




 , Gao Zhang 4,*
, Gao Zhang 4,*1 Department of Thoracic Surgery and Institute of Thoracic Oncology, Frontiers Science Center for Disease-related Molecular Network, West China Hospital, Sichuan University, 610041 Chengdu, Sichuan, China
2 Western China Collaborative Innovation Center for Early Diagnosis and Multidisciplinary Therapy of Lung Cancer, 610041 Chengdu, Sichuan, China
3 Department of Burn and Reconstructive Surgery, Laboratory of Mitochondria and Metabolism, West China Hospital, Sichuan University, 610041 Chengdu, Sichuan, China
4 Faculty of Dentistry, The University of Hong Kong, Prince Philip Dental Hospital, 999077 Hong Kong, China
†These authors contributed equally.
Abstract
Non-small cell lung cancer (NSCLC) accounts for 80–85% of all lung cancers, which has the highest cancer-related mortality worldwide. Regardless of the therapeutic effects of chemotherapy or targeted therapy, drug resistance will occur after 1 year. Heat shock proteins (HSPs) are a class of molecular chaperones participated in protein stability and multiple intracellular signaling pathways. It has been widely reported that HSPs family is over expressed in non-small cell lung cancer, and these molecules are also associated with protein stability and multiple intracellular signaling pathways. The effect of chemotherapy drugs or targeted drugs on cancer cells is usually to induce apoptosis. It is necessary to explore the interaction between heat shock protein family and apoptosis pathway in NSCLC. Here we provide a brief review of how HSPs affect the apoptotic pathway in NSCLC.
Keywords
- Heat shock proteins
- non-small cell lung cancer
- apoptotic
Lung cancer is currently the most cancer-related mortality cause worldwide. In China, the incidence of lung cancer increased yearly of which, non-small cell lung cancer (NSCLC) accounted for 80%–85% of all lung cancers [1, 2, 3]. Classical treatments for advanced NSCLC are chemotherapy and targeted therapy (for tumors harboring activating mutations in cancer driving genes). NSCLC responds well at the beginning, but develops drug resistance after 1–2 years [4, 5, 6, 7]. Therefore, studying the mechanism of the occurrence and development of NSCLC is very important for the development of new adjuvant therapy.
Heat shock proteins (HSPs) are a class of highly conserved molecular chaperones [8]. The presence of HSPs can prevent protein misfolding and over-aggregation by regulating protein assembly, folding and translocation in cells [9, 10]. The expression of most HSPs maintains at low levels under normal conditions but will be significantly upregulated in response to stress or in cancer cells [11, 12, 13, 14]. HSPs are classified as HSP100, HSP90, HSP70, HSP60, and small HSPs (those whose molecular weight under 60 kDa) according to their molecular weights [15]. Several studies showed increased expression of HSPs in a variety of solid tumors, including NSCLC [16, 17, 18, 19, 20]. Multiple articles suggested that HSPs can participate in the apoptotic pathway in vitro and HSP inhibitors enhanced therapeutic efficacy in NSCLC preclinical mouse models treated with chemotherapy or targeted therapy [21, 22, 23, 24, 25], indicating that HSPs may play an important role in the occurrence, development and treatment of NSCLC.
Here, we review current researches of HSPs in NSCLC, emphasizing HSP expression in NSCLC tissues and possible mechanisms for drug resistance.
HSP90 is a class of HSPs with a molecular weight of 90 kDa [26]. Human HSP90
includes four isoforms: HSP90
Recently, it has been found that HSP90 is highly expressed in NSCLC, positively
correlated with age and smoking status, negatively correlated with the 5-year
survival rate [30]. In a recent study, serum HSP90
Multiple studies showed HSP90 participates in apoptotic pathways, especially in caspase pathways. For TRAP1 knockdown NSCLC cells, reactive oxygen species (ROS) production is significantly increased when treated with cisplatin, activating mitochondrial apoptotic pathway. Besides, the pro-apoptotic effect of cisplatin is significantly enhanced in TRAP1 knockdown NSCLC cells [34]. The Grp94 inhibitor PU-H71 induces NSCLC cells apoptosis through the mitochondrial apoptosis pathway by down regulating Bcl-2 and up-regulating Bax, resulting in cytochrome C release [35]. In NSCLC cell lines, HSP90 knockdown causes more cells to enter G2/M phase, inhibiting cell growth, increasing sensitivity to gefitinib, inducing apoptosis [36]. In both mouse NSCLC syngeneic and xenograft models, knockdown of HSP90 followed by treatment with epidermal growth factor receptor (EGFR) inhibitor resulted in enhanced antitumor activity. Meanwhile, immunohistochemistry showed that the combination of HSP90 inhibitor and EGFR inhibitor induced more tumor cell apoptosis in both on-treatment and resistant specimens [37]. In conclusion, HSP90 inhibition activates caspase pathway, leading to NSCLC apoptosis both in vitro and in vivo (Fig. 1).
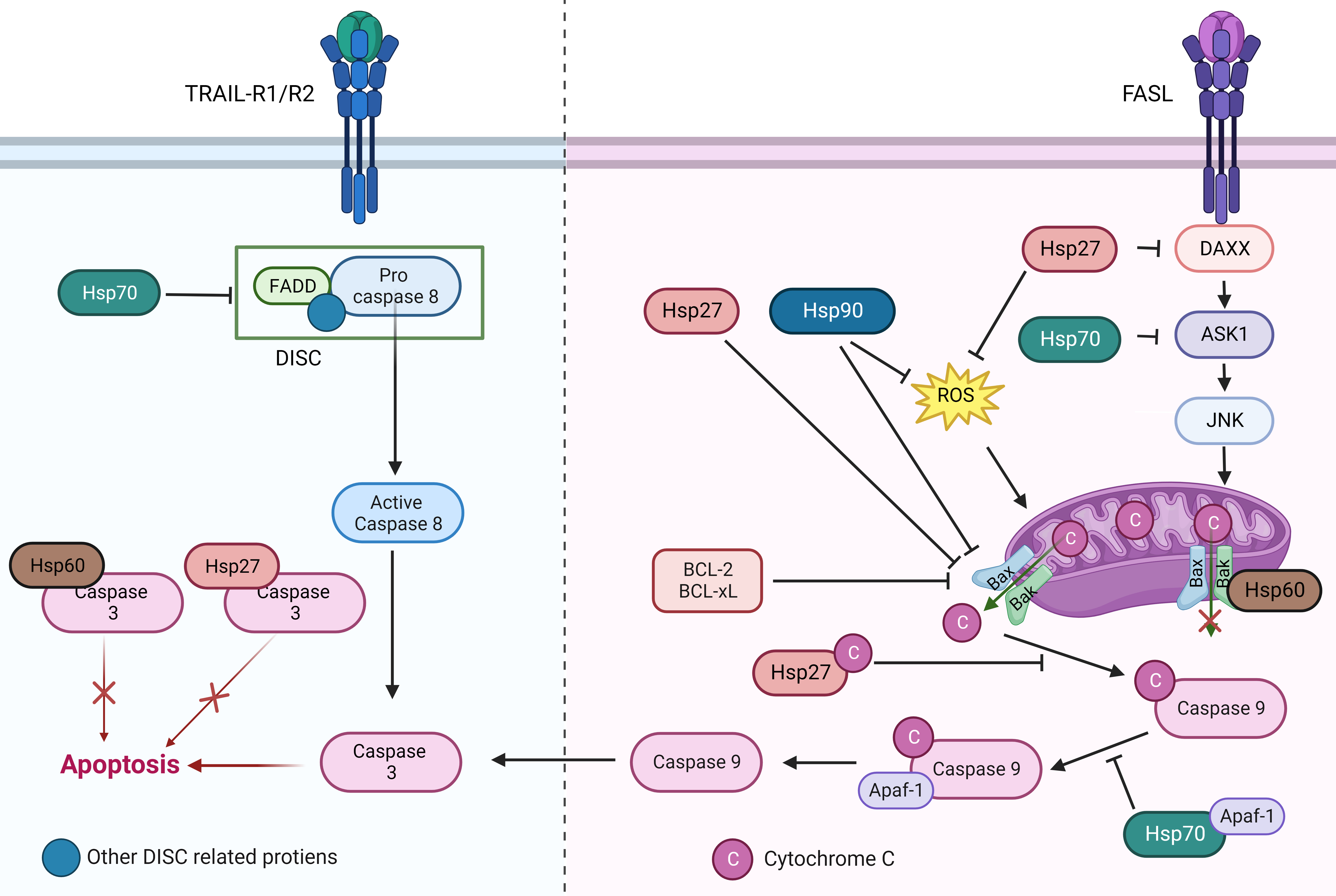 Fig. 1.
Fig. 1.The role of different heat shock protein family members in the apoptotic pathway. HSPs participate in anti-apoptosis mainly in two ways: (1) HSPs can block signal transmissions by competitively binding to apoptotic signaling molecules. (2) HSPs can directly bind to the apoptotic effector molecules to prevent apoptosis.
NSCLC cells often obtain overexpression or activating mutation of
proto-oncogenes, such as human epidermal growth factor receptor (HER2),
serine/threonine kinase (Raf-1), anaplastic lymphoma kinase (ALK), protein kinase
B (PKB or AKT). HSP90 is thought to stabilize above protooncogenes in NSCLC cells
to support cancer initiation and progression [38, 39]. Using HSP90 inhibitor
17-AAG can downregulate EGFR, AKT through decreasing cellular thymidine
phosphorylase via ubiquitin-26S proteasome pathway to inhibit tumor cells growth.
Furthermore, combination of 17-AAG can increase the killing effect of cisplatin
on NSCLC tumors [21, 22, 23]. Another HSP90 inhibitor, AUY922 can inhibit KRAS
signaling and slow down NSCLC cell growth [40]. In KRAS mutant NSCLC cell lines,
HSP90 confers resistance to chemotherapeutic agents and targeted therapy agents
by stabilizing AXL/Elf4e [41]. HSP90 can also activate NF-
Abnormal up-regulation of HSP90 was found in chemotherapy-resistant NSCLC cells. In these chemotherapy-resistant cells, by inhibiting HSP90, the expression of Anti-Müllerian hormone (AMH) and Anti-Müllerian hormone type II receptor (AMHR2) increased, the epithelial–mesenchymal transition (EMT) of NSCLC cells was inhibited, and tumor cell sensitivity to cisplatin restored [43] (Fig. 2).
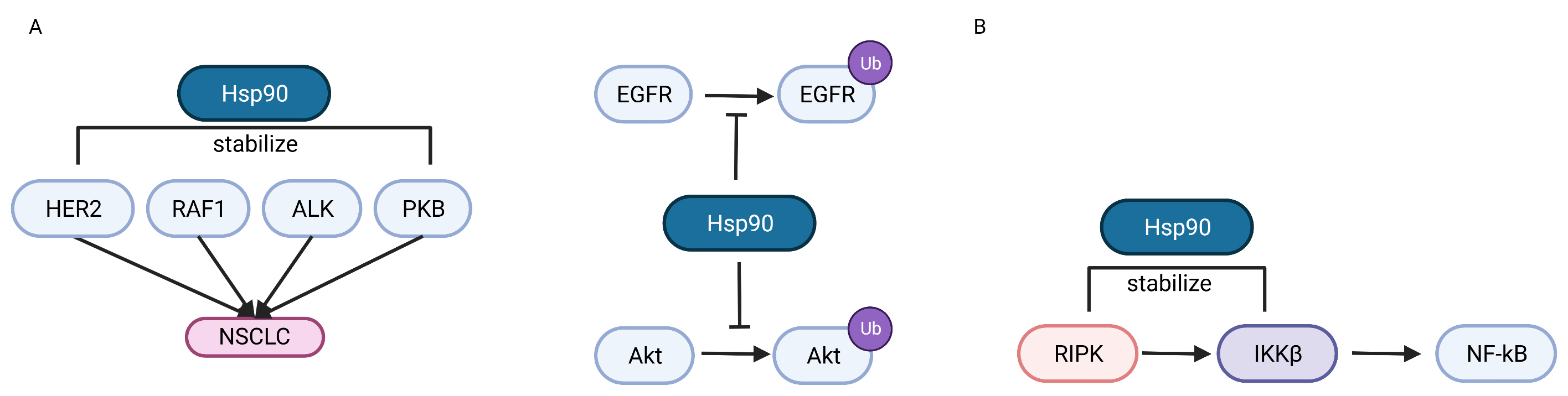 Fig. 2.
Fig. 2.The role of Hsp90 in regulating NSCLC oncogenesis. (A)
HSP90 can promote NSCLC oncogenesis by stabilizing mutated HER2, RAF-1, ALK and
PKB products, or inhibiting the ubiquitin-proteasome pathway of EGFR and AKT. (B)
HSP90 can stabilize RIPK and IKK
The HSP70 family has numerous members, encoded by a multigene family, and is widely distributed in cells, for example, Grp75 is found in mitochondria and Grp78 is in the endoplasmic reticulum [44, 45]. Typically, HSPs1L, HSPs2, HSPs5, HSPs8, HSPs9, HSPs12A, HSPs12B and HSPs13 encode HSP70 proteins that are constitutively expressed in different types of cells, while other HSP70 proteins are expressed in response to cellular stress, such as those encoded by HSPs1A, HSPs1B, HSPs6, HSPs7, and HSPs14 [46]. Although there are many family members and the gene loci and amino acid sequences are different, HSP70s share a similar structure. They have a conserved ATP-binding region at the N-terminus that binds and hydrolyzes ATP [47]. The C-terminus is usually the site where HSP70s bind to target proteins, acting as chaperones. These two functional domains are linked by a highly conserved leucine-rich amino acid sequence [48, 49]. HSP70 itself has weak ATPase activity and usually needs to cochaperone with J-protein (HSP40) to enhance its ATPase activity for function [50].
HSP70 is overexpressed in NSCLC, and sufficient evidence suggests that the overexpression of HSP70 is associated with poor prognosis. Pfister et al. [51] found that HSP70 was expressed on the cell membrane in about 40% of 150 NSCLC tumor samples, revealing that HSP70 may be overexpressed and localized on the membrane in NSCLC patients. The result of a large cohort study in Japan showed that the mean plasma HSP70 level of 189 NSCLC patients was 2.41 ng/mL, which was higher than the mean level of 2.01 ng/mL in healthy cohort [52]. Vostakolaei et al. [53] also detected positive HSP70 in tumor cells from NSCLC patients, and high levels of HSP70 was accompanied with poor prognosis.
HSP70 can also, like other chaperones, enable cancer cells to escape stress for
further progression by inhibiting cell apoptosis. HSP70 can regulate both
intracellular and extracellular apoptotic pathways at the same time. In the
intracellular apoptotic pathway, HSP70 can prevent the combination of Apoptotic
Peptidase Activating Factor 1 (APAF-1) to pro-caspase9 by directly binding to the
caspase recruitment domain of APAF-1, thereby affecting the formation of
apoptotic complexes in the mitochondrial [54]. HSP70 can regulate the level of
Apoptosis signal-regulating kinase 1 (ASK1) protein by binding to CHIP protein in
TNF-
Epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI) can induce phosphorylation of 41 Y residues of HSP70, ubiquitinate and degrade HSP70, increasing the probability of gene mutation, and eventually induce EGFR T790M mutation, leading to drug resistance [58]. Continuous cisplatin stimulation in A549 cells induced cisplatin resistance, and increased expression of HSP70 was detected in the drug-resistant A549 cells after cisplatin stimulation. When stimulating the cisplatin-resistant cell line again with cisplatin, the level of HSP70 was down-regulated, and the apoptotic signal staining was increased by flow cytometry [59]. Studies on NSCLC A549 and H460 cell lines showed that A549 cell lines with high GRP78 expression were less sensitive to cisplatin [60] (Fig. 1).
AIMP2-DX2 is a protein mutant that causes lung cancer. Semi Lim et al. [61] found that AIMP2-DX2 mutant can avoid Siah1-mediated ubiquitination by binding to HSP70 C-terminus, AIMP2-DX2 will eventually be stabilized in cells and cause NSCLC [62].
Recent studies have shown that OTUD3, a deubiquitinating enzyme, promotes NSCLC cell growth and metastasis by stabilizing GRP78 through binding it and co-localizating to the endoplasmic reticulum. It is evident that GRP78 may play an important role in the metastasis of cancer cells [63].
It has been reported that HSP70 modified by Glyoxalase I can activate
TGF-
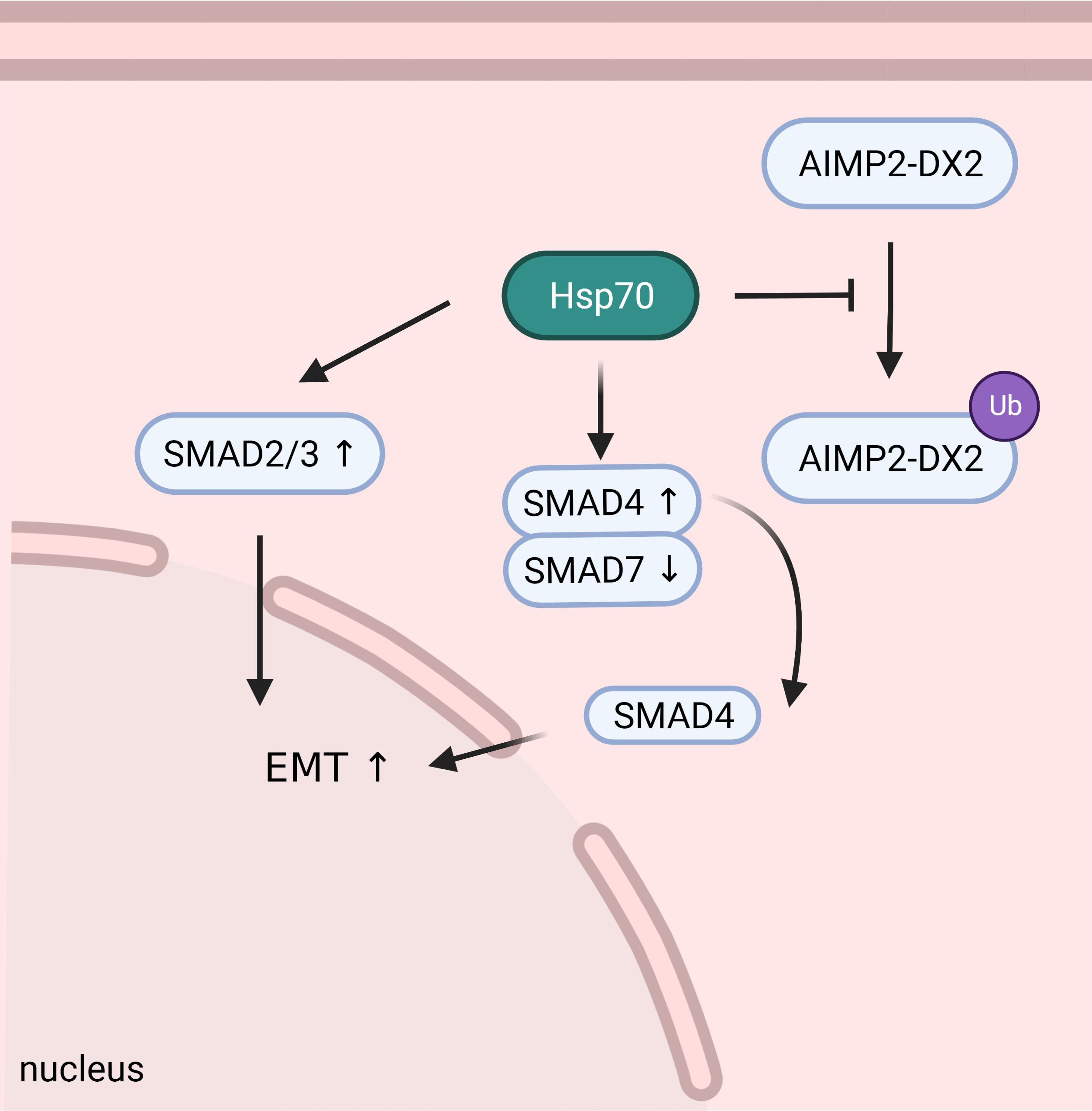 Fig. 3.
Fig. 3.HSP70 can stabilize AIMP2-DX2 to promote tumorigenesis, and participate in the regulation of SMAD2/3/4/7 to induce tumor EMT and further progression and metastasis.
The expression of HSP27, also known as HSPB1, remains at low level under
physiological condition and is induced by stress. The transcriptional regulation
of HSP27 is affected by cell type and cellular environmental factors, such as
heat shock factor 1 (HSF-1) and HIF1
A study using Immunohistochemistry (IHC) stain tested 76 NSCLC tumor samples, the expression of HSP27 was significantly higher in lung cancer than that in normal tissues, proportional to malignancy. Meanwhile, several studies have shown that patients with higher HSP27 expression have a significantly shorter 5-year survival than those with lower HSP27 expression [72, 73, 74, 75]. What’s more, in a multi-institution study, serum phosphorylated HSP27 (S78/S82) was detected in 109 NSCLC patients, and it was found that the level of serum phosphorylated HSP27 (S78/S82) was higher in advanced NSCLC patients than in early-stage NSCLC patients [76].
A report in 2016 showed that HSP27 and caspase-9 colocalize in focal adhesions of lung cancer tissue, and the traditional Chinese medicine YangZheng XiaoJi can block this co-localization by inhibiting the phosphorylation of HSP27 protein S86, making insensitive lung cancer cell lines sensitive to chemotherapeutic agents [24]. Jeroen W J van Heijst and his colleagues [25] showed that in lung squamous cell carcinoma cells, the expression of HSP27 in tumor cells increased after incubation with cisplatin, and the increased HSP27 binded to caspase-3 to inhibit the activation of apoptotic pathway and increased tumor cells’ resistance to cisplatin. The apoptosis induced by TNF-related apoptosis-inducing ligand (TRAIL) was initiated by TRAIL binding to TRAIL-R1/R2. This interaction leaded to cleavage and activation of pro-caspase-8 by the adaptor protein FADD, however this effect is inhibited in A549 cells overexpressing HSP27 [77, 78, 79]. In NSCLC cells with high expression of HSP27 after radiotherapy, inhibition of HSP27 increased the expression of intracellular cytochrome C. After downregulate HSP27 using short interference RNA, it was found that the expression of Bax was up-regulated, the expression of Bcl2 was down-regulated, and pro-caspase-8 was activated to induce cell apoptosis, increasing the sensitivity of NSCLC to radiotherapy [80]. In addition, HSP27 can prevent the formation of apoptotic bodies in NSCLC cells by directly binding to cytochrome C and preventing the release of cytochrome C [81].
HSP27 can reduce the oxidative stress of cells and prevent apoptosis by reducing ROS and increasing reduced glutathione [82]. HSP27 is also able to resist apoptosis by participating in the regulation of the threonine kinase PKB, mimicking a positive survival signal [83]. Phosphorylated HSP27 may directly bind to other pro-apoptotic proteins such as death domain-associated protein (DAXX) to inhibit the apoptotic pathway. For example, it interacted with DAXX to affect the membrane Fas-mediated DAXX-ASK1-JNK apoptotic pathway [84, 85] (Fig. 1).
Zhicheng Huang’s study [86] found that HSP27 expression was increased in A549
cells with TGF-
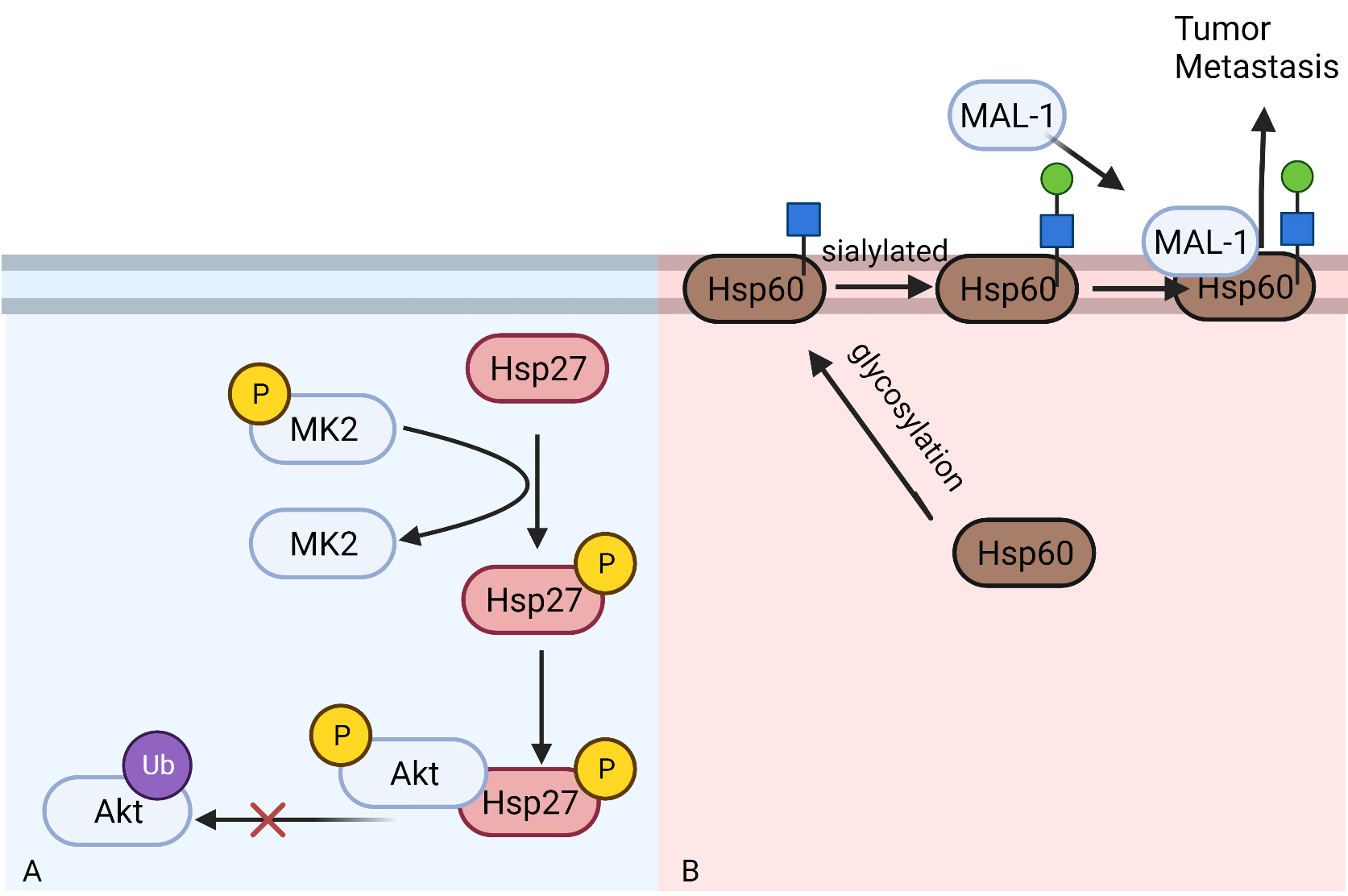 Fig. 4.
Fig. 4.The role of Hsp27 and Hsp60 in regulating NSCLC metastasis and migration. (A) Phosphorylated HSP27 can bind to phosphorylated Akt to prevent its degradation through ubiquitin-proteasome pathway. (B) HSP60 can be sialylated on cell membrane after being glycosylated. Then HSP60 can bind to MAL-1 to promote tumor metastasis.
HSP60 is an important member in heat shock family with a molecular weight of 60 kDa and its structure is highly conserved in a variety of organisms [91]. HSP60 is located in mitochondria under physiological conditions and ensures proper folding, assembly and repairment of mitochondria-associated proteins [92]. However, in tumor cells, HSP60 can be glycosylated, translocated to the cytoplasm or even on the cell membrane, affecting tumor cell migration [93, 94].
Several studies have shown that HSP60 significantly increased in tumor tissues of lung cancer patients compared with healthy control tissues, and is negatively correlated with prognosis, with no significant relationship with Tumor-Node-Metastasis (TNM) classification [95, 96]. It has also been shown that cancer cells located at the center of tumor tissue expressed higher levels of HSP60 in the cytoplasm than those located at the edge of tumor tissue at different invasive sites of tumor cells [97].
HSP60 can bind to and stablize the inactive form of caspase-3, thereby exerting its anti-apoptotic effect in lung cancer. In fact, inhibition of the combination did significantly increase the apoptosis of lung cancer cells [98]. It has also been shown that HSP60 on mitochondria can combine with Bax-Bak-Bcl-XL to form macromolecular complexes and inhibit apoptosis intracellularly [99]. To manipulate the combining of HSP60 and Bax, cells were treated with antisense RNA. With the decrease of HSP60, an increase of unbound Bax and Bak in the cytosol was observed. The presence of HSP60 may be necessary for Bax translocation from the cytosol to the mitochondria, upon which subsequent cytochrome c release and caspase activation can occur in an orderly manner [99, 100]. 6-Shogaol (6-SH) is an anti-cancer active substance found in dried ginger. One study has shown that 6-SH can promote the proteasome-mediated degradation of HSP60, and can induce the reduction of Bcl-2 and survivin expression and the increase of pro-apoptotic protein Bax expression in A549 cells. Then, these 6-SH-treated A549 cells were arrested at G0/G1 phase and were induced to cell apoptosis [101] (Fig. 1).
After N-glycosylation, HSP60 in tumor cells can translocate to tumor cell
membrane [102]. HSP60 on the cell membrane can be further
HSP60 in tumor cells can form a complex with P53, resulting in the reduction of the binding of P53 to molecules related to cell cycle arrest signals, thus making tumor cells to enter a highly active replication state. It has been shown that doxorubicin can attenuate cancer proliferation through acetylation of HSP60, reducing its interaction with P53. Some studies have also shown that P53 is closely related to cell apoptosis, so whether the combination of HSP60 and P53 has a role in this aspect remains to be elucidated by further studies [104, 105, 106] (Fig. 4B). Cancer cells with HSP60 knockdown by HSP60 shRNA showed reduced basal respiration and impaired ATP synthesizing capacity through oxidative phosphorylation (OXPHOS), and accompanied by reduced cancer cell proliferation [107].
HSP is a huge family with lots of family members and complicated functions. In general, HSPs are upregulated in NSCLC, regulating cancer development and drug resistance through apoptotic pathway, p53 pathway. Researches on HSP mechanism may help to seek future therapeutic targets.
In light of the existing research and biological application of HSP family
proteins, there have been a lot of researches in the development of inhibitors
for lung cancer and some have entered clinical trials. For example, phase I
clinical trials of Debio 0932 and preclinical trials of 17-DMAG have been
successfully conducted. In addition, inhibitors for HSP90 like NVP-AUY922,
STA-9090, AT13387 and IPI504 have entered phase II clinical trials. OGX-427, an
inhibitor of HSP27, has also entered phase II clinical trials. However, HSP70
inhibitors, such as Pifithrin-
GuaZ and YP prepared the manuscript and figures. KZ helped with references collection and figures designation. YC and GaoZ designed manuscript outline and revised manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This research was funded by the National Natural Science Foundation of China (No. 31771549 to Y Chen).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.