtQ-binding sites (Q, green
stars) of the 2-oxoglutarate dehydrogenase (OGDh) (O), branched chain
2-oxoacid dehydrogenase (BCODh) (B), 2-oxoadipate dehydrogenase (OADh)
(A), and pyruvate dehydrogenase (PDh) (P) complexes, of complex I
(I, I), complex II (II), the outer mtQ site of complex III
(III), mitochondrial glycerol-3-phosphate dehydrogenase (mtGPDh)
(G), the ETF/ETF:mtQ oxidoreductase system (ETF/ETF:mtQ) (E), and
dihydroorotate dehydrogenase (DHODh) (D). Based on [85].
Site III of complex III and site G of glycerol-3-phosphate
dehydrogenase generate O in both the
mitochondrial matrix and intermembrane space [80, 87, 89]. Other mtROS formation
sites produce O/HO in the
mitochondrial matrix. These different
O/HO formation topologies
are important for intracellular signaling using mtROS as signaling molecules.
4.1 mtQ-Biding Sites of mtROS Production
In the inner mitochondrial membrane, mtQ is a molecule directly involved in the
production of mtROS due to the formation of
O/HO by the leakage of
electrons from the semiubiquinone radical. All four mtQ-binding sites
(I, III, G, D) produce
O and only one of them (D) is
likely to produce HO as well [80, 85].
At I of complex I, O is mainly
generated during reverse electron transport (RET), i.e., a transfer of electrons
against the redox potential gradient supported by a high proton electrochemical
gradient (a high protonmotive force) across the inner mitochondrial membrane and
a high mtQ redox state (highly reduced mtQ pool) [86]. Under these conditions,
mtQH is oxidized to mtQ at the mtQ-binding site (I) to drive the
reduction of NAD to NADH at the FMN site of complex I (I). Inhibition
of mtQ-binding with rotenone inhibits RET and lowers mtROS production.
At the outer mtQ-binding site of complex III (III),
O production is low in the absence of
Q site inhibitors [94]. In the presence of antimycin A, which binds to the
Q site and blocks the oxidation of cytochrome b heme, the
mtQ cycle is disrupted. The resulting accumulation of reduced cytochrome
b heme limits semiubiquinone oxidation at site Q.
Semiubiquinone interacts with oxygen to form
O on both sides of the inner
mitochondrial membrane. In the absence of antimycin A, a high protonmotive force
and highly reduced mtQ pool decreases the electron transfer from the cytochrome
b heme to the cytochrome b heme, leading to more
reduced b heme and thus increased
O production [95].
Site G of mitochondrial glycerol-3-phosphate dehydrogenase produces
O in approximately equal amounts on
each side of the mitochondrial inner membrane, suggesting that the mtQ-binding
site of the enzyme is the major site of
O formation during glycerol-3-phosphate
oxidation [87]. Dihydroorotate dehydrogenase can generate
O and/or HO directly at its
mtQ-binding site (D) at low rates [88]. The enzyme is also capable of
indirect production at higher rates from other sites through its ability to
reduce the mtQ pool.
In a cell, the net O/HOformation rate at mitochondrial mtQ-binding sites depends on the substrates
being oxidized and the antioxidant systems acting on both sides of the inner
mitochondrial membrane. Among the mtQ-binding sites, III and I,
which is mainly active during RET, dominate in
O production and play an important role
in redox signaling and the induction of oxidative damage [80].
4.2 mtQ Reduction Level and mtROS Formation
As a mobile component of the mitochondrial electron transport chain, mtQ acts as
a pro-oxidant in the semiubiquinone state. It is generally accepted that the
production of mtROS depends on the level of reduction of the mtQ pool, which
determines the formation of O from
semiubiquinone. However, there are only a few data relating the mtQ redox state,
i.e., mtQH/Q ratio [96] and mtQH/mtQ ratio [59, 66, 68, 95], with mtROS generation. The formation of mtROS related to the activity of the
respiratory chain, and thus the mtQ redox state, depends on the activity of all
mtQ-reducing dehydrogenases and mtQH-oxidazing pathway(s), and on the
activity of the oxidative phosphorylation process in which ATP is formed [59]. We
have shown that under ADP phosphorylating conditions (state 3), a low mtQ
reduction level and a low m are accompanied by decreased mtROS
production [59]. In a nonphosphorylating state (state 4), a highly reduced mtQ
pool (a high mtQ reduction level) and a high m are accompanied
by increased mtROS production. Thus, the mtQH/mtQ ratio is sensitive to
changes in electron supply from reducing equivalents and to ATP demand. The
activity of uncoupling proteins (UCPs), which mediate proton leakage, can lower
m and the mtQ reduction level, leading to a decrease in mtROS
production [76, 84].
The relationship between mtROS production and mt was
investigated more often than the interplay between mtROS formation and the mtQ
reduction level. For the first time, the dependence of mtROS formation on
mt was described in rat heart mitochondria [97]. Namely, a very
steep dependence of HO formation on mt was observed
in nonphosphorylating rat heart mitochondria after exceeding a threshold value
near mt of ADP phosphorylating mitochondria. However, not under
all conditions lowering mt leads to a decrease in mtROS
production (e.g., in the presence of inhibitors of complexes III and IV),
indicating that mtROS is not a direct function of the mt level
(Fig. 5A) [59]. For example, we have shown that in the mitochondria of the amoeba
Acanthamoeba castellanii, with inhibition of the Q site of complex
III by antimycin A or complex IV by cyanide, m decreases while
the mtQ reduction and HO production increase [59]. Marphy’s group has
shown that the formation of O at the
Q site is associated with the accumulation of reduced cytochrome
b when the Q site is blocked by antimycin A, and this
phenomenon is not dependent on mt [95].
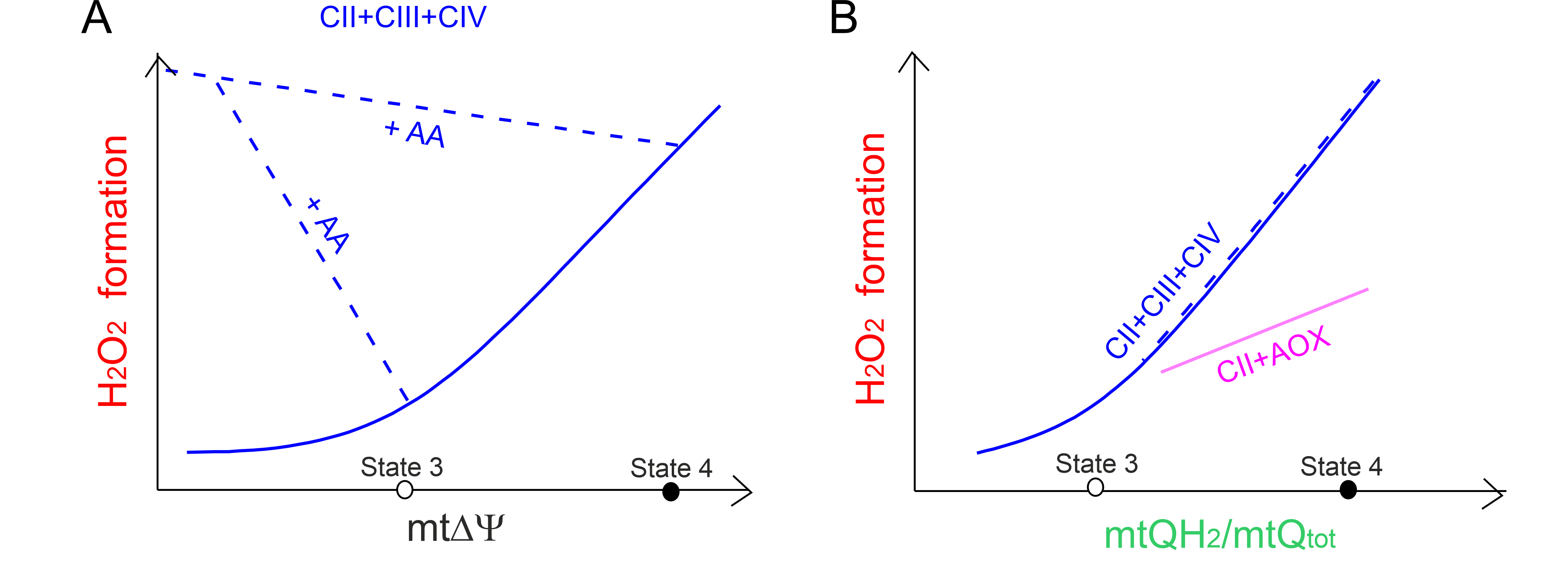 Fig. 5.
Fig. 5.
The relationship between mtROS formation versus
mt and the mtQ reduction level. The relationship between
HO formation versus mt (A) for the cytochrome
pathway (CII + CIII + CIV) fueled by complex II (CII) and the relationship
between HO formation versus the mtQ reduction level
(mtQH/mtQ) (B) for the CII + CIII + CIV pathway (blue curves and
lines) and the CII-fueled alternative oxidase (AOX) (CII + AOX) (purple line).
Based on results described in A. castellanii mitochondria [59]. The
mt level (A) and mtQ reduction level (B) of the CII + CIII +
CIV pathway was varied with the ATP synthesis and CII inhibitors, with the
uncoupler (blue solid curves), and with the inhibitor of CIII (antimycin A, AA)
(blue dashed lines) when AOX inhibitor was present. Measurements were carried out
under nonphosphorylating (state 4) and phosphorylating (state 3) conditions. (B)
For the CII + AOX pathway, the mtQ reduction level was titrated with an activator
of AOX (GMP) and an inhibitor of CII (malonate) in the presence of AA to block
the cytochrome pathway.
We have kinetically described a direct dependence of mtROS production on the mtQ
reduction level in A. castellanii mitochondria [59]. For both
mtQH-oxidizing pathways (the cytochrome and AOX pathways), during the
oxidation of succinate fueling the respiratory chain at the level of complex II,
a higher level of mtQ reduction leads to greater mtROS formation (Fig. 5B) [59].
This relationship is also observed for the cytochrome pathway in the presence of
antimycin A, which by inhibiting the Q site lowers m and
increases mtQ reduction level. In the case of the cytochrome pathway,
HO generation depends nonlinearly on the reduction level of mtQ pool.
At levels of mtQ reduction higher than that of the phosphorylating state (above
35%) there is a steep relationship between HO generation and the mtQ
reduction level. In contrast, AOX is active only when the mtQ reduction level is
high (above 40%) and the dependence between HO generation and mtQ
reduction level is approximately linear (Fig. 5B). We have proposed that the mtQ
pool reduction level (the endogenous redox state of mtQ) may be a useful
endogenous marker that allows for the assessment of total mitochondrial mtROS
production [59].
High levels of mtROS are generated in response to stress via RET through complex
I [61, 98]. RET occurs when the mtQ pool it is strongly reduced by electrons from
the respiratory complex II (succinate dehydrogenase) [95, 98]. In various cell
lines, it has been observed how the respiratory chain is optimized for better
oxidation of various reducing respiratory fuels by the redox state of mtQ
(mtQH/Q ratio) acting as a metabolic detector and mtROS formed by complex I
(via RET) acting as an executor [96]. In the rat heart mitochondria, the
formation of mtROS by RET at complex I has been shown to be dependent on the mtQ
reduction level and mt that indicates the sensitivity of
O formation by the mtQ-binding site of
complex I to these two metabolic variables [95].
4.3 mtQ Content and mtROS Production
It seems that the production of mtROS does not directly depend on the amount of
mtQ but rather on the reduction level of the mtQ pool (mtQH/mtQ
ratio) as a function of the activity of mtQ-reducing pathways (mtQ-reducing
dehydrogenases) and the mtQH-oxidizing cytochrome pathway(s). In the heart
mitochondria of MCLK1 (CoQ7) mutant mice, which contain only
~10% of normal Q content, lower rates of mtROS production from
all known mtROS production sites were detected at respiration levels similar to
those in control mitochondria [99]. One possible explanation for how low mtQ
levels reduce mtROS formation is that low mtQ induces the formation of
supercomplexes that have been proposed to increase electron transport efficiency
from one redox component to another, thus minimizing electron leakage and mtROS
production [100, 101]. In cultured fibroblasts from Q-deficient patients,
increased mtROS production was observed during intermediate Q deficiency
(50–70%) possibly as a result of increased semiubiquinone generation from
increased redox cycling of the restricted mtQ pool, while a more severe loss of Q
(85%) was not accompanied by significant mtROS production [102].
Interestingly, we have shown that in lung mitochondria of endurance-trained rats,
reduced mtQ content (mtQ9 by ~16% and mtQ10 by
~42%) and decreased activity of complex I as well as increased
activity of cytochrome pathway (complex III plus complex IV) may account for the
observed diminished mtQ reduction level during succinate and/or malate oxidation,
resulting in a general decrease of mtROS formation by mitochondria [66].
Moreover, endurance training downregulates complex I in supercomplexes and
upregulates complex III in the CIII + CIV supercomplex in the inner
membrane of lung mitochondria. In addition, comparison of the effects of
endurance training on mtQ amount and mtROS generation in rat tissues with high
energy demand, i.e., in the brain, liver and heart, indicates that endurance
training may induce various mitochondrial and tissue responses related to mtQ
acting as an electron carrier in the respiratory chain and as an antioxidant
[67]. Changes in the formation of mtROS observed in the mitochondria of
individual rat tissues may result from changes in the size of the pool of
oxidized mtQ, which acts as an electron carrier, as well as the amount and
activity of individual complexes of the oxidative phosphorylation system and its
molecular organization.
A decrease in the mtQ pool size and mtQ reduction level may results from drug
treatment. We have shown that in cultured endothelial cells, chronic exposure to
atorvastatin, an inhibitor of HMG-CoA reductase led to a significant decrease in
mtQ10 content (~23%) [68]. The activity of complex III and its
amount in a supercomplex with complex IV was also diminished. The statin-induced
changes at the respiratory chain level of the endothelial mitochondria led to a
decrease in succinate and/or malate oxidation accompanied by a decreased
mt, as well as to an increase in the mtQ10 reduction level and
consequently increased mtROS production.
Thus, mtQ redox homeostasis is a key factor in modulating mtROS production. As
mentioned above, depending on the activity of mtQ-reducing pathways and
mtQH-oxidizing pathways, a decreased amount of mtQ can lead to either an
increased reduction level of mtQ (a higher mtQH/mtQ ratio) and
thus to an increased production of mtROS or to a decreased reduction level of mtQ
(a lower mtQH/mtQ ratio) and hence decreased production of mtROS.
Superphysiological mtQ levels could possibly have the same effect by altering the
level of mtQ reduction. Thus, changing the size of the mtQ pool and its redox
state is an important physiological adaptation. The metabolic response to the
altered mtQ pool appears to depend on whether it is more important to maintain an
efficient oxidative phosphorylation process or whether it is more important to
defend against oxidative stress.
The level of Q, including mtQ, varies with the type of membrane, tissue and
organism [4, 103]. It is highest in organs with a high metabolic rate and hence a
high energy requirement, such as the heart, muscles, and liver. However, despite
high metabolic activity, total Q levels and the QH/Q ratio in the human
brain are very low compared to other tissues [4]. The mtQ pool is in excess
compared to other components of the respiratory chain [100]. However, with age
and in many disease states, including primary and secondary Q deficiencies,
associated with increased production and action of mtROS, the level of cellular
and thus mitochondrial Q decreases [23, 60, 71, 72, 104]. mtQ deficiency may
impair oxidative phosphorylation and therefore respiratory chain activity and ATP
synthesis [105, 106, 107, 108], and may induce oxidative stress (mtROS overproduction)
leading to oxidative modification of macromolecules, and ultimately to apoptosis.
However, the question of how Q deficiency due to disease or aging affect the
redox state of mtQ has not yet been thoroughly investigated.
Increasing mtQ levels must also result in a change in mtQ redox homeostasis and
mtROS production. For example, we have observed an increase in mtQ9 content (by
~20%) in skeletal muscle mitochondria after eight-week endurance
training [54]. In these mitochondria, HO production and mtQ reduction
level were elevated under nonphosphorylating conditions, and decreased under
phosphorylating conditions, while the efficiency of oxidative phosphorylation
increased with unchanged components of the respiratory chain. Recently, it has
been shown that elevated levels of mtQ caused by Q10 delivery (via by BPM31510)
lead to an increase in ROS generation and consequently, an imbalance in cellular
redox homeostasis that activates mtROS-mediated cell death pathways in pancreatic
cancer cells [109]. Similarly, cell death activation due to increased mtQ pool
was observed in vitro in pancreatic cancer cells, and both human
patient-derived organoids and tumor xenografts. Interestingly, in other studies,
chemotherapy induced increased production of ROS and increased levels of Q10
(especially its reduced form Q10H) in all cancer cell lines tested [110].
But again, we do not know how mtQ redox state changes under these conditions and
what role it plays in mtROS production.
5. mtQ as an Antioxidant
The redox state of the mtQ pool is an important indicator of the bioenergetic
and antioxidant status of the mitochondria. The fully reduced form of mtQ
(mtQH) can act as a nonprotein lipophilic antioxidant [111, 112]. The level
of mtROS depends not only on their production in the mitochondria and thus on the
amount of mtQ and its redox state in the inner mitochondrial membrane, but also
on the mtROS scavenging by reduced forms of mtQ. Additionally, by acting as an
antioxidant, mtQ protects against oxidative damage caused by mtROS. mtQH
regenerates other antioxidants (vitamin C or vitamin E) as well as reduces and
neutralizes oxidants or free radicals, including mtROS [11, 22, 112]. As an
effective free radical scavenger, mtQH prevents oxidative modifications of
mitochondrial proteins and DNA and inhibits the initiation and propagation of
membrane lipid peroxidation. In lipid peroxidation reactions, one-electron
oxidation of mtQH to semiubiquinone is linked to the reduction of perferryl
radicals or lipid peroxyl radicals [12]. Reactive semiubiquinone radicals are
neutralized by reduction to QH by -tocopherol (vitamin E).
However, mtQH is still a capable antioxidant molecule when vitamin E is
absent [113]. On the other hand, the indirect antioxidant effect of mtQH
can regenerate -tocopherol from -tocopheroxyl radicals
(-TO) [114]. Interestingly, in addition to the plasma
membrane and intracellular membranes, Q is also a component of low-density
proteins (LDL), suggesting its potential antioxidative role along with
-tocopherol in the prevention of atherosclerosis [115].
Thus, as a molecule with antioxidant and prooxidative properties, mtQ
contributes both to oxidative damage to mitochondria and to their antioxidant
defense. The antioxidant action of mtQH is associated with deprotonation
(and electron donation), leading to the formation of semiubiquinone radicals and
ultimately oxidized mtQ, which is then reduced back to mtQH by the
mitochondrial respiratory chain [112, 116].
It is widely believed that the antioxidant properties of Q may have beneficial
effects on health and length of life [117]. Q has been widely introduced as an
antiaging supplement as well as a potential treatment for several human diseases
related to mitochondrial dysfunction-induced oxidative stress. However,
understanding the complex effect of Q on the human body and its relationship to
lifespan is still incomplete in view of inconsistent literature reports on Q
supplementation, describing its beneficial or nil effects [23, 29, 60, 71, 73].
Paradoxically, some Q biosynthesis defects result in an increase in life span in
animal models, most likely due to a mild reduction in respiratory chain activity
and thus a reduction in mtROS production or due to a physiological adaptation to
early mitochondrial dysfunction [118]. On the other hand, Q10 supplementation may
have positive effects in the treatment of certain cancers in patients with
reduced plasma and tissue Q levels [109]. Similarly, Q10 supplementation may have
beneficial effects on aging-related disorders accompanied by a reduced Q content,
especially in cardiovascular and metabolic diseases [119, 120, 121, 122]. However, effect
of Q10 supplementation on reversing functional decline of mitochondrial
bioenergetics is unclear. Moreover, it is unclear if this improvement is always
related to the increase in mtQ levels and the change in mtQ redox state after Q10
supplementation. Supplementation with high doses of Q10 may increase both
circulating and intracellular levels, but there are conflicting results regarding
bioavailability [71, 123, 124]. Although it has been shown that endogenous levels
of mtQ in the mitochondria of the rodent brain, skeletal muscle, and heart can be
increased after Q10 administration [125, 126].
Nonoxidizable mtQH Pool as an Mitochondrial Antioxidant
We have proposed that the antioxidant effect may also be exerted by the reduced
mtQH pool in the inner mitochondrial membrane, which is not oxidized by the
respiratory chain [74]. Interestingly, in mitochondria isolated from different
rat tissues with different energy requirements and different mitochondrial
oxidative activity heart, brain, liver and lungs, the size of the mtQH pool
that was not oxidized by the respiratory chain (measured in the absence of
respiratory substrates) was associated with the mtROS production capacity during
the oxidation of respiratory substrates [74]. Thus, the size of the reduced
nonoxidizable pools of mtQ (nonoxidizable mtQH pool) may reflect the need
for mtQ as a mitochondrial antioxidant. Therefore, it is possible that both the
mtQH pool, which participates in the mtQ cycle of the electron transport
chain, and the mtQH pool, which is not oxidized by the chain, contribute to
the overall antioxidative activity of mitochondria.
6. Conclusions and Perspectives
As a redox active cofactor of mitochondrial respiratory chain complexes and
related enzymes, mtQ influences mtROS formation and contributes to oxidative
damage to mitochondria. On the other hand, thanks to its redox properties, mtQ
acts as an antioxidant that protects the mitochondria from oxidative
modifications. Mitochondria contain the most Q in a cell but are also the most
dependent on its content. A number of physiological and pathophysiological
factors affect the amount of mtQ and thus its redox state and mtROS production
level (Fig. 6), indicating that mtQ homeostasis is essential for metabolic
adaptation and the maintenance of mitochondrial function.
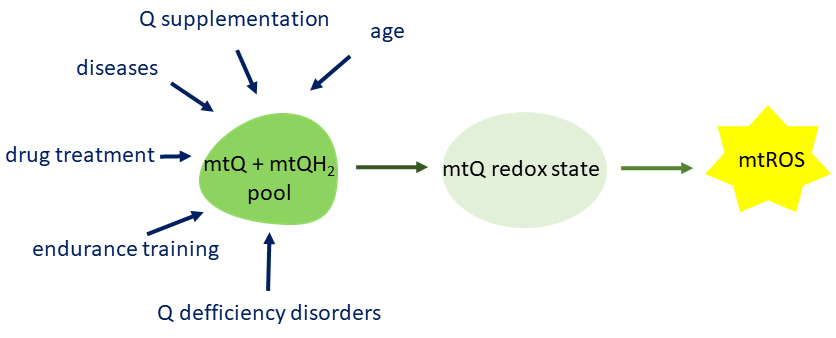 Fig. 6.
Fig. 6.
Factors modulating the mtQ pool content which influences the
redox state of mtQ and thus the formation of mtROS. mtQ, mitochondrial coenzyme
Q; mtROS, mitochondrial reactive oxygen species.
The purpose of our review was to highlight the rather little-studied and
discussed crucial role of mtQ redox homeostasis in modulating mtROS production.
As we proposed, the level of reduction (the endogenous redox state) of mtQ may be
a useful marker to assess total mtROS generation. However, there are few studies
describing a direct relationship between the mtQ redox state and mtROS production
under physiological and pathological conditions. mtQ redox ratio has rarely been
measured, especially under conditions of Q deficiency. Undoubtedly, measuring the
Q redox state in cells, tissues or isolated mitochondria is technically demanding
and not as popular as measuring mtROS formation. Determination of the QH/Q
ratio is more complicated than determining the total Q pool because ubiquinol
(QH) is particularly sensitive to oxidation. The difficulty in determining
the redox state of mtQ in biological samples and in vivo has been a
significant obstacle in characterizing mtQ function in mitochondria under
physiological and pathophysiological conditions. An extraction technique followed
by high performance liquid chromatography (HPLC) detection [127, 128] is usually
used to determine the concentration of cellular and mitochondrial Q and QH,
and to calculate the level of Q reduction (QH/Q) or Q redox state
(QH/Q) in cells/tissues and isolated mitochondria, respectively. However,
this technical approach requires a large amount of biological material, is time
consuming and, importantly, results in a non-kinetic point measurement.
Presumably, the recent advancement in the technique of simultaneous kinetic
measurement of oxygen consumption and mtQ redox state by combining a Clark-type
oxygen electrode and a Q-type electrode [28, 129] will contribute to the
widespread measurement of the redox state of mtQ in isolated respiring
mitochondria. In addition, a sensitive approach to assess the Q redox state by
liquid chromatography-tandem mass spectrometry (LC-MS/MS) in isolated
mitochondria, cells and tissues in vivo has recently been developed
[130]. This technical approach allows the analysis of both the redox state of Q
(non-kinetic point measurements) and the size of the Q pool with negligible
changes in the redox state due to isolation and extraction from small amounts of
biological material. Understanding metabolism linked to the mtQ pool is
challenging but new experimental models and methodologies will undoubtedly help.
Determining the redox state of Q under physiological and pathological conditions
is of great importance for better understanding how Q contributes to the
maintenance of cellular and mitochondrial homeostasis.
Author Contributions
WJ, KD, AB, KW and LG wrote the manuscript. All authors contributed to editorial
changes in the manuscript. All authors read and approved the final manuscript.
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
Not applicable.
Funding
This research was funded by National Science Centre, Poland, OPUS
2020/37/B/NZ1/01188. KD was funded by PRELUDIUM 2019/N/NZ1/01366.
Conflict of Interest
The authors declare no conflict of interest.







 , Karolina Dominiak 1,†, Adrianna Budzinska 1,†, Krzysztof Wojcicki 1, Lukasz Galganski 1
, Karolina Dominiak 1,†, Adrianna Budzinska 1,†, Krzysztof Wojcicki 1, Lukasz Galganski 1