






 , Wei Jiang 1,†, Junsen Cheng 1, Wei Guo 1, Yongquan Li 1, Chenlei Li 1
, Wei Jiang 1,†, Junsen Cheng 1, Wei Guo 1, Yongquan Li 1, Chenlei Li 11 College of Horticulture and Landscape Architecture, Zhongkai University of Agriculture and Engineering, 510225 Guangzhou, Guangdong, China
†These authors contributed equally.
Abstract
Background: Salinity is the main abiotic stress that affects seed germination, plant growth and crop production. Plant growth begins with seed germination, which is closely linked to crop development and final yields. Morus alba L. is a well-known saline-alkaline tree with economic value in China, and the most prominent method of expanding mulberry tree populations is seed propagation. Understanding the molecular mechanism of Morus alba L. salt tolerance is crucial for identifying salt-tolerant proteins in seed germination. Here, we explored the response mechanism of mulberry seed germination to salt stress at physiological and protein omics levels. Methods: Tandem mass tag (TMT)-based proteomic profiling of Morus alba L. seeds germinated under 50 mM and 100 mM NaCl treatment for 14 days was performed, and the proteomic findings were validated through parallel reaction monitoring (PRM). Results: Physiological data showed that salt stress inhibited the germination rate and radicle length of mulberry seeds, decreased the malondialdehyde (MDA) content and significantly increased superoxide dismutase (SOD), peroxidase (POD), and catalase (CAT) activities. Then, a TMT marker technique was used to analyze the protein groups in mulberry seeds with two salt treatment stages, and 76,544 unique peptides were detected. After removing duplicate proteins, 7717 proteins were identified according to TMT data, and 143 (50 mM NaCl) and 540 (100 mM NaCl) differentially abundant proteins (DAPs) were screened out. Compared with the control, in the 50 mM NaCl solution, 61 and 82 DAPs were upregulated and downregulated, respectively, and in the 100 mM NaCl solution, 222 and 318 DAPs were upregulated and downregulated, respectively. Furthermore, 113 DAPs were copresent in the 50 mM and 100 mM NaCl treatments, of which 43 were upregulated and 70 were downregulated. Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis revealed that the DAPs induced by salt stress during mulberry seed germination were mainly involved in photosynthesis, carotenoid biosynthesis and phytohormone signaling. Finally, PRM verified five differentially expressed proteins, which demonstrated the reliability of TMT in analyzing protein groups. Conclusions: Our research provides valuable insights to further study the overall mechanism of salt stress responses and salt tolerance of mulberry and other plants.
Keywords
- Morus alba L.
- proteomic
- seed germination
- salt stress
- tandem mass tag
- parallel reaction monitoring
Soil salinization is among the most serious abiotic factors responsible for
decreasing crop yields worldwide; the damage is mainly due to high sodium salt
concentrations (NaCl or Na
Morus alba L. is an important perennial woody plant, and its leaves are the main food source for silkworms. Mulberry has high nutritional and economic value and has various uses in ecology, food and medicine [22]. Its high content of active ingredients and high resistance to adversity stresses, such as drought, salinity and flooding, make mulberry an excellent material for researching stress in woody plants [23] and one of the ecological tree species with great potential for afforestation, soil and water conservation, bioremediation of pollutants, land desertification and saline land management [24, 25]. In depth research on genes related to mulberry stress signaling has been conducted. For example, under ultraviolet-B and dark stress, mulberry leaves activate the cell redox and ER quality control systems, enhancing protein synthesis and weakening N-glycan biosynthesis in the ER to resist damage [26]. The activities of superoxide dismutase (SOD), catalase (CAT) and peroxidase (POD) in mulberry can eliminate Cd stress-induced oxidative stress [27]. After abscisic acid (ABA) biosynthesis is activated in mulberry, some drought-responsive genes (such as CBP6A, MGST3, ERF1B, etc.) are also activated [28]. In recent years, mulberry planting has been performed for the ecological restoration of land, which is of great significance for restoring and reconstructing vegetation in saline-alkali land and to improve the ecological environment. Seed sowing propagation is an important agricultural production method for mulberry propagation. Mulberry seeds in the germination stage are extremely sensitive to salt stress, and China contains a large area of saline-alkali land, which urgently needs to be utilized. It is necessary to study the response mechanism of mulberry seeds to salt stress. Proteins are directly involved in the stress response of plants. Studying the response of mulberry to salt stress conditions at the protein level can provide a powerful tool for revealing the physiological mechanism of mulberry stress resistance.
Furthermore, physiological responses in cells are produced by proteins, and
proteomics, a global analysis strategy, is the key to studying gene functions
[29]. Proteomic analysis can provide information on multiple processes in complex
events, such as seed germination and seed filling. Seed germination is the very
beginning of plant life and is among the most critical and decisive stages in the
plant growth cycle, which determines the formation of adult plants and the final
crop yield [30, 31]. Analyzing seed germination through proteomics is mainly
carried out by sequencing plants, such as Oryza sativa [32],
Brassica napus [33], Populus [34], Jatropha curcas [35], Araucaria [36] and Platanus orientalis [37]. Relatively
few studies have focused on the germination of seeds from Morus alba. In
recent years, the molecular basis of salt responses and tolerance during seed
germination has been investigated using proteomic approaches in some crops and
woody plant species, including Triticum aestivum [38], Zea mays [39], Sorghum bicolor [40] and Ulmus pumila [41]. The results
showed that the abundance of H
The experimental material of this study is “Yuesang No. 11” provided by Botanical Garden of Zhongkai University of Agriculture and Engineering. First, the mature mulberries were harvested; the seeds were washed; mature, full and uniform seeds were selected; and the seeds were stored at 4 °C for subsequent experiments. The seeds were soaked at room temperature for 24 h and the soaking solution was discarded. The seeds were then surface disinfected with 75% ethanol and sodium hypochlorite solution (1% effective chlorine content). 75% ethanol was soaked for 5 min, rinsed with sterile water 3 times [17], sodium hypochlorite solution was soaked for 30 min, rinsed with sterile water 3 times, and the moisture on the surface of the seeds was absorbed with filter paper. The samples were placed in a sterile 90 mm diameter Petri dish covered with three layers of filter paper. The control group (CK) was treated with a proper amount of sterilized water (approximately 12 mL/dish) and 50 and 100 mM NaCl solutions. Each dish was placed with 100 seeds for germination, and biological reproduction was carried out three times. After 14 days, the seedlings were sampled for physiological analysis and protein extraction.
Mulberry seeds germination began after 2 days of treatment (seeds were
considered germinated when radicles appeared), and germinating seeds were
observed and counted every 48 hours to calculate the germination percentage.
After 14 days, mulberry seedlings from different treatments were collected and
rinsed with deionized water. The germination rate and radicle length of mulberry
seedlings were determined, and the final germination rate was the number of seeds
germinated divided by the total number of seeds used for the test
In this experiment, SOD, POD, CAT activities as well as H
Mulberry seedling samples that were stored at –80 °C were ground into a
fine powder, and four volumes of phenol extraction buffer (1% protease inhibitor
and 2 mmol/L EDTA, 10 mmol/L dithiothreitol) were added for ultrasonic cracking.
Add the same amount of Tris balanced phenol, centrifuge at 5500
The extracted proteins were subjected to trypsin (317107 Promega) hydrolysis
(protein/trypsin, 50/1) using the method of filter-aided proteome preparation
(FASP) described by Wisniewski et al. [45]. Add the total protein
solution into a protein concentrator (10K MWCO, 0.5 mL Pierce™
Protein Concentrator, Thermo Scientific, Waltham, MA, USA), dilute the sample
with 8 mol/L urea, and after reduction and alkylation, digest it with trypsin at
37 °C overnight. After digestion, centrifuge at 10,000
100
Each set of labeled peptide fragments was uniformly mixed and fractionated using the Reverse-Phase High pH Peptide Fractionation Kit (Thermo Scientific, USA). The TMT-labeled digested sample was fractionated into 10 fractions using a gradient of acetonitrile elution using the Pierce™ High pH Reverse-Phase Peptide Fractionation Kit (Thermo scientific, USA) according to the manufacturer’s instructions.
Each fraction was injected and analyzed by Nano LC-MS/MS. The peptide mixture
was loaded onto a reversed-phase trap column (Thermo Scientific Acclaim
PepMap100, 100
Raw data for the Q-Exactive spectrometry analysis were obtained as RAW files.
Identification and quantitative analyses were performed by searching the database
using Mascot version 2.2 and Proteome Discoverer version 1.4. The
UniProt_Lycopersicon_esculentum Mulberry_35921_20211214.fasta (35921
sequences, downloaded on 14 December 2021) database was searched. Trypsin was
selected as a random cleavage enzyme, and the parameter of max missed cleavages
was set to 2. Carbamidomethyl (C), TMT 9 plex (N-term), and TMT 9 plex (K) was
selected as fixed modifications. The variable modifications were set to oxidation
(M), TMT 9 plex (Y). The peptide mass tolerance was set to
All annotation and DAP classification of Gene Ontology (GO) features were performed using the Blast2GO (https://www.blast2go.com/) program. The Automatic Annotation Server was used to perform Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotations [46]. The significance of protein enrichment of GO functional annotations and KEGG pathway annotations was evaluated by Fisher’s exact test.
The peptide information suitable for PRM analysis was imported into Xcalibur
software (Thermo scientific, USA) to establish the PRM method. An equivalent
amount of digested peptide of approximately 1
PRM analysis of samples separated by HPLC was performed for 60 min using a
Q-Exactive HF mass spectrometer (Thermo Scientific, USA). The analytical column
was a homemade tip column (75
Analysis and examination of the obtained experimental data were performed using
Excel (Microsoft office professional plus 2010; Microsoft corporation, Redmond, WA, USA) and SPSS software 17.0 (SPSS
Inc., Chicago, IL, USA). All data are the mean
The germination percentage reflects the resistance of the seed germination process to abiotic stress. As shown in Fig. 1A, the germination rate of mulberry seeds increased significantly with increasing incubation time. Different concentrations of the same treatment days had different effects on the germination of mulberry seeds. With 0 mmol/L salt concentration, the germination rate continued to increase and reached the highest rate of 94.33% at day 14. With the 50 mmol/L salt treatment, the germination rate of seeds increased slowly at 4 days, increased rapidly from 4 to 8 days, and then increased slowly again from 8 to 14 days, and the germination rate of seeds at day 14 was 50%, which decreased by 44.33% compared with that of the control. With the 100 mmol/L salt treatment, the seed germination rate increased slowly from 0 to 8 days, but both were slower than the control and 50 mM NaCl stress treatments, and the seed germination rate on day 14 was 36.67%, which was decreased by 57.66% compared to that of the control. The germination rate of mulberry seeds was significantly lower than the germination rate of the control on the same day when the salt concentration was 50 mmol/L and 100 mmol/L. The growth of embryonic roots is crucial for plants to properly develop into seedlings out of the soil. As shown in Fig. 1B, the embryo root length increased with increasing embryo root growth days at the same concentration. Under 0 mmol/L salt, the embryo root length of mulberry seeds increased significantly, and the longest radicle length was 32.20 mm on the 14th day. With the 50 mmol/L salt stress treatment, the radicle length increased slowly from 0 to 6 days and rapidly from 6 to 14 days and basically stopped increasing after day 14. After 14 days, the length basically stopped increasing. At a salt concentration of 100 mmol/L, mulberry embryo roots basically did not grow. The length of the mulberry radicles varied at the same time for different concentrations. Compared to the control, both the 50 mmol/L and 100 mmol/L salt treatments significantly reduced the radicle length on the same day.
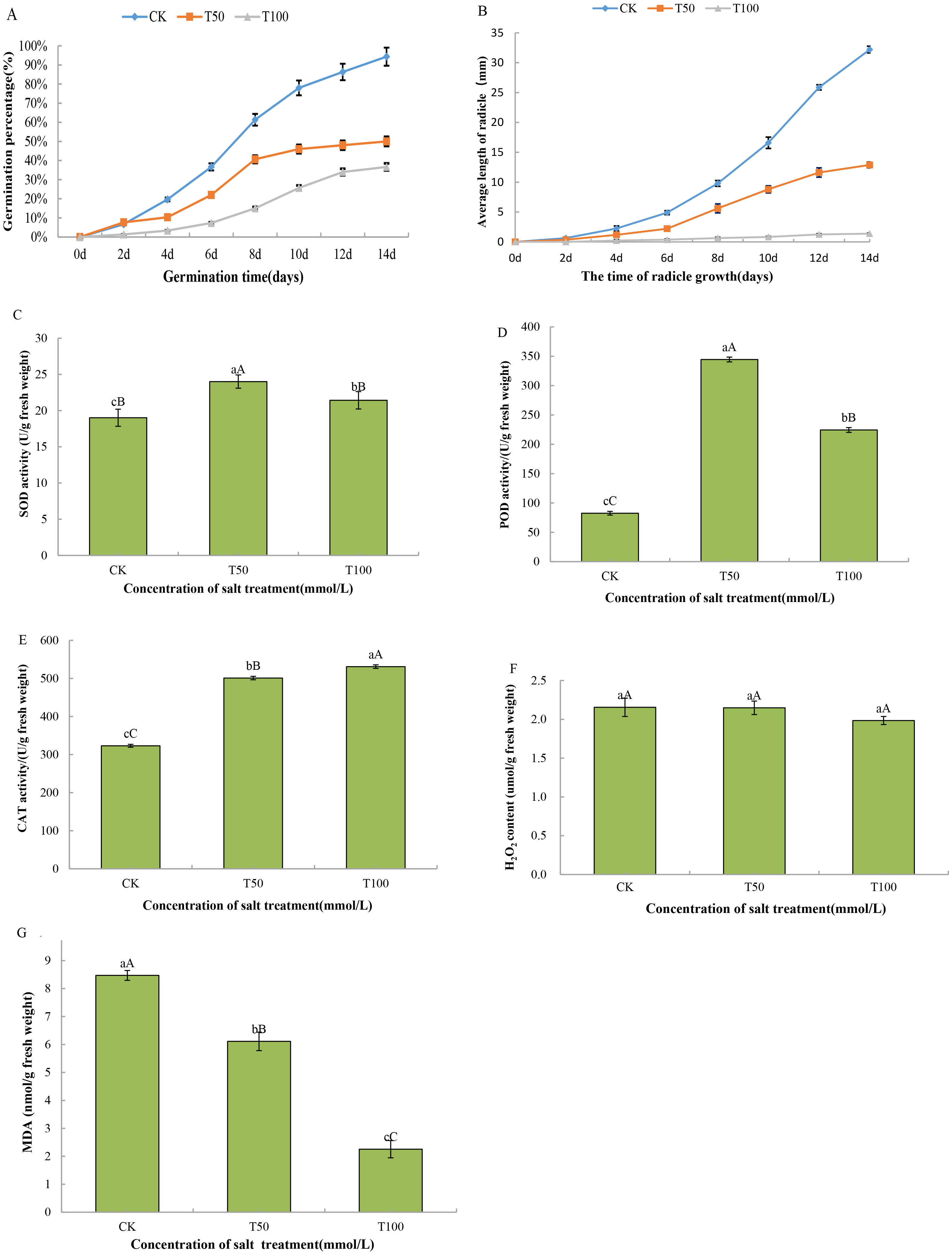 Fig. 1.
Fig. 1.Detection of the physiological parameter of mulberry seeds at
different salt treatment stages. (A) 14-day germination rate of mulberry seeds.
(B) Radicle length of mulberry seeds in 14 days. (C) Superoxide dismutase (SOD)
activity. (D) Peroxidase (POD) activity. (E) Catalase (CAT) activity. (F)
H
In response to salt stress, antioxidant enzymes (GST, POD, SOD, DHAR, APX and
CAT) were differentially accumulated during seed germination of mulberry.
Therefore, we studied the physiological indicators of salt stress, including the
determination of SOD, POD and CAT activities and the content of H
TMT-based quantitative proteomics was used to analyze the dynamic changes in
proteins in mulberry seeds. A total of 76,544 unique peptides were detected, and
7717 proteins were identified based on TMT data after removing duplicate
proteins. We counted the molecular weights of the identified proteins (Fig. 2A),
and the results showed that there were 811 (10.51%) of 10.0–20.0 kD, 1123
(14.55%) of 20.0–30.0 kD, 1193 (15.46%) of 30.0–40.0 kD, and 1067 (13.83%)
of 40.0–50.0 kD, 959 (12.43%) were 50.0–60.0 kD, and 90 (1.17%) were larger
than 200.0 kD. The numbers of proteins with 1 peptide, 2–5 peptides, 6–10
peptides and more than 11 unique peptides were 2224, 3207, 1351 and 935,
respectively. In addition, the number of peptides corresponding to almost 70% of
the identified proteins was
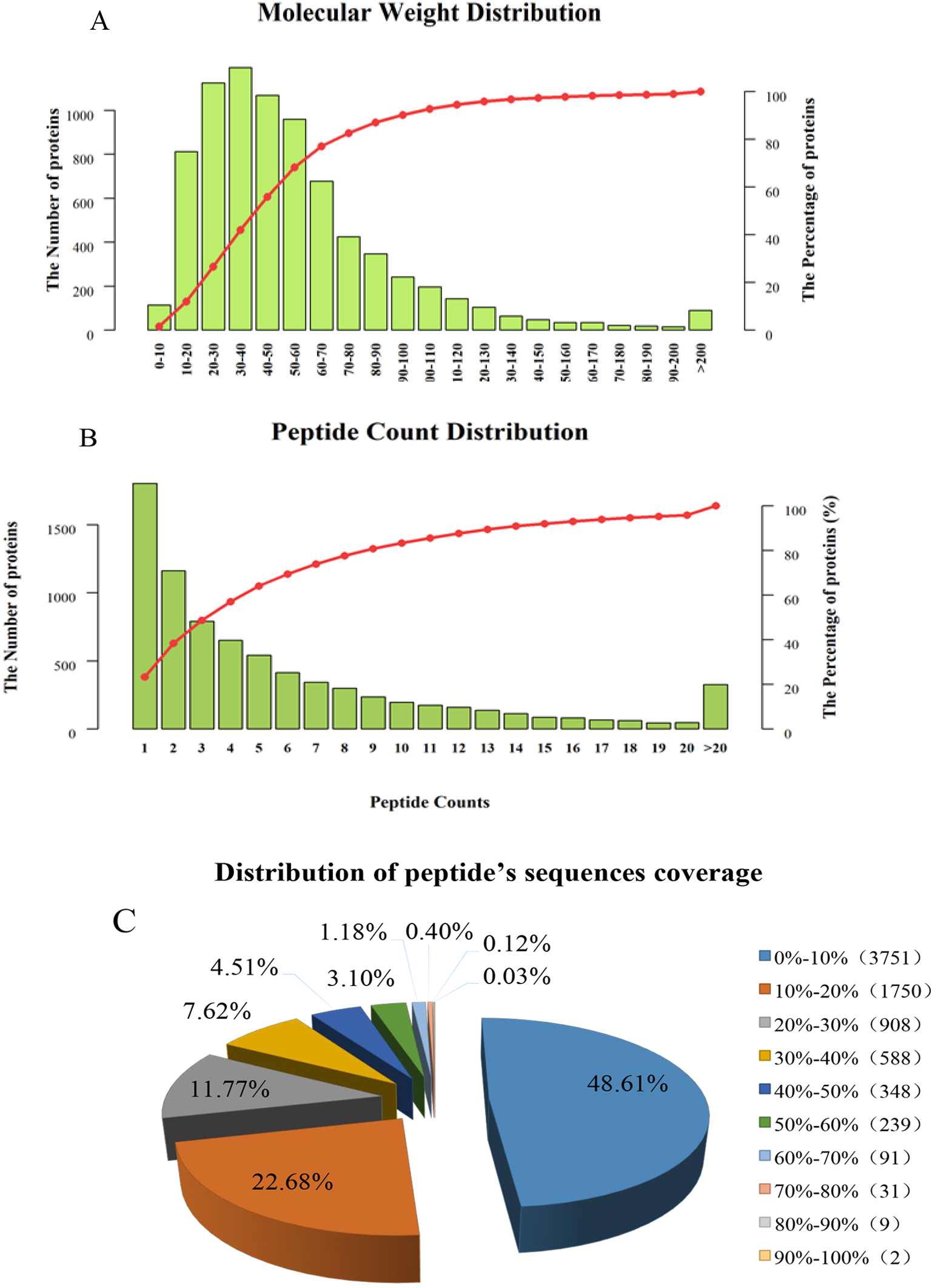 Fig. 2.
Fig. 2.The overall distribution of mulberry seed proteins identified by TMT. (A) Protein molecular weight distribution. (B) Peptide count distribution. (C) Distribution of peptide sequence coverage.
In this study, with the mulberry seedling protein sample (CK group) as the
control, the quantitative intensity of mass spectrometry of the mulberry seedling
protein sample in the 50 mmol/L NaCl and 100 mmol/L NaCl salt-treated groups was
compared with the protein sample in the CK group. The mean value of three
biological replicates was used as the fold difference of protein expression. A
fold-change cut off of
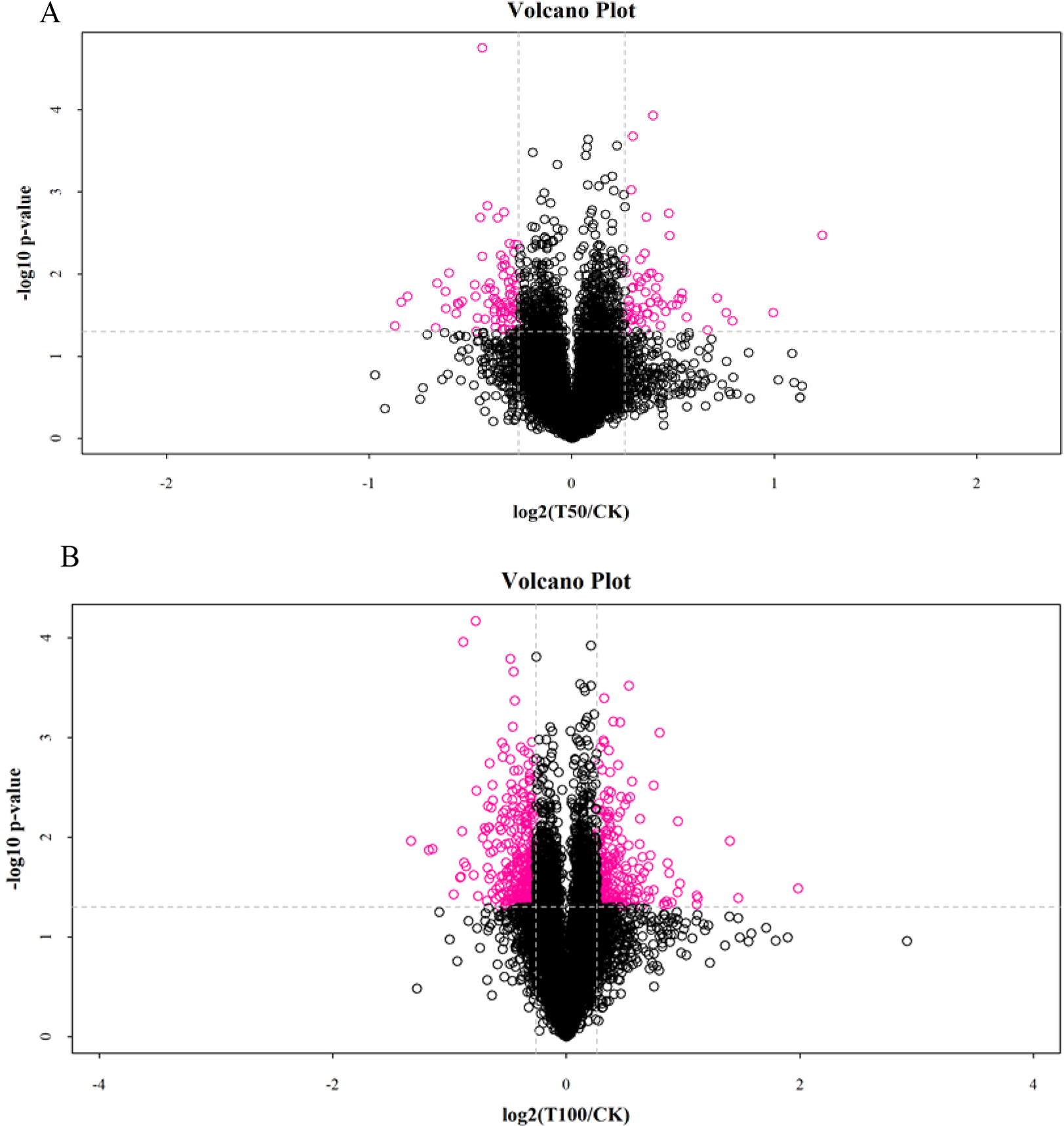 Fig. 3.
Fig. 3.Analysis of DAPs in mulberry seeds. (A) The volcano plots of DAPs in T50 vs. CK. (B) The volcano plots of DAPs in T100 vs. CK.
A total of 113 DAPs were screened in both salt treatments, of which 43 DAPs were upregulated and 70 DAPs were downregulated. By analyzing the hierarchical clustering analysis of DAP in different treatments, every three replicates of CK and salt-treated samples were clustered together separately with high reproducibility. Furthermore, the protein expression abundance and expression levels in mulberry seeds differed significantly between the CK and salt-treated samples (Fig. 4). At the same time, the functional analysis of the differentially abundant proteins screened from the two salt-stressed mulberry seeds showed that the differentially expressed proteins mainly belonged to reactive oxygen species (ROS) scavenging, stress defense, energy production, carbohydrate metabolism, transcription and translation, growth and development, signal transduction, substance transport, protein synthesis, folding and degradation categories (Table 1). Statistics of differential proteins include molecular weight, fold difference and p value.
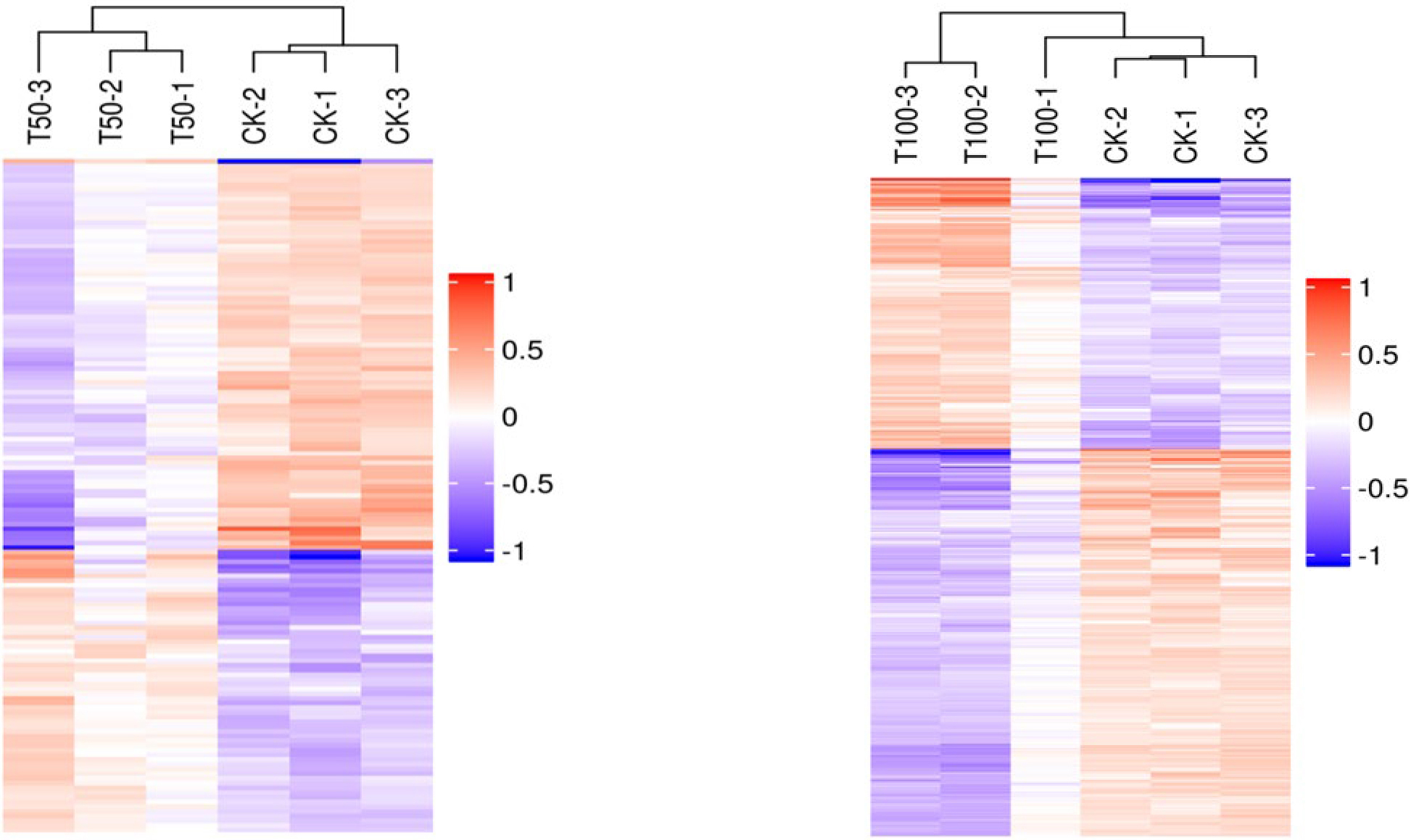 Fig. 4.
Fig. 4.Hierarchical clustering analysis of the DAPs in different treatments.
| Function classification | Accession | Description | MW [kDa] | Fold Change | p-value | |
| T50/CK | T100/CK | |||||
| Reactive oxygen scavenging and stress defense | W9QU60 | Peroxidase | 34.19 | 1.379 | 1.678 | 0.024 |
| W9RZP5 | Superoxide dismutase | 26.41 | 1.276 | 1.405 | 0.036 | |
| A0A1D8KVU3 | Dihydroflavonol reductase 1 | 38.30 | 0.793 | 0.775 | 0.008 | |
| W9QH65 | Glutathione peroxidase | 26.50 | 1.268 | 1.361 | 0.024 | |
| W9T1J8 | SHSP domain-containing protein | 24.41 | 1.206 | 1.323 | 0.003 | |
| W9RN30 | Jacalin-type lectin domain-containing protein | 15.71 | 1.321 | 1.740 | 0.017 | |
| W9RJC9 | Putative disease resistance protein RGA4 | 128.22 | 0.833 | 0.800 | 0.030 | |
| W9SE40 | Sulfiredoxin | 13.76 | 0.827 | 0.773 | 0.002 | |
| W9RK87 | Spermine synthase | 43.61 | 1.258 | 1.284 | 0.024 | |
| W9SCX0 | Lycopene beta cyclase | 56.26 | 0.768 | 0.806 | 0.029 | |
| W9QK12 | Lycopene epsilon cyclase | 59.98 | 0.827 | 0.725 | 0.026 | |
| W9RBR2 | Allene oxide synthase | 57.48 | 0.649 | 0.538 | 0.042 | |
| W9RAN9 | Anthocyanin 5-aromatic acyltransferase | 52.24 | 0.795 | 0.685 | 0.028 | |
| Energy production | W9S8G4 | Lipase | 45.45 | 1.458 | 1.866 | 0.030 |
| W9QX55 | Lipoxygenase | 86.97 | 0.821 | 0.758 | 0.009 | |
| Q09X21 | Photosystem II CP43 reaction center protein | 51.79 | 0.827 | 0.783 | 0.048 | |
| Q09X17 | Photosystem I P700 chlorophyll a apoprotein A1 | 83.06 | 0.807 | 0.724 | 0.029 | |
| W9QPP1 | Oxygen-evolving enhancer protein 3-2 | 24.70 | 0.628 | 0.545 | 0.019 | |
| W9R2T4 | Glucose-1-phosphate adenylyltransferase | 59.46 | 1.249 | 1.299 | 0.020 | |
| W9RFS1 | Short-chain type | 64.75 | 1.397 | 1.413 | 0.011 | |
| W9SKC6 | Flavin-containing monooxygenase | 47.43 | 0.736 | 0.543 | 0.034 | |
| W9R035 | Frataxin | 22.18 | 1.292 | 1.279 | 0.035 | |
| W9R2T4 | Glucose-1-phosphate adenylyltransferase | 59.46 | 1.249 | 1.299 | 0.020 | |
| W9RCL2 | Lipase_3 domain-containing protein | 58.76 | 1.288 | 1.437 | 0.036 | |
| W9QJ14 | Sphingoid base hydroxylase 2 | 30.37 | 1.290 | 1.458 | 0.015 | |
| W9RFT7 | Alpha-carbonic anhydrase domain-containing protein | 30.62 | 0.718 | 0.644 | 0.023 | |
| Carbohydrate metabolism | W9QF53 | Cellulose synthase | 120.43 | 0.829 | 0.830 | 0.024 |
| W9RH68 | Endoglucanase | 55.19 | 0.802 | 0.741 | 0.019 | |
| W9SJJ0 | Alpha-L-fucosidase 1 | 58.36 | 1.218 | 1.277 | 0.003 | |
| W9S5L8 | Fructose-1,6-bisphosphatase | 44.87 | 0.810 | 0.744 | 0.029 | |
| W9R8E3 | Putative polygalacturonase | 63.38 | 1.442 | 1.426 | 0.033 | |
| Transcription and translation | W9R0R0 | Histone | 53.57 | 0.721 | 0.748 | 0.013 |
| W9SN28 | Histone H1 | 23.17 | 0.687 | 0.628 | 0.023 | |
| W9RWW0 | Transcriptional corepressor SEUSS | 108.92 | 0.785 | 0.583 | 0.023 | |
| W9SG17 | BHLH domain-containing protein | 39.45 | 0.719 | 0.658 | 0.021 | |
| W9QUN0 | Transcription initiation factor IIF subunit alpha | 52.71 | 0.817 | 0.801 | 0.025 | |
| W9RKQ5 | HMG1/2-like protein | 15.88 | 0.736 | 0.762 | 0.002 | |
| W9RJ53 | High mobility group B protein 2 | 16.04 | 0.755 | 0.735 | 0.001 | |
| W9QWT1 | Peptidylprolyl isomerase | 24.58 | 0.794 | 0.731 | 0.025 | |
| W9QWT8 | Glyceraldehyde-3-phosphate dehydrogenase B | 147.81 | 0.795 | 0.713 | 0.034 | |
| W9STM4 | Btz domain-containing protein | 73.82 | 0.771 | 0.695 | 0.016 | |
| W9SN10 | AT-hook motif nuclear-localized protein | 31.51 | 0.631 | 0.621 | 0.045 | |
| W9QMK8 | TCP domain-containing protein | 38.21 | 0.752 | 0.679 | 0.015 | |
| W9RVK1 | SWI/SNF chromatin-remodeling complex subunit snf22 | 246.07 | 0.803 | 0.750 | 0.008 | |
| W9SBM7 | U3 small nucleolar RNA-associated protein 15-like protein | 58.89 | 0.788 | 0.725 | 0.028 | |
| Growth and development | W9R8E1 | GDSL esterase/lipase | 38.53 | 1.695 | 1.822 | 0.010 |
| W9RZ71 | Aspartic proteinase nepenthesin-2 | 63.51 | 1.457 | 1.571 | 0.030 | |
| W9RMA6 | Beta-galactosidase | 88.18 | 1.644 | 1.785 | 0.022 | |
| W9RY25 | Subtilisin-like protease | 87.92 | 1.310 | 1.359 | 0.011 | |
| W9QYC8 | GDSL esterase/lipase | 41.61 | 1.216 | 1.203 | 0.023 | |
| W9SHN8 | GDSL esterase/lipase | 40.75 | 1.409 | 1.446 | 0.011 | |
| W9RD56 | GDSL esterase/lipase 6 | 39.63 | 1.432 | 1.543 | 0.002 | |
| W9RG64 | Purple acid phosphatase | 50.47 | 1.344 | 1.368 | 0.039 | |
| W9QMY5 | Purple acid phosphatase | 50.11 | 1.345 | 1.382 | 0.025 | |
| W9S1A8 | Pectinesterase | 62.59 | 0.813 | 0.784 | 0.013 | |
| W9SKF4 | Caffeic acid 3-O-methyltransferase | 29.05 | 0.657 | 0.647 | 0.016 | |
| Signal transduction | W9RBX0 | Putative receptor-like protein kinase | 91.76 | 1.333 | 1.388 | 0.007 |
| W9RZ35 | Putative L-type lectin-domain containing receptor kinase S.5 | 76.53 | 1.204 | 1.248 | 0.037 | |
| W9QVJ6 | Protein kinase domain-containing protein | 31.02 | 0.796 | 0.702 | 0.002 | |
| W9RA31 | Systemin receptor | 127.65 | 0.806 | 0.811 | 0.046 | |
| W9R8I4 | G-type lectin S-receptor-like serine/threonine-protein kinase | 134.05 | 0.794 | 0.800 | 0.048 | |
| Transport proteins | W9R5Q9 | Putative cyclic nucleotide-gated ion channel 8 | 85.60 | 0.790 | 0.783 | 0.037 |
| W9SAJ0 | ABC transporter G family member 28 | 112.05 | 1.247 | 1.334 | 0.049 | |
| W9RS36 | ABC transporter G family member 8 | 66.42 | 0.790 | 0.746 | 0.048 | |
| W9RGK8 | Nitrate transporter 1.1 | 29.38 | 0.755 | 0.680 | 0.023 | |
| W9SDQ8 | Nitrate transporter 1.3 | 66.41 | 0.768 | 0.639 | 0.020 | |
| W9QFD5 | Protein DETOXIFICATIO | 59.23 | 1.213 | 1.301 | 0.031 | |
| W9RWS8 | Transmembrane ascorbate ferrireductase 1 | 25.28 | 1.232 | 1.226 | 0.047 | |
| W9QQQ4 | Putative sulfate transporter 3.3 | 74.52 | 0.806 | 0.789 | 0.046 | |
| W9QN95 | Protein translocase subunit SecA | 118.21 | 0.829 | 0.750 | 0.016 | |
| W9S950 | CMP-sialic acid transporter 2 | 44.79 | 1.355 | 1.335 | 0.006 | |
| W9SK42 | t-SNARE coiled-coil homology domain-containing protein | 56.65 | 1.394 | 1.414 | 0.017 | |
| Protein synthesis, folding and degradation | W9SIU2 | 30S ribosomal protein S21 | 20.85 | 0.824 | 0.735 | 0.012 |
| W9QWL4 | 50S ribosomal protein L34 | 16.60 | 0.680 | 0.683 | 0.030 | |
| W9SXH8 | Carboxypeptidase | 58.05 | 1.335 | 1.451 | 0.012 | |
| W9SPH4 | Ribonuclease 3 | 23.25 | 2.357 | 2.638 | 0.015 | |
In the GO enrichment analysis of DAP in T50 vs. CK, DAP was significantly enriched into 66 functional GO terms, of which 40 belonged to biological processes (BPs), 25 to molecular functions (MFs), and 1 to cellular components (CCs). Among the top 20 enriched GO terms (Fig. 5A), carotenoid biosynthesis, tetraterpenoid biosynthesis, tetraterpene metabolism, carotenoid metabolism, lipid metabolism, and transcription elongation from the RNA polymerase II promoter were the most significantly enriched GO terms in the BP category. In the MF category, hydrolase activity (acting on ester bonds), polygalacturonase activity, secondary activity transmembrane transporter activity, nuclease activity and hydrolase activity were the most significantly enriched GO terms. Moreover, the most important term for GOs in the CC category is the extracellular region. GO enrichment analysis of the DAPs identified in T100 vs. CK resulted in a significant enrichment of 122 functional GO terms, of which 50 belonged to BPs, 46 to MFs, and 26 to CCs. In the top 20 enriched GO terms (Fig. 5B), cellular glucan metabolism, glucan metabolism, cellular polysaccharide metabolism, carotenoid biosynthesis, and abscisic acid-activated signaling pathway were the most significantly enriched GO terms in the BP category. Protein dimerization activity, protein heterodimerization activity, DNA binding, isoprenoid binding, alcohol binding, and abscisic acid binding were the most significantly enriched GO terms in the MF category. The most significantly enriched GO terms in the CC category were protein‒DNA DNA packaging complex, DNA packaging DNA packaging complex, nucleosome, chromatin and chloroplast part.
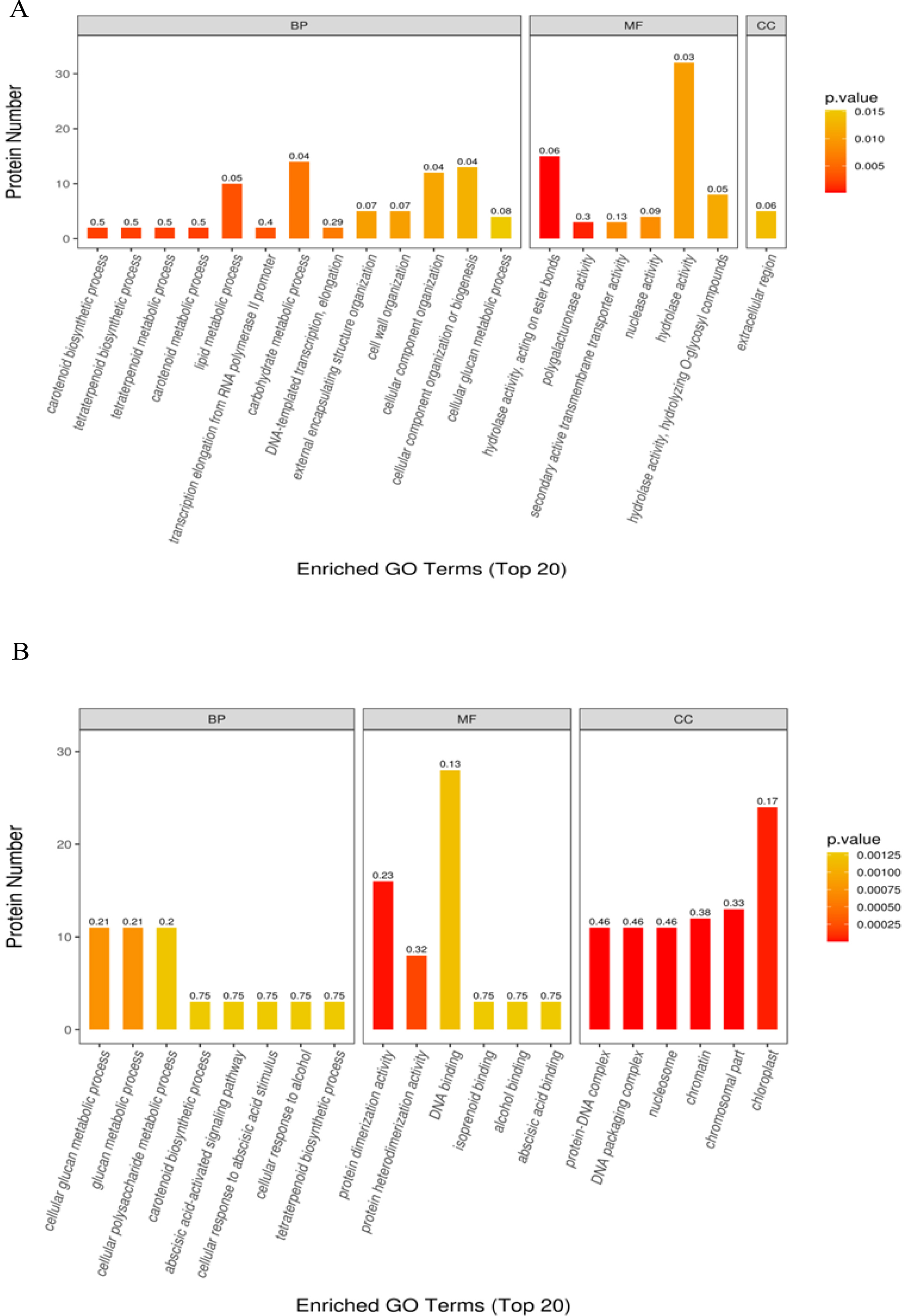 Fig. 5.
Fig. 5.Gene ontology annotation of all differentially abundant proteins identified in mulberry seeds. The top 20 significantly enriched GO terms in (A) T50 vs. CK and (B) T100 vs. CK.
Furthermore, DAPs were also annotated according to the KEGG pathway. KEGG enrichment analysis of T50 vs. CK DAPs showed that 55 pathways were enriched, including photosynthesis, chlorocyclohexane and chlorobenzene degradation, fluorobenzoate degradation, toluene degradation, and prodigiosin biosynthesis. Notably, only one pathway, carotenoid biosynthesis, was significantly enriched among these enriched pathways (Fig. 6A). In the KEGG pathway enrichment analysis of DAPs of T100 vs. CK, a total of 147 pathways were enriched, of which 7 were significantly enriched; these pathways mainly related to photosynthesis, systemic lupus erythematosus, carotenoid biosynthesis, alcoholism, mineral absorption, plant hormone signal transduction and necroptosis (Fig. 6B).
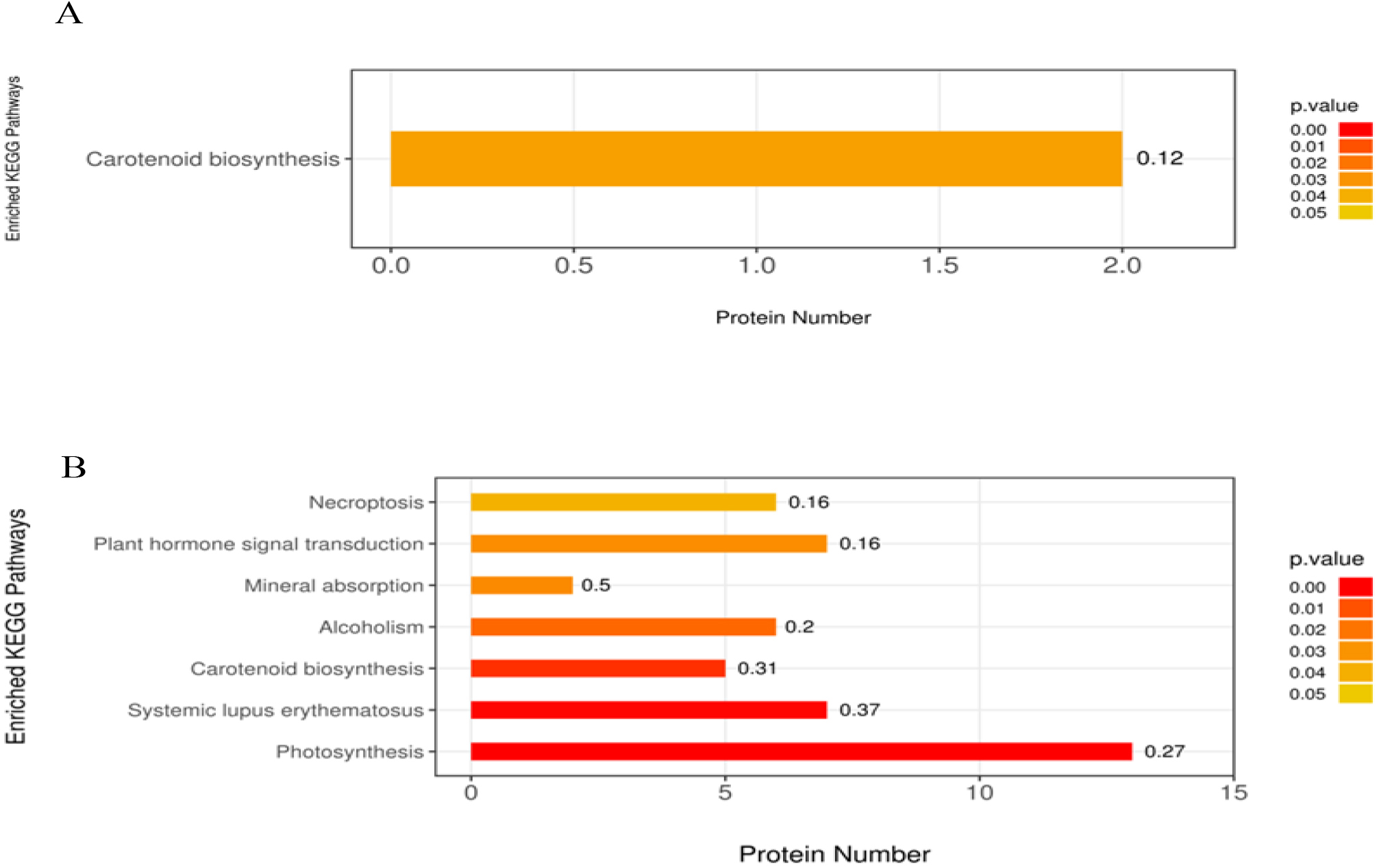 Fig. 6.
Fig. 6.KEGG pathway enrichment analysis of DAP in mulberry seeds. The significantly enriched KEGG pathways in DAPs from (A) T50 vs. CK, (B) T100 vs. CK. The abscissa is the number of proteins involved in metabolic pathway; the ordinate is metabolic pathway. The color gradient represents the p-value below 0.05. The darker the color (closer to red), the smaller the p-value, and the more significant is the enriched KEGG pathways. The number on the bar graph indicates the rich factor, referring to the percentage of differential proteins that are involved in this pathway.
To validate the TMT proteomic data, we selected five differentially abundant proteins for PRM quantification (Table 2). PRM identified five candidate differentially abundant proteins, including cellulose synthase, glyceraldehyde-3-phosphate dehydrogenase B, superoxide dismutase, glutathione peroxidase, and photosystem II CP43 reaction center protein. The PRM results exhibited consistency in the differential expression patterns of the selected proteins with the TMT data. In 50 mmol/L NaCl solution, the expression levels of glutathione peroxidase and superoxide dismutase increased significantly in PRM and TMT analysis, while the expression levels of cellulose synthase, glyceraldehyde 3-phosphate dehydrogenase B and photosystem II CP43 reaction center decreased significantly in PRM and TMT analysis. In the 100 mmol/L NaCl solution, the expression of cellulose synthase, glyceraldehyde-3-phosphate dehydrogenase B and photosystem II CP43 reaction center protein was downregulated in the PRM data and did not change according to the TMT results. Overall, the consistency of PRM and TMT results validates the credibility of TMT in proteomic analysis.
| Accession | Gene name | Description | TMT | PRM | ||
|---|---|---|---|---|---|---|
| T50/CK | T100/CK | T50/CK | T100/CK | |||
| W9QF53 | L484_012515 | Cellulose synthase | 0.829 | 0.830 | 0.835 | 0.455 |
| W9QWT8 | L484_024833 | Glyceraldehyde-3-phosphate dehydrogenase B | 0.795 | 0.713 | 0.682 | 0.175 |
| W9RZP5 | L484_013984 | Superoxide dismutase | 1.276 | 1.405 | 1.363 | 1.868 |
| W9QH65 | L484_024293 | Glutathione peroxidase | 1.268 | 1.361 | 1.016 | 1.508 |
| Q09X21 | psbC | Photosystem II CP43 reaction center protein | 0.827 | 0.783 | 0.648 | 0.196 |
Seed germination and seedling growth are among the most sensitive stages in plant life, representing the first contact with water and soil, whereas salt stress affects seed germination and seedling growth [47]. In the current study, we identified salt stress-responsive proteins during mulberry seed germination at the proteomic level by the TMT method. A total of 143 DAPs were identified when treated with 50 mmol/L NaCl solution, of which 61 were upregulated and 82 were downregulated, and 540 DAPs were identified when treated with 100 mmol/L NaCl solution, of which 222 proteins were upregulated and 318 were downregulated. A total of 113 DAPs were screened in the two salt treatments, of which 43 proteins were upregulated and 70 proteins were downregulated. Functional analysis of DAPs screened from mulberry seeds treated with salt stress showed that the differentially expressed proteins mainly belonged to ROS scavenging, stress defense, energy production, carbohydrate metabolism, transcription and translation, growth and development, signal transduction, substance transport, protein synthesis, folding and degradation. In the following section, we will discuss the differential proteins that fall into these functional categories.
During seed germination, respiration produces large amounts of ROS, and salt stress accelerates the accumulation of ROS, resulting in an imbalance between ROS production and detoxification. In response to this oxidative stress, an effective antioxidant system can scavenge excess ROS [48]. ROS can also contribute to the endosperm weakening during germination as a result of the cell wall loosening, but uncontrolled ROS production can lead to cell damage, resulting in seed deterioration, retarded germination and early seedling development, and the production of some antioxidants, such as SOD, POD, CAT, glutathione-related enzymes and dehydrogenases, to reduce ROS-induced cell damage in plants [49]. In this study, physiological data showed that the activities of SOD, POD, and CAT in mulberry seedling samples continued to increase under NaCl-induced salt stress. It has been reported that the activities of antioxidant enzymes, such as SOD, POD, CAT, and glutathione-related enzymes, generally increase in plants under oxidative stress conditions, resulting in enhanced tolerance to stress [50]. Proteomics research in this study showed that antioxidant-related proteins, such as peroxidase (W9QU60), superoxide dismutase (W9RZP5) and glutathione peroxidase (W9QH65), were significantly upregulated during seed germination under salt stress. Moreover, the changes in peroxidase, superoxide dismutase and glutathione peroxidase were higher under 100 mM NaCl treatment than under salt stress of 50 mM NaCl. These results may indicate that the plants suffer from relatively severe oxidative stress and need to initiate antioxidant enzyme systems to scavenge excessive ROS. Under the induction of 50 mM NaCl and 100 mM NaCl, the MDA content decreased from 1.39-fold to 3.81-fold compared with the control. MDA is mainly produced by polyunsaturated fatty acid peroxides and is an important indicator of cell membrane lipid peroxidation and plasma membrane damage caused by biotic and abiotic stresses [51]. In general, the lower the MDA content is, the less damage the plasma membrane incurs. The results of this study showed that the damage to the plasma membrane in mulberry seeds was relatively mild, indicating that mulberry seedlings exhibit significant salt tolerance under salt stress.
Salt stress can induce the production of ROS and damage plant cells, and to
regulate ROS levels, many defense proteins are induced during plant responses to
biotic or abiotic stresses [52]. In this study, in response to changes in ROS
scavenging and stress defense induced by salt stress, 15 DAPs (7 upregulated and
8 downregulated) were screened under salt stress (50 mM NaCl), while under 100 mM
NaCl salt stress, 40 DAPs were screened (24 upregulated and 16 downregulated); a
total of 13 DAPs were screened in both salt treatments (5 upregulated and 8
downregulated). Heat shock proteins (HSPs), also known as heat stress proteins,
play an important role in regulating the disease resistance and stress response
of plants [53]. sHSP is the plant heat shock protein with the smallest molecular
weight, and this protein mainly protects other proteins from various stresses in
plants through molecular chaperones. A large number of studies have shown that
sHSP members can respond to various external stresses [54]. Overexpression of the
rice sHSP member Os HSP16.9 can improve the salt resistance and drought
resistance of rice plants [55]. Overexpression of the sHSP member Ms HSP17.7 in
alfalfa can improve the defense ability of alfalfa plants against heat, drought,
and antioxidant stresses [56]. In this study, the expression of sHSP
domain-containing protein (W9T1J8) was upregulated, and the fold change ratio
increased with increasing salt concentration, which indicated that the
upregulated expression of sHSP domain-containing protein was a defense mechanism
of mulberry seed germination in response to salt stress. Furthermore, KEGG
enrichment analysis of DAPs showed that seed germination under salt stress was
mainly enriched in carotenoid biosynthesis. Carotenoids are natural pigments that
exhibit antioxidant activity, play an important role in the production of plant
hormones and can effectively protect plant cells [57]. Lycopene cyclization is an
important part of the carotenoid biosynthetic pathway in plants and is catalyzed
by both lycopene
In plants, glycolysis and the tricarboxylic acid (TCA) cycle are the main features of carbohydrate and energy metabolism, which not only satisfy energy demands but also produce many essential cofactors and other substrates of metabolism [60]. In this study, to examine changes in energy production and carbohydrate metabolism induced by salt stress, 26 DAPs (9 upregulated and 17 downregulated) were screened under a salt stress of 50 mM NaCl, 77 DAPs (31 upregulated and 46 downregulated) were screened under a 100 mM NaCl salt stress, and a total of 17 DAPs (8 upregulated and 9 downregulated) were screened under both salt treatments. Glyceraldehyde 3-phosphate dehydrogenase catalyzes the conversion of 3-phosphoglyceraldehyde to 1,3-diphosphoglyceric acid and plays an important role in sugar metabolic pathways in plants [61]. Phosphoglycerate translocase plays an important role in glycolysis by catalyzing the conversion of phosphate from 3-phosphoglycerate to a second carbon to form 2-phosphoglycerate. In this study, the proteomic analysis results showed that related proteins involved in glycolysis and the tricarboxylic acid cycle, including short-chain type (W9RFS1), 2,3-diphosphoglycerate nondependent phosphoglycerate translocase (W9SCE4), 3-phosphoglyceraldehyde dehydrogenase (W9SF99), and phosphoenolpyruvate carboxylase 2 (W9RNP2), were significantly upregulated, while fructose diphosphate aldolase (W9RMK1) was significantly downregulated, indicating that when most identified glycolytic enzymes exhibited increased expression, the production of more NADH was promoted during mulberry seed germination under salt stress; then, the oxidative phosphorylation process by mitochondria began for ATP synthesis, providing the necessary energy for normal cell metabolism under salt stress and playing an important role in the process of improving salt tolerance of mulberry. Moreover, seeds require energy for germination and growth, and lipids are the most efficient form of energy storage in plant seeds [62]. Plant lipases are responsible for the hydrolysis of acylglycerides during lipid processing and can hydrolyze the oil stored in the seeds to provide energy and protection [63]. The expression of lipase (W9S8G4) was significantly upregulated in this study, indicating that this lipase may have enhanced the salt tolerance of mulberry seeds by providing energy and thus promoting germination. It has been found that the overexpression of Arabidopsis lithium-tolerant lipase increased salt tolerance in transgenic Arabidopsis plants, allowed seed to germinate in the presence of NaCl, and stimulated nutritional growth, flowering, and fruit set in the presence of NaCl [64]. Photosynthesis is a fundamental process that promotes the nutritional growth and development of plants, providing the necessary energy for seed germination and early seedling establishment [65]. It has been shown that during seed development, photosynthesis plays an important role in seed maturation; furthermore, inhibition of photosynthesis in seeds results in delayed germination and reduced seed storability, as well as reduced seedling vigor [66]. In this study, proteins related to photosynthesis cytochrome f (Q09X04), chloroplast heme oxygenase (A0A076K090) and oxygen precipitation enhancing protein 3-2 (W9QPP1) were significantly downregulated, indicating that photosynthesis-related proteins may be inhibited by salt stress during seed germination.
In addition, carbohydrate metabolism regulates the synthesis and conversion of
sugars, and environmental stresses can disrupt carbohydrate metabolism in plant
species [67]. It has been reported that glycoside hydrolase is among the key
enzymes in carbohydrate metabolism, which can affect cellulose biosynthesis, such
as
In this study, the changes in transcription, translation and growth and
development caused by salt stress were examined, and 32 DAPs were screened under
50 mM NaCl stress treatment (11 upregulated and 21 downregulated), 116 DAPs (50
upregulated and 66 downregulated) were screened under 100 mM NaCl salt stress
treatment, and a total of 22 DAPs were screened in both salt treatments (10
upregulated and 12 downregulated). Seed germination is a complex trait influenced
by many genetic, endogenous and environmental factors and is driven by many
cellular processes, including transcription and translation [71]. Proteins
involved in the regulation of transcriptional and translational activities are
important because oxidative stress induced by high NaCl concentrations may affect
protein integrity, indicating that the production of new proteins is essential to
maintain plant growth [72]. In this study, we found that various transcription
and translation factor-related proteins were significantly upregulated,
especially under 100 mM NaCl stress, including transcription factor VIP1
(W9RA57), nuclear transcription factor Y subunit C-9 (W9QV28), translation
initiation factor IF-2 (W9RQB9), and eukaryotic translation initiation factor 5A
(W9S2P4). This indicates that salt stress may affect the process of initiating
protein translation during seed germination in Morus alba. It has been
shown that under stress conditions, epigenetic changes (i.e., DNA methylation and
histone modifications can regulate the expression of important genes associated
with stress tolerance and can activate the transcription of genes that respond to
abiotic stresses [73]. Histones can be involved in promoting the unwinding,
replication and transcription of DNA as well as stabilizing chromatin structure
under salinity [60]. It has been reported that SRT-like histone deacetylases in
Arabidopsis inhibit transcription, thereby reducing excessive
H
Studies have shown that GDSL esterase/lipase is widely distributed in plants, is mainly involved in the regulation of growth and development, and plays an important role in seed germination [76, 77]. It has been reported that GDSL esterase/lipase is overexpressed in transgenic plants [78]. The overexpression of Arabidopsis LTL1, a salt-inducible gene encoding a GDSL motif lipase, also improved salt tolerance in yeast and transgenic plants [79]. The expression levels of GDSL esterase/lipase (W9R8E1) and GDSL esterase/lipase 6 (W9RD56) were significantly upregulated in this study, indicating that these proteins may promote the tolerance of mulberry seed germination to salt stress. Aspartic proteinases (APs) are involved in the degradation of storage proteins, hydrolyze seed storage proteins to provide amino acids for seed germination, and play an important regulatory role in plant growth and development, disease resistance, and adversity stress processes [80]. Studies have shown that the Fagopyrum esculentummoench AP member Fe AP9 is upregulated in response to darkness, drought and salicylic acid stress [81]. Overexpressing the grape AP gene VlAP17 can improve salt tolerance and drought tolerance by protecting the integrity of the plasma membrane and increasing the root quality of seedlings [82]. In this study, the expression of aspartic proteinase nepenthesin-2 (W9RZ71) and bark storage protein A (W9RDI0) were significantly upregulated, which indicated that the salt tolerance of mulberry seeds might be enhanced by hydrolyzing and storing protein during germination.
In this study, we studied the changes in signal transduction and substance
transport caused by salt stress. We screened 14 DAPs (7 upregulated and 7
downregulated) under salt stress (50 mM NaCl) and 51 DAPs (16 upregulated and 35
downregulated) under salt stress (100 mM NaCl), while 17 DAPs (7 upregulated and
10 downregulated) were screened in two salt treatments. Seed germination
determines the beginning of the seed plant life cycle, which is a complex process
that involves various signal transduction pathways [83]. Receptor-like kinase is
a common key signaling molecule in many stress reactions that can mediate the
response of plants to development and environmental stimuli [84]. Receptor-like
protein kinase may play a role in transducing extracellular information into
cells. The kinase consists of an extracellular domain, transmembrane domain and
cytoplasmic-located serine/threonine protein kinase domain [85]. Studies have
shown that the plant hormone abscisic acid (ABA) regulates seed germination,
plant growth and development, while protein kinase may positively regulate ABA
signal transduction in seed germination and seedling growth [86]. In this study,
many protein kinases related to signal transduction were found to be
significantly upregulated, including putative receptor-like protein kinase
(W9RBX0), receptor-like protein kinase HERK 1 (W9RFT8), putative serine/threonine
protein kinase (W9RNL9), serine/threonine protein kinase PRP4-like protein
(W9RJN3) and putative LRR receptor-like serine/threonine protein kinase (W9SN33),
suggesting that these proteins may play a critical role in the germination of
mulberry seeds. Under stress, plants induce transporters to transport signal
molecules to express some stress resistance genes and cope with adverse
environments [87]. In wheat roots under NaCl stress, several K
In this study, to examine the changes in protein synthesis, folding and
degradation caused by salt stress, we screened 7 DAPs (2 upregulated and 5
downregulated) under salt stress (50 mM NaCl) and 39 DAPs (9 upregulated and 30
downregulated) under salt stress (100 mM NaCl), while 5 DAPs (3 upregulated and 2
downregulated) were screened under two salt treatments. Proteins enable life
activities, and when seeds germinate, the change in soluble protein content can
reflect various information on protein synthesis, denaturation and degradation in
cells. In many plants, such as Arabidopsis [97], Ricinus
communis [98] and Phoenix dactylifera [99], protein folding and
stability involve protein synthesis, including ribosomal proteins. Ribosomes
consist of two parts, namely, ribosomal protein (RP) and ribosomal RNA. RP not
only maintains the configuration of RNA but also participates in the synthesis,
transportation and localization of proteins [100]. Studies have shown that under
Na
Studies have shown that ubiquitin-binding enzymes are highly involved in proteolysis, protein modification and protein degradation, including abiotic stress, and play an important role in all aspects of plant growth, development and cell processes [105]. It has been reported that overexpressing the soybean ubiquitin-binding enzyme gene GmUBC2 can enhance salt tolerance by regulating the expression of abiotic stress-responsive genes in Arabidopsis thaliana [106]. DnaJ plays an important role in the NaCl tolerance of Arabidopsis thaliana and is a molecular chaperone involved in many cellular processes, including the regulation of protein folding, protein degradation, protein activity and protein aggregation prevention [107]. In this study, proteins related to protein folding and degradation were significantly upregulated, including ubiquitin-binding enzyme E2 27 (W9RHB5), DnaJ-like protein (W9QJ41) and peptidyl prolyl cis-trans isomerase (W9RAD0). This result indicated that the folding and stability of these proteins might be enhanced to cope with the salt tolerance of mulberry seed germination.
This study explored the response mechanism of mulberry seed germination to salt stress at the physiological and proteomic levels. Physiological data showed that salt stress inhibited the germination rate and radicle length of mulberry seeds, decreased the content of MDA, and significantly increased the activities of SOD, POD and CAT. Then, the TMT marker technique was used to analyze the protein group of mulberry seeds in two salt treatment stages, and 76,544 unique peptides were detected. After removing duplicate proteins, 7717 proteins were identified according to TMT data. Through proteomic analysis, we obtained 143 (50 mM NaCl) and 540 (100 mM NaCl) DAPs associated with mulberry seed germination. Furthermore, a total of 113 DAPs were treated with 50 mM NaCl and 100 mM NaCl solutions, of which 43 were upregulated and 70 were downregulated. KEGG analysis showed that the proteins involved in photosynthesis, carotenoid biosynthesis and plant hormone signal transduction were usually enriched in mulberry seeds. Finally, five differential proteins were verified by PRM, which validated the credibility of TMT in proteomic analyses. The data presented herein provide new insights into plant salt-responsive proteins, with potential implications for enhancing the salt tolerance of mulberry plants. Future research requires further analysis at the RNA and RT-PCR levels for deeper molecular level studies to provide a more in-depth functional study of the genes and proteins involved in salinity tolerance.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
YW conceived and designed this study; YW, WJ, JC, WG and YL performed the experiments, interpreted the results and wrote the article; YW, WJ, WG, YL and CL revised the article and analyzed the data. All the authors read and approved the final version.
Not applicable.
Not applicable.
This work was supported by Guangdong Provincial Department of Education Youth Innovative Talents Project (No. 2019KQNCX050), Guangzhou Basic and Applied Basic Research Foundation (No. 202201011164), Forestry Scientific technology Innovation Project of Guangdong Province (No. 2021KJCX015) and Scarce and Quality Economic Forest Engineering Technology Research Center (No. 2022GCZX002). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.