
- Academic Editors
†These authors contributed equally.
§ These authors contributed equally.
Alpha-synuclein inclusions are the distinctive trait of brain areas affected by neurodegeneration in Parkinson’s disease (PD). Nevertheless, PD is now considered as a multisystemic disorder, since alpha-synuclein pathology has been described also outside the central nervous system. In this regard, the early, non-motor autonomic symptoms point out an important role for the peripheral nervous system during disease progression. On this basis, we propose a review of the alpha-synuclein-related pathological processes observed at peripheral level in PD, starting from molecular mechanisms, through cellular processes to systemic modifications. We discuss their relevance in the etiopathogenesis of the disease, suggesting they are concurrent players in the development of PD, and that the periphery is an easily-accessible window to look at what is occurring in the central nervous system.
Parkinson’s disease (PD) is a common progressive neuropathological condition clinically characterized by resting tremors, bradykinesia, postural instability and general rigidity. These cardinal manifestations are due to a depletion in the dopaminergic neuronal subpopulation of the Sӧmmering’s substantia nigra, a basal ganglia structure located in the midbrain that projects to the striatum, establishing the so-called nigro-striatal pathway, a trait crucially involved in the circuits that ensure control and modulation of movement [1]. At brain level, PD is characterized by the presence of proteinaceous inclusion bodies within neuronal soma and neuropil, named Lewy bodies and Lewy neurites, respectively, which constitute the hallmark of the pathology and whose main component was found to be alpha-synuclein [2]. The discovery of the A53T mutation in the SNCA gene encoding for alpha-synuclein as the cause of a familial form of the disease also supported the crucial involvement of this protein in PD pathogenesis [3]. Alpha-synuclein is a natively unfolded protein that owes its name to its pre-synaptic and nuclear localization [4]. Human alpha-synuclein belongs to the synuclein family, which also includes beta- and gamma-synuclein. It is constitutively expressed in the nervous system, accounting for 1% of total neuronal cytosolic proteins, and in the brain, where it is predominantly found in the neocortex, hippocampus, substantia nigra, and corpus striatum [5, 6]. Although the physiological role of alpha-synuclein has not been completely discovered, the known list of its interacting partners is abundant and includes lipid membranes, synaptic vesicles, tubulin, SNARE complex protein and proteins involved in calcium regulation and dopamine homeostasis [7] thus suggesting it could be involved in the modulation of neurotransmission, working on vesicle release and trafficking.
Together with PD, further pathologies were found to feature alpha-synuclein-positive inclusions, namely multiple system atrophy and Lewy body dementia, and thus they were classified as synucleinopathies [8]. Nevertheless, it is still a matter of debate whether alpha-synuclein aggregates are toxic, protective, or are an epiphenomenon related to progressive failure of cellular clearance mechanisms for aggregated proteins [9, 10].
The proposed molecular mechanisms involved in PD pathogenesis include alteration of the microtubular cytoskeleton, synaptic dysfunctions, mitochondrial impairment, defective autophagy, impaired protein turnover and clearance, oxidative stress, and inflammation [11, 12, 13]. Interestingly, alpha-synuclein pathology was reported in multiple body regions and organs of patients affected by synucleinopathy, both in autoptic and living individuals [14, 15], suggesting that it could also affect peripheral tissues. In addition, the early-onset non-motor symptoms that feature the pathology include a wide range of autonomic manifestations such as hyposmia, hypotension, sleep disorder and gastrointestinal symptoms [16]. This paved the way to redefining PD as a multisystemic disorder and, in such a scenario, the study of the pathology in the periphery provides an easily-accessible window to the central nervous system, making it possible to evaluate pathological processes that may be mirrored in the brain, thus allowing to investigate the mechanisms underlying the disease’s pathogenesis. Consequently, in this review we describe the pathological processes reported at the peripheral level in PD, microtubules, synapses, mitochondria, autophagy, protein clearance and inflammation, and we discuss their link to alpha-synuclein pathology and relevance to the etiopathogenesis of the disease.
Microtubules (MTs) are highly dynamic polymers of
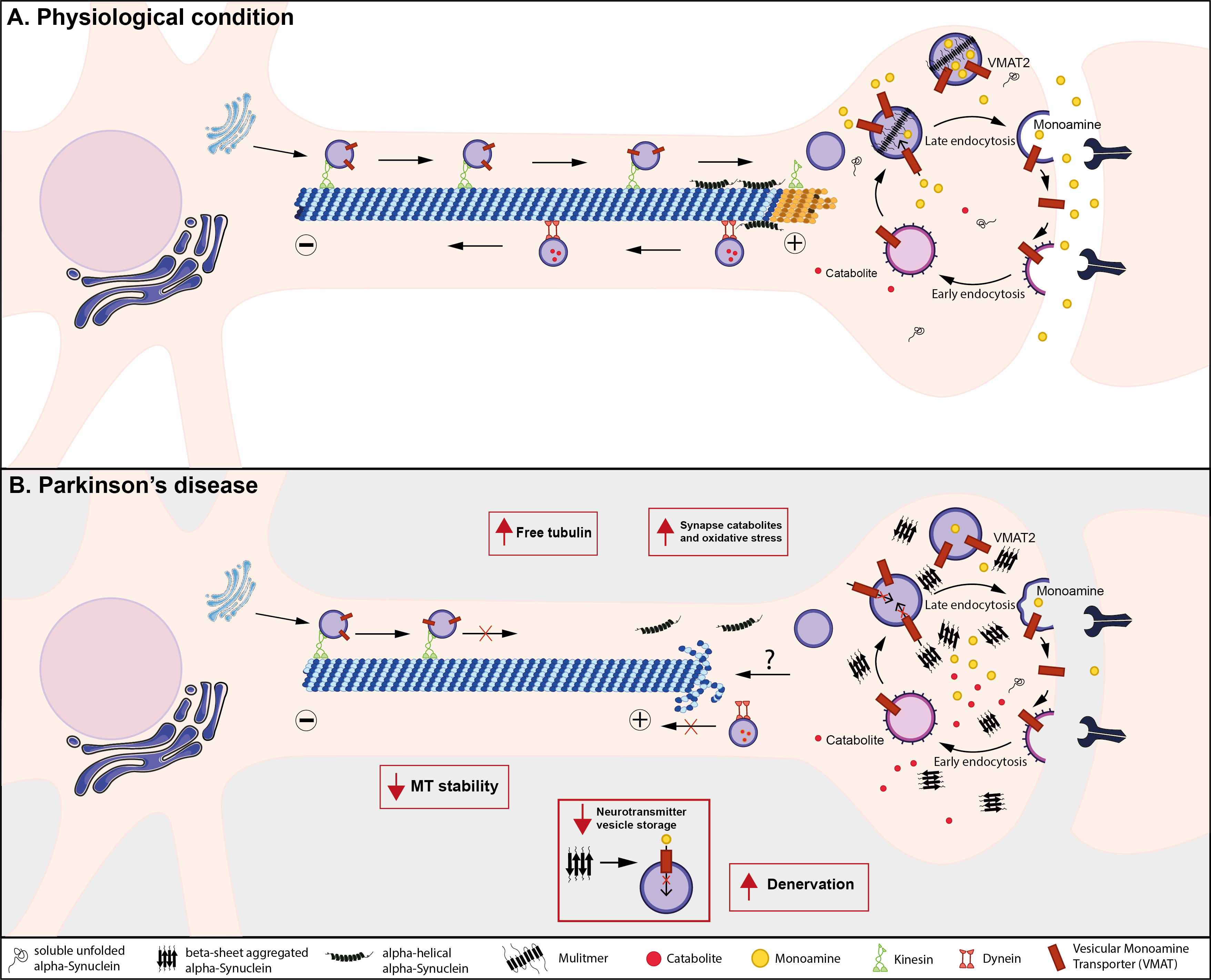 Fig. 1.
Fig. 1.Suggested model for alpha-synuclein impact on microtubule homeostasis and synaptic functioning in PD. (A) Under physiological conditions, MTs are conventional and well-organized; vesicles use MTs as tracks to reach the synaptic boutons from the cell body, moving toward the distal tip of the axon in an anterograde movement. Alpha-helical alpha-synuclein interacts with MTs at the neuronal growth cone. In the synaptic compartment, vesicular monoamine transporter 2 (VMAT2) loads presynaptic vesicles with monoamines, maintaining the releasable pool as well as the recycling pool. The retrograde movement sustains catabolite clearance, avoiding harmful interactions with the synaptic components. (B) In Parkinson’s disease, alpha-helical alpha-synuclein may lose its interaction with MTs and alpha-synuclein aggregation leads to MT destabilization, thus impairing transport towards and away from the synapses. This could trigger the accumulation of reactive catabolites and/or aberrant proteins, likely increasing oxidative stress damage at the synaptic level. Additionally, the alteration of VMAT2 functioning by a possible direct interaction with alpha-synuclein aggregates could reduce the extent of the neurotransmitter vesicular storage and releasable pool which lead to synaptic transmission defects and, eventually, to neuronal death.
Given their importance in neuron physiology, numerous studies have demonstrated
how microtubule impairment can lead to neurodegenerative disorders and in
vitro and in vivo studies have indicated that microtubule dysfunction
is linked to the pathogenesis of PD (Fig. 1B) [7, 18]. First of all, Lewy bodies,
the pathological hallmark of PD, have been found to contain a large number of
cytoskeletal proteins, including tubulin, MAPs, and neurofilaments, suggesting
cytoskeletal alterations in PD [19, 20]. More recent studies have offered novel
insights into the molecular composition of Lewy bodies. Using STED (Stimulated emission depletion microscopy)-based super
resolution microscopy and correlative high-resolution imaging and biophysical
approaches, the presence of crowded membranous materials including membrane
fragments, dysmorphic mitochondria and structures resembling lysosomes, together
with cytoskeletal elements were revealed in Lewy bodies [21]. Interestingly, the
analysis of the subcellular rearrangement of the aggregated forms of
alpha-synuclein unraveled the presence of cytoskeletal elements such as tubulin
and neurofilaments at the periphery of Lewy bodies and suggested their role in
the morphogenesis of Lewy bodies [22]. Further evidence as to the role of
microtubules in PD pathogenesis come from genome-wide association studies in
familial events of the pathology, that identified mutations in
microtubule-related protein genes including MAPT [23], which is also
independent of SNCA mutations. Looking at toxin-based models of PD, the
parkinsonism-induced neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
(MPTP) and its toxic metabolite 1-methyl-4-phenylpyridinium (MPP
The interplay between microtubules and alpha-synuclein have gained increasing
interest over time. In 2001, Payton and colleagues [33] first reported the
co-immunoprecipitation of alpha-synuclein with both alpha- and beta-tubulin from
zebra finch and mouse brain homogenates, which was later confirmed also in rat
and hamster brain [34, 35, 36]. Besides, a study in HeLa cells reported that
alpha-synuclein co-localizes with MTs [36]. More recently, Cartelli and
colleagues demonstrated that alpha-synuclein forms a complex with an
Few data are actually reported on microtubule dysfunction in the periphery with
respect to PD. Toba and co-workers [39] demonstrated that bi-directional axonal
transport is severely affected in alpha-synuclein depleted rat dorsal root
ganglion neurons, and that, alpha-synuclein interaction with
Synaptic homeostasis is crucial to maintain correct neuronal functioning as well as to guarantee the proper transmission of information within the nervous system. Even though the exact physiological role of alpha-synuclein in neuronal cells has not yet been elucidated, its interaction with different proteins in the synaptic compartment and its prevalence in the pre-synapse point to a possible biological role as a wide regulator of synaptic activity in the central nervous system (CNS) [45, 46]. Among the physiological interactions proposed for alpha-synuclein, its activity as a mediator of the neuronal trafficking in the synapse stands out [47]. A series of discoveries have indeed outlined a role of alpha-synuclein as a Soluble N-ethylmaleimide Sensitive Factor Attachment Protein (SNAP) receptor (SNARE)-chaperone that helps SNARE-mediated vesicle-membrane fusion, thus promoting exocytosis in a physiological context. Further evidence found in the CNS indicates that other alpha-synuclein putative interactions with synaptic proteins are certainly worth noting, including the reuptake neurotransmitter proteins such as dopamine transporter (DAT) and the vesicular monoamine transporter 2 (VMAT2) [48, 49]. It has also been demonstrated that alpha-synuclein interacts with the family of the synapsins, a group of proteins largely implicated in synaptic plasticity, exocytosis and in the regulation of the vesicle releasable pool [50] (Fig. 1A). Experimental results were obtained by analyzing these pathways in a pathological background of alpha-synuclein mutation/overexpression or in the presence of misfolded aggregates, namely oligomers and fibrils, to elucidate how this could induce synaptic dysfunction throughout the CNS.
In a PD-related experimental setup, Garcia-Reitböck and colleagues [51] carried out a double staining immunohistochemistry between alpha-synuclein and the t-SNARE proteins SNAP-25 or syntaxin-1, and demonstrated their co-localization in the cerebral cortex of a PD-mouse model expressing full-length human mutant A30P alpha-synuclein. They found an accumulation and redistribution of SNARE proteins in the same transgenic mice compared to controls. In addition, to correlate this finding with the disease in humans, SNARE proteins repositioning was unraveled in the striatum of patients affected by early onset PD [47], hinting that alpha-synuclein mutations may have a harmful effect on synaptic vesicle exocytosis. Evidence in primary mouse cortical neurons and adult mouse hippocampal neurons revealed a dose-dependent reduction of pre-synaptic markers levels, and synaptic vesicle recycling upon recombinant alpha-synuclein administration [52]. These results are in line with the loss of synaptic terminals reported in the brain [53]. As mentioned, multiple interactions with synaptic transporters of monoamine have also been proposed as physiological functions of alpha-synuclein. Alterations in these putative modulation roles were suggested to be concurrent enhancers in the onset and progression of synaptic dysfunction. Indeed, the reduced total amount and the simultaneous clustered pattern of DAT, which co-localized in the caudate-putamen with alpha-synuclein and phospho-alpha-synuclein, was demonstrated by immunofluorescence labeling [54]. DAT/alpha-synuclein complexes were also detected by means of “in situ” proximity ligation assay (PLA), which showed a marked DAT redistribution in the caudate-putamen of PD patients [53]. Likewise, interactions with dopamine membrane transporters were reported after adeno-associated virus type 6 (AAV6) injection in the caudate-putamen of adult rats overexpressing human alpha-synuclein. Immunohistochemical analysis revealed axonal and synaptic swellings after 8–16 weeks, as well as inclusions positive for both alpha-synuclein and VMAT2 [55], suggesting an impairment in dopamine vesicle uptake and a cytosolic accumulation of this neurotransmitter, which may produce DA-derived, cytotoxic reactive species (Fig. 1B). Accordingly, in a similar model alpha-synuclein preformed fibrils (PFFs) were injected in the substantia nigra, and VMAT2 expression resulted significantly lower in those neurons where the phospho-alpha-synuclein pathology was more severe compared to controls who had not received PFFs injection [56]. Since alpha-synuclein is regarded to interact directly with lipid membranes [57], the effect of alpha-synuclein aggregation on membrane damage was also investigated. A covalent modification of alpha-synuclein by a dopamine catabolite (DOPAL) which provokes its oligomerization was noticed in vitro as well as in cellular models. Such aggregates were shown to induce dopamine leak through synaptic vesicle disruption or permeabilization and their effect was correlated with increased cell death [58]. All these effects, reported to happen when alpha-synuclein switches to create pathological small inclusions, further foster the idea of a putative detrimental role of oligomers and/or amyloid fibrils in membrane homeostasis that may ultimately contribute to neurodegeneration by causing increased oxidative stress, altered neurotransmitter storage and vesicle integrity (Fig. 1B).
Alpha-synuclein pathology seems to progressively move from the periphery to the spinal cord and brainstem in a centripetal movement [59, 60] by exploiting a prion-like diffusion through the neuronal network [61, 62]. The hypothesis was supported by analyzing patients’ peripheral tissues clearly reporting denervation [63, 64, 65] and alpha-synuclein or phospho-alpha-synuclein inclusions in autonomic neurons [15, 66, 67, 68, 69]. This peripheral degeneration may reflect the autonomic symptoms starting in the early stages of synucleinopathies. Although most synucleinopathies ultimately affect the CNS, little is known regarding the role that alpha-synuclein aggregation might play in the PNS. Recently, Challis and colleagues [70] showed gut dysfunction and motor symptoms in aged mice after PFFs duodenal seeding, showing a significant increase in brainstem phospho-alpha-synuclein and a reduced striatal dopamine in aged mice inoculated with PFFs, compared to PFF-inoculated younger WT mice or monomer-inoculated aged mice. Axonal phospho-alpha-synuclein aggregates and degeneration of the cardiac sympathetic nerve were already known to anticipate neuronal cell loss in the paravertebral sympathetic ganglia of patients affected by incidental Lewy body dementia [71], thus supporting the idea of a centripetal distal-driven degeneration in the peripheral nervous system in PD. This fits in the dying-back mechanism of cell death suggested for synucleinopathies, in which cell body damage occurs following synaptic dysfunction in a retrograde axonal degeneration [72, 73]. Interestingly, the impairment of the synaptic compartment in peripheral tissues from PD patients was linked to synuclein oligomers in skin biopsies by Mazzetti and colleagues [74]. The authors managed to evaluate the synaptic density, described as the ratio between the area of synaptic terminals and the total area of the targeted structure, recalling the procedure used for the assessment of denervation in PD samples [75]. By highlighting the synaptic compartment with synaptophysin, a reduced synaptic density was reported in 26% of PD patients, whereas only 3% of consecutive controls presented this feature, thus indicating an increased degeneration of peripheral synapses.
Additionally, in vivo non-invasive imaging techniques, such as
Since synaptic dysfunction was reported to precede neuronal death in patients, the impairment caused by alpha-synuclein aggregation in the synaptic compartment may assume a pivotal role in determining the onset of PD in the peripheral nervous system (PNS) and ultimately in the CNS. Together with the emerging evidence of synaptic dysfunction in human tissues, peripheral synapses might be considered as putative initiators or supporters of alpha-synuclein aggregation in the early stages of the pathology.
Impaired mitochondrial functioning has been associated with several disorders such as cancer and diabetes type-II, but also ageing and neurodegenerative diseases, including PD [82]. Multiple groups have reported the presence of alpha-synuclein in mitochondria [83, 84, 85], and, more specifically, localized at the mitochondria-associated endoplasmic reticulum membranes (MAM), a distinct domain of the endoplasmic reticulum [86] (Fig. 2A).
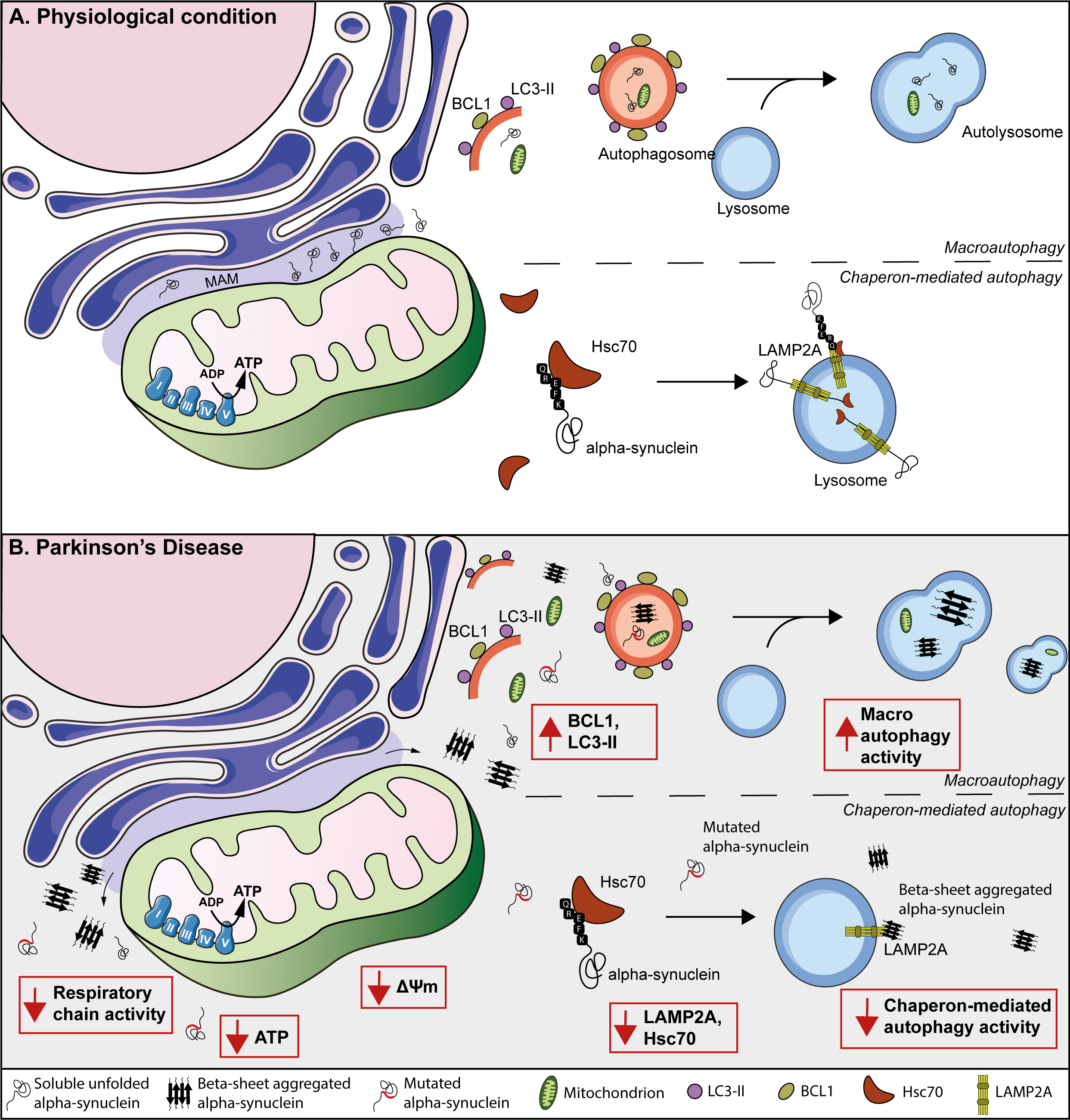 Fig. 2.
Fig. 2.Suggested model for mitochondria and autophagy contribution to PD evolution and progression in peripheral tissues. (A) Representation of the endoplasmic reticulum, the mitochondrial associated endoplasmic reticulum membrane (MAM), and a mitochondrion containing the mitochondrial respiratory chain. Physiologically, alpha-synuclein (black tangles) is localized at the MAM. At the same time, the macroautophagy machinery forms double-membrane organelles, called autophagosomes, mediated by LC3-II and BCL1. Then, autophagosomes incorporate cytosolic cargo, (including i.e., alpha-synuclein, mitochondria), fuse with lysosomes, and form autolysosomes to achieve cargo degradation. Besides, during chaperon-mediated autophagy, the KFERQ domain of the alpha-synuclein protein is recognized by Hsc70, which interacts with LAMP2A, and promotes its degradation via lysosomes. (B) In PD patients, mutated and/or oligomeric variants of alpha-synuclein affect its localization in MAM and move towards the cytosol. Moreover, these alpha-synuclein species causes mitochondrial dysfunction, including reduced mitochondrial complex I activity, decreased ATP production and, finally, a reduction in the mitochondrial membrane potential. PD peripheral tissue demonstrated an increase in autophagic structures (i.e., autophagosomes, lysosomes, and autolysosomes), suggesting that peripheral tissue of PD patients relies on an auto-activated compensatory mechanism of degradation. However, it might eventually be difficult to sustain this compensatory increase of autophagy, causing an elevation of alpha-synuclein aggregates, toxicity, and neuronal death. Moreover, reduced chaperone-mediated autophagy, including LAMP2A and Hsc70, has been observed in peripheral tissue derived from PD patients.
The first evidence of a link between mitochondrial dysfunction and PD came from
an observation in young drug addicts who developed parkinsonism after
accidentally injecting themselves with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [87]. Its toxic metabolite MPP
Mitochondrial dysfunction in PD is not only reported in the CNS but has also been shown and widely investigated in the periphery (Fig. 2B). A reduced activity of the respiratory chain’s complexes has been unraveled in multiple cell types from PD patients, including platelets, fibroblasts, lymphocytes and myocytes, suggesting a widespread reduction of mitochondrial activity in PD [97, 98, 99]. Chen and colleagues [100] revealed increased oxidative stress and elevated levels of leukocyte 8-hydroxydeoxyguanosine (8-OHdG) and malondialdehyde in blood plasma derived from PD patients. Likewise, Baumuratov and colleagues [101] reported aberrant mitochondrial morphology in colon biopsies from PD patients, reporting that colon ganglia from patients contained significantly more mitochondria per ganglion and exhibited a higher total mitochondrial mass compared to controls. Interestingly, expression of recombinant wild-type alpha-synuclein has also been shown to cause impaired mitochondrial complex I activity in human foetal enteric neurons [102].
Recently, morphometric mitochondrial changes have been demonstrated also in skin fibroblasts from individuals with idiopathic PD [103] together with an impaired mitochondrial membrane potential. Finally, the effect of alpha-synuclein mutations on mitochondrial dysfunction has also been investigated in human fibroblasts derived from a patient with parkinsonism carrying a triplication in the alpha-synuclein gene, which revealed a reduced complex I activity, decreased ATP production and a reduction in the mitochondrial membrane potential compared to controls [104].
In conclusion, these studies provide substantial evidence that mitochondrial dysfunctions are features of PD both in the CNS and in a broad range of different peripheral samples, including skeletal muscle, lymphocytes, platelets, blood plasma, skin fibroblasts and colon ganglia. This makes peripheral samples, naturally easily-accessible and less-invasive, a reliable option for the investigation of mitochondria- related dysfunctions in PD, considering that further research needs to be conducted into the role of alpha-synuclein in the physiology and pathology of mitochondria.
The main cellular mechanisms responsible for the degradation of large, long-lived proteins and cytoplasmic organelles, and, particularly, those that are toxic or damaged, are the ubiquitin-proteasome pathway (UPP) and autophagy [105, 106].
The impairment of both UPP and the autophagy machinery has been associated with the pathogenesis of neurodegenerative diseases, including PD [107]. Under physiological conditions, alpha-synuclein can be degraded via UPP in which it is tagged with ubiquitin molecules and transferred to the proteosome complex for ATP dependent degradation [106] as well as via the autophagic pathways (Fig. 2A) [108]. UPP failure has been reported in PD patients. In fact, in post-mortem human brains significantly decreased chymotrypsin-like and trypsin-like proteasome activity in the substantia nigra of PD patients compared to controls has been reported, together with a reduction in proteasome subunit levels. The observation that alpha-synuclein staining was more sensitive and specific than ubiquitin, indicates that alpha-synuclein aggregation precedes ubiquitination, that in turn promotes degradation by proteasome [8]. Proteasome dysfunction together with alpha-synuclein aggregates induce the involvement of both macroautophagy (Fig. 2A) and chaperone-mediated autophagy [109]. Previous studies have demonstrated that intracellular degradation of alpha-synuclein via the macroautophagic pathway is modulated, at least partly, by the autophagic regulator protein Beclin-1 [110, 111]. For chaperone-mediated autophagy, the alpha-synuclein KFERQ domain is recognized by hsp70, interacts with LAMP2A, and is subsequently degraded in the lysosome by proteases [111]. In 1997, Anglade and colleagues [112] already described autophagic alterations in PD, reporting an accumulation of autophagic vacuoles in nigral dopaminergic cells from post-mortem PD patients. Since then, growing evidence from pathological studies suggests that autophagy is dysregulated in PD. Key proteins, Beclin-1 and LC3II, involved in the formation and recognition of autophagosomes, respectively, are found to be increased in the substantia nigra of PD patients [113] and in brain samples from patients with Lewy body dementia [114]. Additionally, disrupted autophagosomes and endolysosomes have recently been found within Lewy bodies, thus suggesting the involvement of autophagic mechanisms in the formation of intracellular inclusions [115]. In contrast, proteins involved in chaperone-mediated autophagy, including chaperone Hsc70 and LAMP2A, are reduced in several brain areas of individuals diagnosed with PD [116, 117], suggesting that autophagy pathways are defective in PD brains. Substantial evidence has shown a link between autophagy and alpha-synuclein toxicity in synucleinopathies, including PD, Lewy bodies dementia, and multiple system atrophy [108]. First of all, LC3 is shown to co-localize with alpha-synuclein in Lewy Bodies and Lewy neurites in the substantia nigra of PD patients [116], and with Lewy bodies in the hippocampus [118] and temporal cortex [119] of people with Lewy body dementia. Co-localization of LC3 with phosphorylated variants of alpha-synuclein has been observed in glial cytoplasmic inclusions in patients affected by multiple system atrophy [120]. Moreover, histone deacetylase-6 (HDAC6), involved in the transport of misfolded proteins to the aggresome, also localizes with alpha-synuclein in Lewy bodies from PD and Lewy body dementia patients and in Papp Lantos bodies in multiple system atrophy [121, 122]. Together, these findings suggest an impaired clearance of autophagosomes in synucleinopathies. Notably, in neuronal cultures, wild-type alpha-synuclein can be degraded by autophagy [123, 124, 125], whereas mutated or structurally-changed variants impact on the degradation pathways, blocking the chaperone-mediated autophagy pathway, thereby, affecting protein degradation [123]. Interestingly, the blockage of chaperone-mediated autophagy by mutant A53T increases the activation of macroautophagy, although this compensatory result appears to have a negative effect causing neuronal cell death [124]. Furthermore, the inhibition of chaperone-mediated autophagy by 3-Methyladenine induces the formation of high molecule weight or detergent-insoluble oligomeric species of alpha-synuclein [125], thus suggesting that proper chaperone-mediated autophagy is important for the prevention and degradation of alpha-synuclein variants. Likewise, the failure of the autophagic machinery in clearing misfolded and/or aggregated alpha-synuclein has been proposed as a key mechanism in the formation of Lewy bodies [126]. More controversial results have been published concerning the pharmacological inhibition of macroautophagy. A study in PC12 cells demonstrated an expected increase in wild-type alpha-synuclein after inhibiting macroautophagy with 3-Methyladenine, whereas others reported that 3-Methyladenine macroautophagy inhibition results in a significant increase of mutant A53T but not wild-type alpha-synuclein [127]. Furthermore, pharmacological stimulation of macroautophagy, using rapamycin, significantly increases the clearance of wild-type, A30P, and A53T alpha-synuclein. Together, these somewhat controversial results suggest that macroautophagy is involved in the degradation of both wild-type and mutated variants of alpha-synuclein. A more recent study proved that the accumulation of alpha-synuclein also led to increased damage to the integrity of lysosomal membranes, and that a mutation of the pore-forming lysosomal protein TMEM175 is associated with an increased loss of dopaminergic neurons and an increased formation of alpha-synuclein pathological variants [128]. As regards studies on human-derived cells, few data are available regarding peripheral aspects of UPP impairment. In blood mononuclear cells (PBMCs) and blood lymphocytes collected from PD patients a reduced activity of the 20S proteasome subunit was observed, with an inverse correlation with disease duration and severity, with no differences in ubiquitin levels [129].
Prigione and colleagues [130] identified the induction of the macroautophagy response in PBMCs of PD patients compared to controls. Moreover, in line with results from the CNS, studies reported elevated levels of LC3II, and reduced levels of Hsc70 and LAMP2A in PBMCs obtained from individuals with PD [131, 132, 133, 134], suggesting a potential dysregulation of autophagy also in the periphery of PD patients (Fig. 2B). Furthermore, a whole-transcriptome assay revealed mRNA downregulation of 6 regulators involved in the formation of the autophagosome (i.e., ULK3, ATG2A, ATG4B, ATG5, ATG16L1, HDAC6), whereas protein levels of upstream autophagy markers (i.e., ULK1, Beclin-1, autophagy/beclin-1 regulator 1) were elevated in PD-derived PBMCs [135]. The levels of some autophagy-related proteins were significantly correlated with mutant alpha-synuclein, but, even more importantly, all autophagy-related proteins demonstrated a strong correlation with oligomeric alpha-synuclein. This suggests that alpha-synuclein, and, in particular, the oligomeric variants, may influence the macroautophagic pathway. Besides PBMCs, one study investigated the role of macroautophagy on PD in skin fibroblasts from patients with late-onset PD [136]. They displayed a striking increase of autophagosomes and autolysosomes in PD fibroblasts compared to healthy controls. Interestingly, pharmacological blockage of macroautophagy resulted in an even greater autophagic flux, with a widespread collection of macroautophagic structures.
Supporting the hypothesis that clearance mechanisms are key players in the etiopathogenesis of the disease, some familial forms of PD can be ascribed to mutations in genes involved in the autophagic machinery. Mutations in the LRKK2 gene are known to be the most common cause of dominant familial PD (2% of sporadic PD events) [137], affecting the autophagic pathway [138] and increasing the propensity to accumulate alpha-synuclein [139]. Ho and colleagues [140] demonstrated that the LRRK2 R1441G mutation in mouse embryonic fibroblasts induces more perinuclear clustering of lysosomes, whose redistribution indicates impaired degradation of aberrant proteins. Similarly, the same redistribution was also observed in aged LRRK2 R1441G knock-in mouse brain. Furthermore, the rate of cellular clearance of alpha-synuclein using the photoactivatable fluorescence-based cell assay in knock-in mouse embryonic fibroblasts was significantly lower than the wild type one [140].
According to genome wide association studies, mutations in the GBA1 gene, encoding for glucocerebrosidase, are the most common genetic risk factor for PD. This mutation is causative for Gaucher disease, a lysosomal storage disorder [141], and a decreased enzyme activity has been reported in PD patients with or without GBA1 mutations [142]. Emerging experimental evidence both in the central and peripheral nervous systems suggests a correlation between this decreased activity and alpha-synuclein accumulation [143, 144, 145]. In particular, the L444P mutation resulted in the exacerbation of both motor and gastrointestinal deficits found in the A53T mouse model of Parkinson’s disease [145].
In conclusion, these studies suggest that the impairment of the autophagy machinery, characterising PD affected brains is also present at the peripheral level.
A neuroinflammatory environment is a common feature shared by neurodegenerative diseases, including PD. Indeed, mutations in immune system-related genes, namely LRRK2, DJ-1 and HLA-DR, are known to be responsible for familial forms of PD [146]. Initial signs of neuroinflammation found in brains affected by synuclein-related pathologies include an extensive activation of microglial cells and a high density of astrocytes, as well as the infiltration of blood-derived mononuclear phagocytes and T lymphocytes. In addition, a significant increase in levels of pro-inflammatory cytokines (e.g., IL-1, IL-6 and TNF) has been described in the brain and CSF of PD patients [147, 148]. In support of this, the use of non-steroidal anti-inflammatory drugs (NSAIDs) has been associated with a decreased risk of developing PD, despite the fact they are inefficient in the treatment of the late stages of the disease, thus suggesting an early involvement of neuroinflammatory processes in its pathogenesis [149]. However, the topic is controversial and neither epidemiological studies nor meta-analyses have demonstrated that NSAIDs decrease the risk of PD or modify disease progression [150].
Universal consensus as to the actual contribution of inflammation to neurodegeneration is still lacking, and it remains unclear whether alpha-synuclein pathology is a cause or consequence of a neuroinflammatory environment. Indeed, microglia activation is reported to be triggered by misfolded, nitrated and aggregated alpha-synuclein, while inflammatory processes are known to elicit an oxidative environment that can, in turn, enhance protein misfolding, aggregation and cell death [151]. Studies in vitro and in animal models suggest that the extracellular aggregated and misfolded alpha-synuclein is recognized by microglial Toll-Like receptors, triggering protein uptake, microglial activation and inflammatory response prior to neuronal loss [152].
Intriguingly, an unexpected relationship seems to exist between peripheric inflammation and neurodegenerative diseases. In particular, there is a noteworthy overlap between genetic variants linked to PD and gut disorders and, likewise, inflammatory processes of the gastrointestinal tract have been reported to increase the risk of PD [153]. In addition, dysbiosis of gastrointestinal microbiota has been described in synucleinopathies as well as in several CNS disorders, and linked to the inflammatory environment observed in disease [154, 155, 156]. Similarly, population studies have shown that PD patients exhibit an increased prevalence of dermatological inflammatory disorders associated with bacterial colonization [157, 158]. However, no studies have questioned the correlation between skin microbiota and neurodegenerative diseases, and the few investigations into other vulnerable sites like oral and nasal mucosa showed no significant association [159]. On the contrary, a study on wild type mice reports that intraperitoneal injection of bacterial lipopolysaccharides (LPS) together with alpha-synuclein strains results in leukocytes infiltration and increased microglial population at the central level [160]. In the model proposed by the authors, LPS-activated inflammatory monocytes could internalize alpha-synuclein and move to the brain promoting its propagation at the central level. Similar indications arose from studies in synuclein-overexpressing mouse models. Sampson and colleagues [161] described an enhanced neuroinflammation as well as worsened motor deficits and increased synuclein pathology in mice colonized with microbiota derived from PD patients. In addition, in a study by Kishimoto and colleagues [162], mild gut inflammation is reported to reduce the age of PD onset and increase motor deficits and inflammation markers in mouse brain. At the same time, both synuclein and phospho-synuclein immunoreactivity are increased in the brain and gut, while a selective decrease in the neuronal dopaminergic population has been found in the substantia nigra. Consistently, alpha-synuclein pathology has been proven to spread from the gut to brain through the vagal nerve and reach the dorsal motor nucleus of the vagus [163] and, at the same time, LPS-mediated inflammation seems to enhance blood-brain barrier permeability, facilitating alpha-synuclein transit towards the brain [164]. The putative involvement of peripheral inflammation in neurodegenerative disease has been investigated also in living patients. Devos and colleagues [165] collected ascending colon biopsies from PD patients and age/sex matched healthy controls, who were tested for inflammatory status and alpha-synuclein deposition. The authors reported a significant increase in mRNA levels of pro-inflammatory cytokines (TNF-alpha, IL-6 and IL-1-beta) and a greater extent of glial markers, glial fibrillar acid protein and sox-10 in PD samples. Furthermore, two-thirds of the biopsies from PD patients resulted positive for phospho-alpha-synuclein immunostaining. Interestingly, levels of mRNAs displayed a negative correlation with disease duration and no correlation with disease severity (evaluated using the UPDRS scale) or enteric synuclein pathology, thus suggesting that inflammatory processes are involved predominantly in the early phases of PD and that the gastrointestinal tract might be the priming site of disease onset. An interesting study by Stolzenberg and colleagues [166] investigated the relationship between alpha-synuclein and gut inflammation in a pediatric cohort of 42 children with upper gastrointestinal pathologies (i.e., duodenitis, gastritis, Helicobacter pylori, reactive gastropathy) with no CNS disease. They evaluated by immunohistochemistry the extent of synuclein pathology and inflammatory status in endoscopic biopsies. Interestingly, the severity of inflammation, rated as neutrophil and mononuclear leukocyte density, was found to correlate with the enteric alpha-synuclein burden and, in addition, both monomeric and aggregated species of alpha-synuclein were proven to be chemotactic towards neutrophils and monocytes and to promote dendritic cell maturation. In support of their data, the authors reported that biopsies collected before the onset of infections were devoid of synuclein deposits and that follow-up in patients with acute and non-chronic infection showed a decreased gastrointestinal alpha-synuclein. The association between peripheral inflammation and neurodegeneration was directly tested in a noteworthy study by Moghaddam and colleagues [167], who investigated a cohort 388 drug-naïve PD patients and 148 controls. The authors assessed nigrostriatal dysfunction by DAT Single Photon Emission Computed Tomography (SPECT) imaging and evaluated the inflammatory status, rated as neutrophil to lymphocyte ratio, in patients’ blood samples. As expected, the inflammatory status was significantly higher in PD subjects compared to controls and, intriguingly, it was negatively correlated with the severity of nigrostriatal dysfunction. This is coherent with the observation that PD progression is reported to not follow a linear model, and striatal neurons depletion is faster in the early stages of the disease, while a way slower progression is observed in advanced stages [168]. Taken together these results endorse the hypothesis that an inflammatory environment is required in the initial phase of neurodegenerative diseases. Alpha-synuclein, in turn, could act as a mediator for immune cell recruitment and trigger an enhanced inflammatory response, both in the periphery and in the CNS, through microglial activation. In addition, the finding of blood-derived leukocytes in affected brain suggests an increased blood brain barrier permeability which increases the transport of misfolded and aggregated alpha-synuclein towards the brain. These pathways, summarized in Fig. 3, can potentially act together in the neurodegenerative process in a non-exclusive fashion, resulting in a systemic inflammatory state and alpha-synuclein deposition both in the periphery and in the CNS.
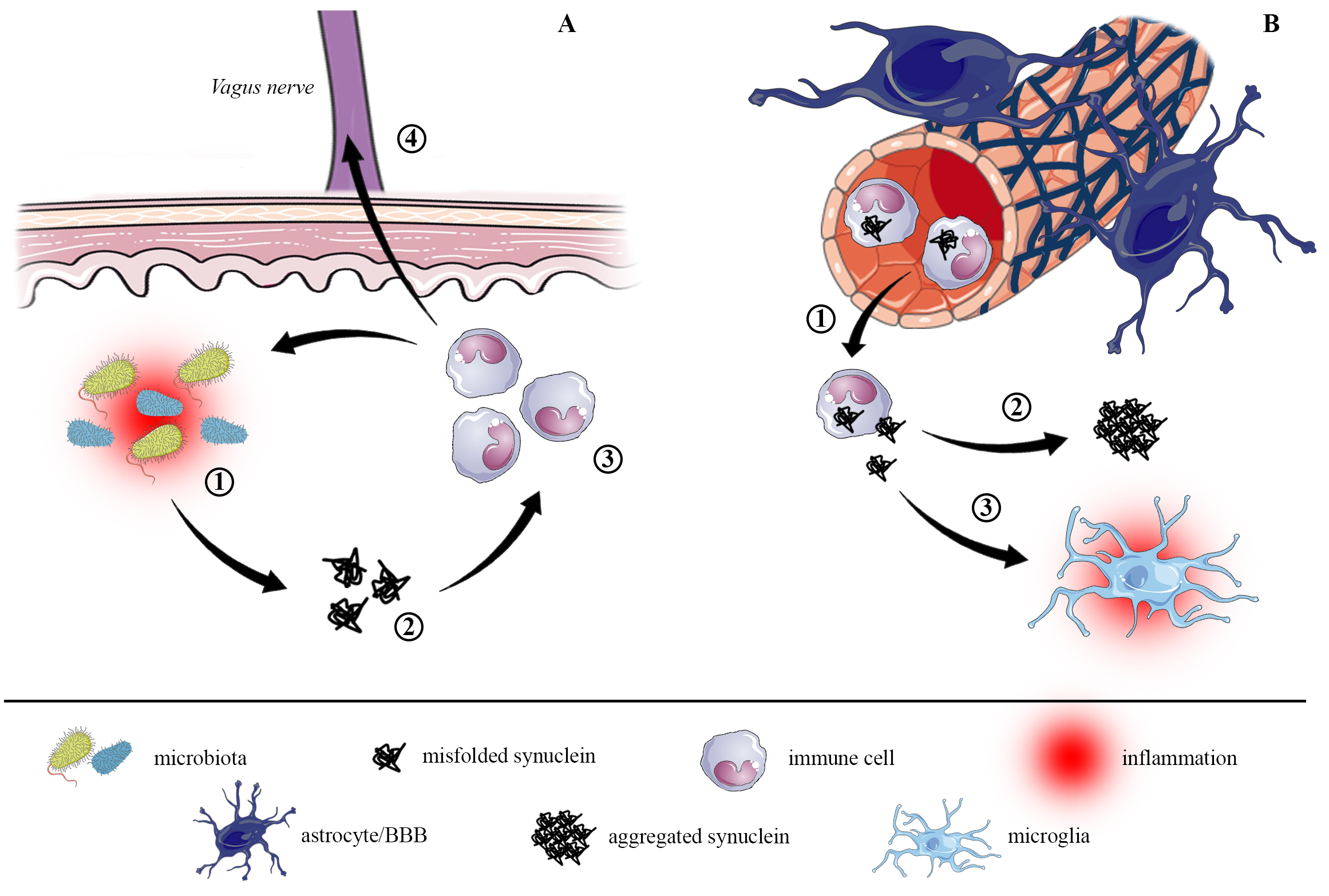 Fig. 3.
Fig. 3.Proposed pathways for alpha-synuclein dissemination from gut to brain. (A) The inflammatory environment in gastrointestinal regions, probably triggered by external stimuli and sustained by dysbiosis (1), increases alpha-synuclein expression and misfolding, leading to the formation of small alpha-synuclein aggregates (oligomers, 2). Misfolded and oligomeric synuclein species activate the peripheral immune response by recruiting immune cells (mostly monocytes and neutrophils, 3), which are primed by the inflammatory environment. The immune cells uptake alpha-synuclein and, in turn, further promote inflammation and protein misfolding and aggregation. The increased intestinal permeability allows immune cells to migrate through the vagus nerve (4) directly towards the dorsal motor nucleus of vagus in the brainstem, from where it starts to spread in the brain. (B) At the same time, chronic local inflammation promotes a systemic inflammatory status that can affect permeability of the blood brain barrier, allowing alpha-synuclein to be transported to the brain (1). There, alpha-synuclein fosters the formation of bigger aggregates (2), resulting in the neuropathological lesions hallmark of synucleinopathies, and it triggers microglial activation (3) that perpetuates neuroinflammation.
Perceiving PD as a pathology confined to the CNS is now an outdated picture. The non-motor symptoms include several autonomic, non-dopaminergic manifestation affecting multiple body districts and, interestingly, these symptoms characterize the prodromal phase of the pathology and can be discrete predictors for the development of clinical PD. This evidence endorses the hypothesis that the pathology can even start at the periphery, likely through susceptible sites and with a multifactorial etiology that embodies genetic predisposition and environmental factors [169]. The neuropathological staging proposed by Braak and colleagues seems to confirm this hypothesis, on the grounds that the first brain areas affected by Lewy pathology are the dorsal motor nucleus of vagus and olfactory bulb, both receiving afferences from the peripheral nervous system and associated with body regions acting as an interface with the outside and so more susceptible to environmental triggers (namely the gastrointestinal tract and olfactory mucosa). According to this hypothesis, alpha-synuclein aggregates enter the brain simultaneously via two routes: nasal, with a progression to the amydgala, and gastric, with retrograde diffusion to the dorsal motor nucleus. Recently, it has been suggested that rather than this “dual-hit” scenario, the “single-hit olfactory bulb (OB)/brain or autonomic” hypothesis is taking place [170]. The dual-hit hypothesis is supported by the fact that constipation, REM sleep behavior disorder (RBD), and hyposmia prior to diagnosis are present in the prodromal phase. However, clinical observation of this phase could be more complicated, as constipation is manifested in fewer than half of PD patients in the early stage, RBD affects approximately only a third, and hyposmia around 70% before diagnosis. These data suggest that not all patients fit into the dual-hit hypothesis, including a clinical “OB/brain-first” or “body first” subtype. This is also supported by a very recent analysis of two datasets (Vanta and Tokyo) in which it is emerged that alpha-synuclein pathology in the enteric nervous system or lower brainstem very rarely present OB pathology, while patients with mild amygdala-prevalence nearly always present OB pathology.
On this basis, looking at the processes that occur at the periphery may represent an easily-accessible window to look at what is occurring in CNS. In this review, we have provided a comprehensive report of the pathological mechanisms described in extra-brain districts, ranging from molecular to systemic levels (Fig. 4).
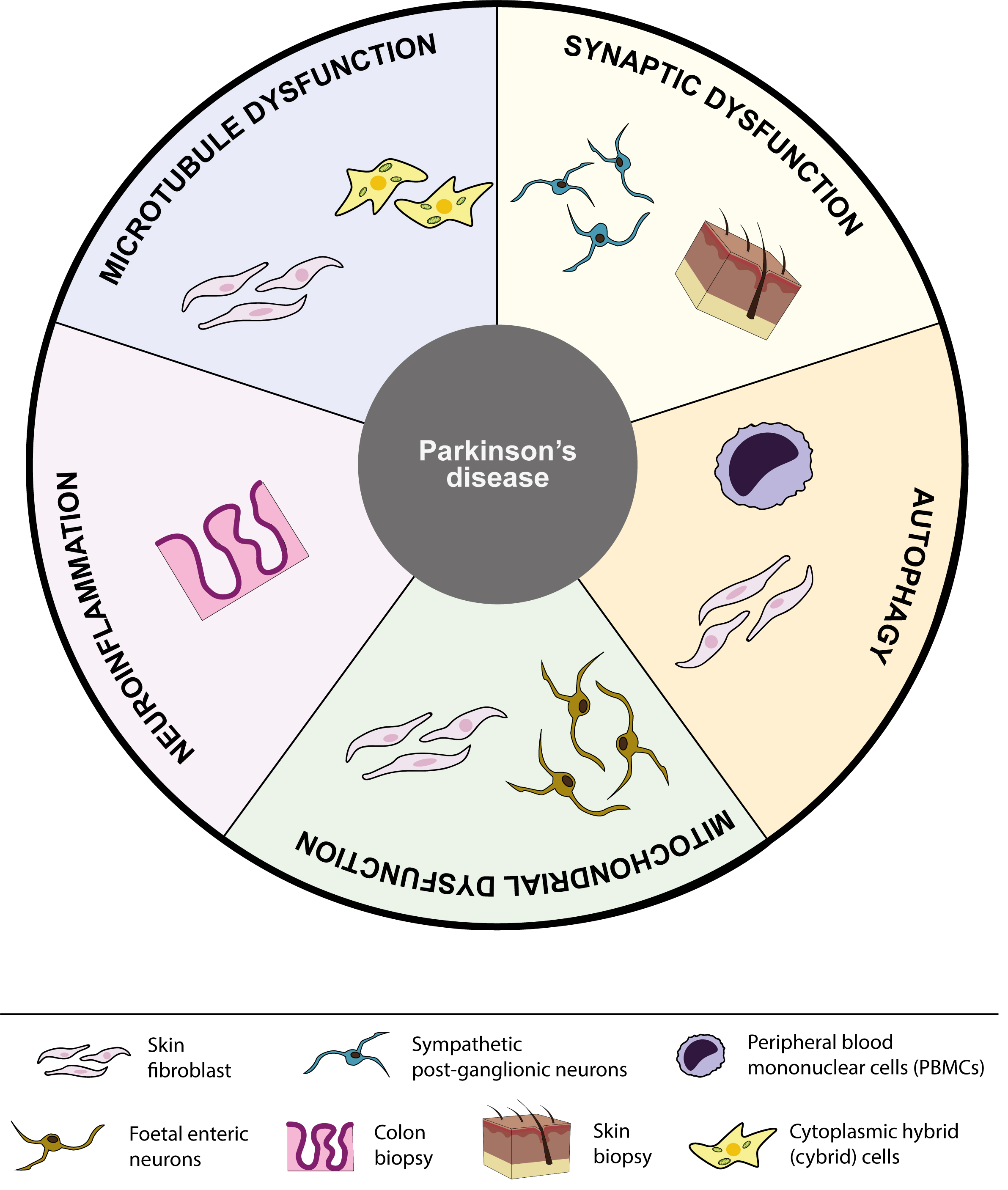 Fig. 4.
Fig. 4.Schematic summary of peripheral tissues linked to each pathological pathway involved in PD. Overview of the five different pathological pathways discussed in this review, including microtubule dysfunction, synaptic dysfunction, autophagy, mitochondrial dysfunction and neuroinflammation. Each pathological pathway shows a schematic representation of all peripheral tissues investigated for that particular pathway to date.
Many of the studies performed on peripheral samples collected from patients involved blood mononuclear cells and skin fibroblasts that have proven to be easily-accessible models. They revealed impaired microtubular and mitochondrial physiology, as well as a defective autophagic machinery at the periphery in PD. However, the relevance of these data is limited, as the models lack the organizational complexity of a tissue and, in addition, the intracellular high specialization of a neuron, where the microtubule-mediated connection between synapses and soma is complex and highly regulated. Despite the lack of molecular insights, the collection of body fluids and tissue biopsies from skin and gut could be useful and provide an overview of the systemic modifications occurring in the periphery in the pathological environment of PD, as demonstrated for the increased inflammatory state and a progressive loss of synaptic terminals. Therefore, this paper wants to draw attention to the importance of the periphery as a field of investigation into CNS diseases, aiming to study the mechanistic features of the pathology. Each of the reported processes could potentially trigger PD, as suggested by familial forms of the pathology, and it may impact reciprocally on the others due to the close interconnection between these processes. To conclude, we emphasize how the peripheral aspects of PD warrant further studies as they may become a reliable predictive element as well as an accessible target for disease modifying agents, especially in light of a possible peripheral onset.
The pathological mechanisms we have discussed in this review are potentially linked to each other. The dying back neurodegeneration that starts from the synapses could be fostered by microtubule dysfunction, inducing mitochondrial impairment and autophagic failure as a consequence of the defective transport towards the cell body. This degeneration then elicits inflammation, especially through microglia activation. Additionally, all these pathological processes may have a mutual impact on the others, since synaptic functioning could be impaired firstly by autophagy or mitochondrial mutations, such as Parkin, LRRK2, GBA and TMEM175 mutations, known to be factors for PD, which increase during aging. Lastly, the pivotal emerging role of the microtubule system, as suggested by MAPT mutations, definitely warrants more in depth investigation, since it connects the synapses and the cell bodies and could play an additional pivotal role both in axonal transport and synaptic activity. Notably, the peripheral aspects of PD are undoubtedly worthy of further studies as they may represent the onset of CNS pathology and, if this were to be proven, they could become reliable predictors PD development as well as an accessible target for disease modifying agents.
MJB, JMK, and AC wrote the manuscript and designed the figures. GP, GC and SM critically reviewed it. All the authors read and approved the final manuscript.
Not applicable.
The authors thank “Fondazione Grigioni per il Morbo di Parkinson” (Milan-Italy) for long-lasting support to GC. We are grateful to all the members of our team for stimulating discussion and to Karen Doyle for reading and editing the paper.
This research was funded by Italian “5
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.