
 , Bin Zeng 1,2,3,*
, Bin Zeng 1,2,3,*1 College of Pharmacy, Shenzhen Technology University, 518118 Shenzhen, Guangdong, China
2 Jiangxi Key Laboratory of Bioprocess Engineering and Co-Innovation Center for In-Vitro Diagnostic Reagents and Devices of Jiangxi Province, College of Life Sciences, Jiangxi Science and Technology Normal University, 330013 Nanchang, Jiangxi, China
3 College of Chemistry and Chemical Engineering, Jiangxi Science and Technology Normal University, 330013 Nanchang, Jiangxi, China
4 School of Materials and Mechanical & Electrical Engineering, Jiangxi Science and Technology Normal University, 330038 Nanchang, Jiangxi, China
5 Research Centre of Printed Flexible Electronics, School of Materials Science and Engineering, Harbin Institute of Technology, 518055 Shenzhen, Guangdong, China
6 Sauvage Laboratory for Smart Materials, Harbin Institute of Technology, 518055 Shenzhen, Guangdong, China
7 State Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, 999078 Macau, China
Academic Editor: Jae Man Lee
Abstract
The liver is the most significant metabolic organ in the body and plays an important role in lipid metabolism. Liver lipid metabolism disorders cause hepatic diseases such as hepatitis, hepatic cirrhosis, and hepatoma. Autophagy is a process of generating energy and building blocks by degrading redundant or damaged proteins and organelles. Thus, it helps in the maintenance of cellular homeostasis. Recent discoveries revealed that lipophagy plays a vital role in hepatic cellular homeostasis and lipid metabolism. Its imbalance is always associated with the perturbation of lipid metabolism in the liver. This article reviewed the molecular mechanisms involved in lipophagy and the interaction between lipophagy and hepatic lipid metabolism. Increasing evidence suggests that lipophagy is an effective method to resolve liver diseases.
Keywords
- autophagy
- lipophagy
- liver
- lipid metabolism
The liver is the central organ of lipid metabolism and plays a vital role in cellular metabolisms, such as lipid digestion, absorption, transport, and decomposition [1]. Chronically disturbed hepatic metabolism may lead to obesity and metabolic syndrome, causing non-alcoholic fatty liver disease (NAFLD) [2]. Approximately 1 in 30 patients diagnosed with NAFLD develops cirrhosis or a liver-associated complication [3]. Autophagy plays a crucial role during hepatic lipid metabolism. Diet, environment, and drugs strongly affect hepatic lipid metabolism through autophagy [4]. Therefore, a further understanding of the specific molecular mechanisms of autophagy and various cell types involved has been deeply explored. The current article reviews the main pathways and regulatory mechanisms of hepatic autophagy and the related effects on lipid metabolism.
The liver is one of the main organs metabolizing all lipid types. There are many
molecular mechanisms controlling the homeostasis of lipid metabolism in the
liver, such as lipoprotein synthesis, lipid uptake and transport, triglyceride
synthesis, ab initio fatty acid synthesis, and oxidative metabolism [5].
Intracellular lipid droplets (LDs) constitute the main energy reserve in the
liver. The imbalance of LDs and cellular triglycerides (TGs) causes various
diseases of the liver, including fatty liver disease and type 2 diabetes [6]. The
dynamic balance of liver TG is regulated by lipid input, secretion, synthesis,
lipolysis, and oxidation [7]. Neutral lipase is considered the primary regulator
of the degradation of liver TGs. Nevertheless, the expression of neutral lipases,
lipase, hormone-sensitive (LIPE/HSL), and patatin-like phospholipase domain
containing 2 (PNPLA2/ATGL) is very low [7]. There are two known lipid degradation
pathways. There is also a lysosome-mediated acid lipolysis (lipophagy) in
addition to the classical cytosolic lipase-driven neutral lipolysis. Lipophagy
selectively degrades LD, and the released free fatty acids and glycerol are
transported inside the mitochondria for
Lipophagy is an alternative lipid metabolism pathway via the lysosomal degradative autophagy pathway [8]. During lipophagy, TGs and cholesterol are absorbed by autophagosomes and then delivered to lysosomes for degradation using acidic hydrolases [9]. Fat autophagy controls intracellular lipid storage, cellular free lipids (including fatty acids), and energy homeostasis [10]. The quantity of lipids metabolized by lipophagy varies with the extracellular nutrient intake and the cell’s ability to degrade lipids by lipophagy.
The liver is rich in lysosomes and high basal levels of autophagy [11]. Autophagy plays an essential role in regulating the homeostasis of liver lipid metabolism. Enhanced autophagy increases liver fat consumption and decreases liver lipid accumulation [12]. Therefore, the inhibition of autophagy causes liver lipid accumulation and leads to disease in severe cases [13].
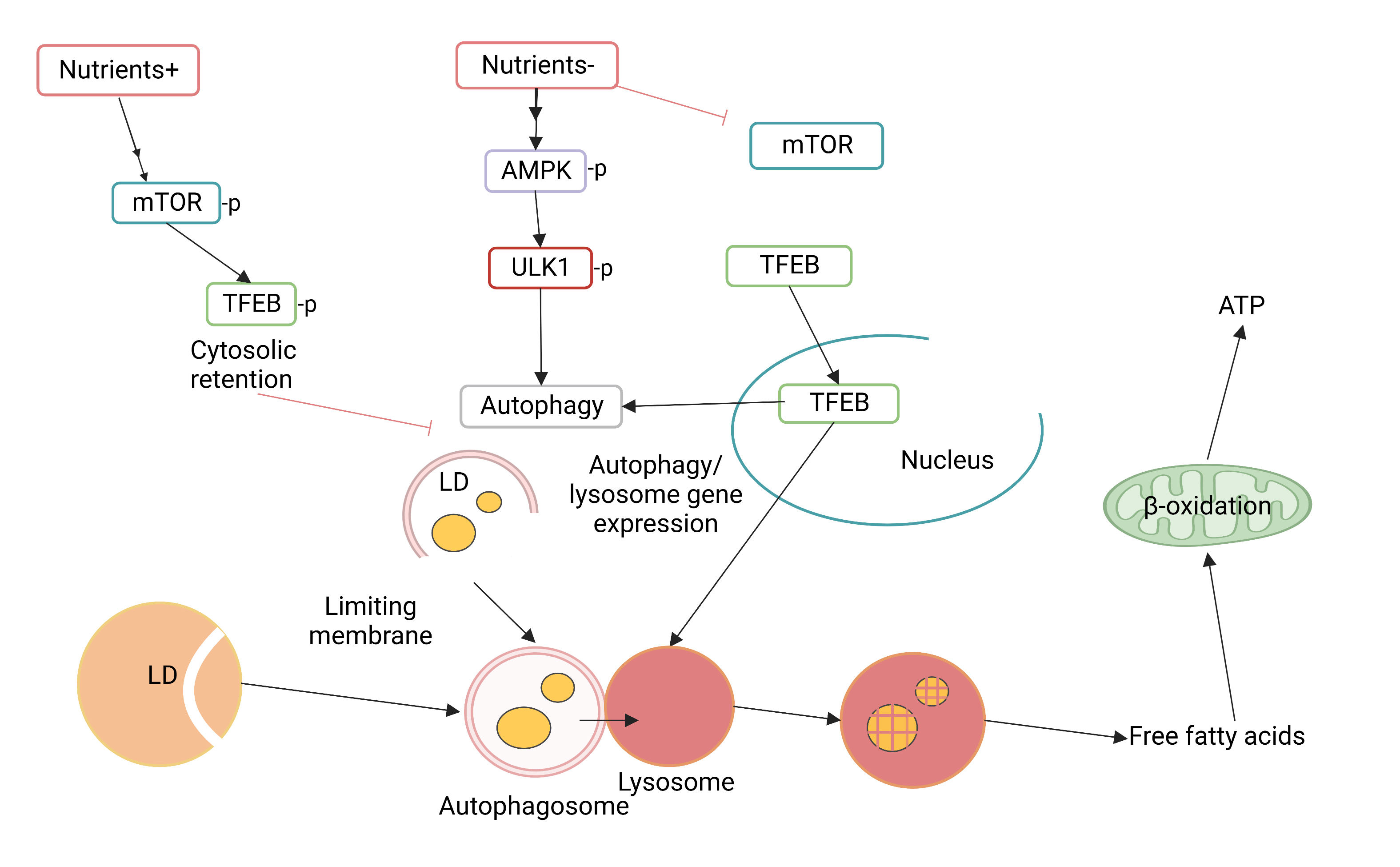
The formation and turnover of autophagosomes include the evolutionarily conserved autophagy-related gene Atg [14]. It can be divided into different stages, such as autophagosome initiation, nucleation, expansion, and extension of the autophagy membrane, closure of the autophagy membrane and fusion using lysosome, and autophagy content degradation [15]. Microtubule-associated protein light chain 3 (LC3) in selective autophagy interacts with the selective autophagic receptor p62 as a connector molecule. It selectively recruits transporters into autophagosomes and enters the subsequent lysosome degradation process (Fig. 1) [16]. Moreover, autophagy is regulated at different levels, from gene transcription to post-translational modification of proteins. These factors control the activity of autophagy and affect the homeostasis of liver lipid metabolism [17]. This section summarizes the main autophagy pathways associated with lipid metabolism.
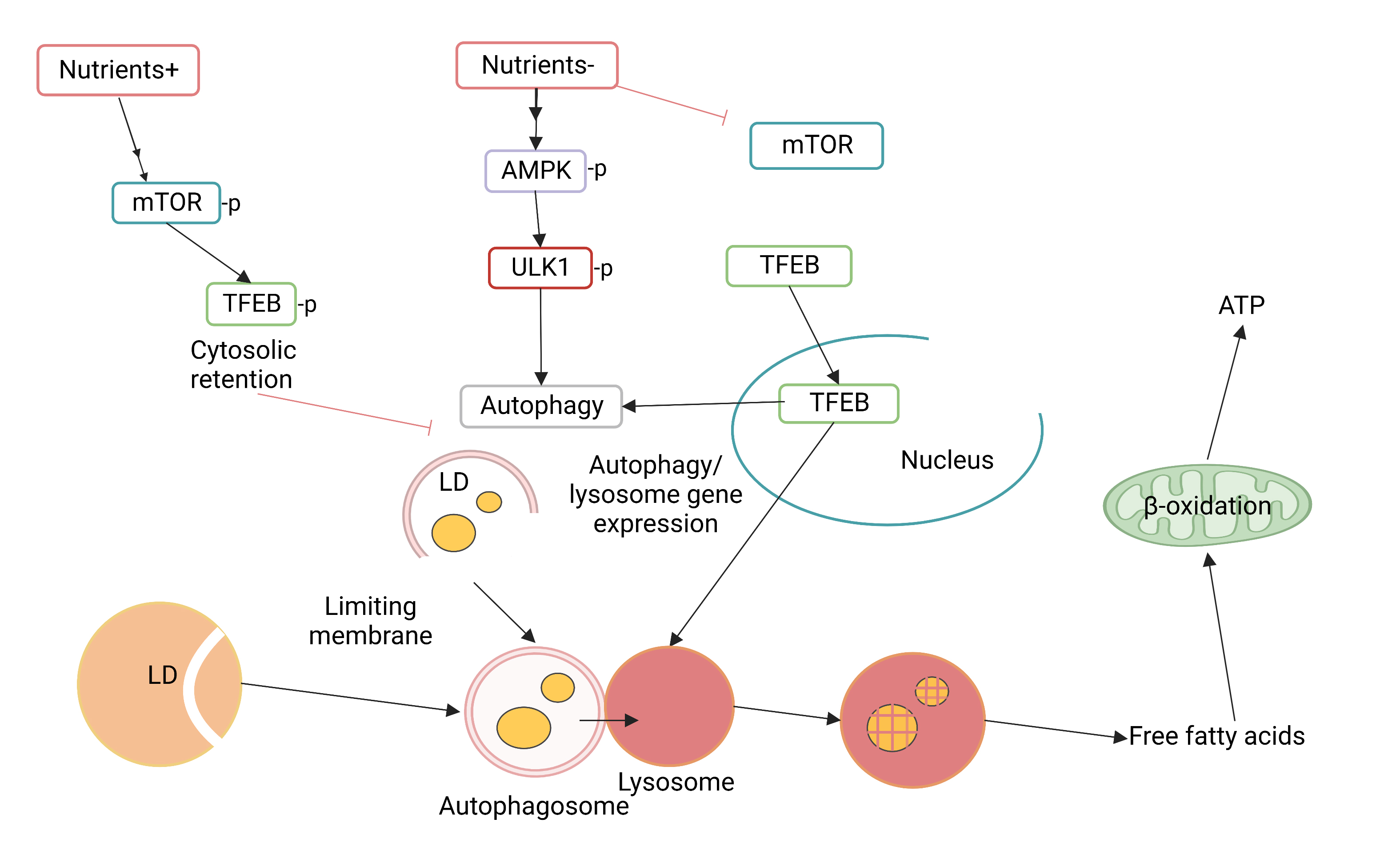 Fig. 1.
Fig. 1.Schematic diagram of main autophagy pathways affecting lipid metabolism.
Autophagy is induced by two key kinases, mechanistic target of rapamycin kinase (mTOR) complex 1 (mTORC1) and protein kinase AMP-activated catalytic subunit (PRKA)/adenosine monophosphate-activated protein kinase (AMPK). It integrates upstream molecular signals, activates the unc-51 like autophagy activating kinase 1 (ULK1/Atg1) kinase complex, and ultimately induces autophagy [18].
AMPK is a critical kinase regulating energy homeostasis and one of the central regulators of eukaryotic cell and organism metabolism. It is responsible for the control of the input and output of cells and the regulation of the stability of cellular physiological activities [19]. Various substances in the body activate AMPK, which in turn inhibits lipogenesis, increases fatty acid oxidation, and plays a crucial role in the synthesis of cholesterol and total TG [20]. Additionally, a study revealed that AMPK regulates autophagy through the autophagy-related protein 1 (Atg1), participating in autophagy initiation and enhancing autophagosome maturation, thereby enabling the fusion of autophagosomes and lysosomes [21].
AMPK is activated through the phosphorylation by the upstream AMPK kinases (AMP-KKs). Activated AMPK regulates multiple metabolic processes like autophagy [22]. AMPK enhances autophagy through the phosphorylation of autophagy-related proteins in the mTORC1, ULK1, and PIK3C3/VPS34 complexes or indirectly by controlling the expression of autophagy-related genes downstream of transcription factors, including Forkhead box O3 (FOXO3), Transcription Factor EB (TFEB), and Bromodomain-containing protein 4 (BRD4) [23, 24].
A study revealed that zinc is an effective lipophagy promoter. Zinc-induced
autophagy and lipid metabolism include the up-regulation of protein
kinase-
A study indicated that stearoyl-CoA desaturase 1 (SCD1) inhibits hepatic steatosis by inducing AMPK-mediated lipophagy. SCD1 is a potential therapeutic target for NAFLD. SCD1 activity is enhanced in NAFLD individuals, and the deletion of the SCD1 gene decreases liver lipid synthesis [26]. AMPK activity is influenced by SCD1: the inhibition of SCD1 enhances AMPK phosphorylation and alters the expression of genes associated with adipogenesis [27]. A study showed that SCD1 inhibition induces hepatocyte lipophagy and apoptosis of human hepatocellular carcinoma (HCC) through the AMPK signaling pathway [28].
A study showed that the phosphorylation of AMPK is increased, and the expression of hepatic autophagy markers is also increased in the livers of mice fed with barley sprout extracts (BSE). Saponin is a major flavonoid in BSE and an activator of AMPK [29]. Saponins enhance the degradation of LD in the liver and the subsequent activation of fat oxidation by activating AMPK, thereby inhibiting hepatic steatosis in alcohol-fed mice [30].
Sirtuin3 (SIRT3), a nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase, is an important biomolecule regulating macroautophagy and lipid metabolism [31]. SIRT3 overexpression activates macroautophagy using the AMPK-ULK1 pathway, primarily acting on LDs. Thus, it results in smaller LD and decreases lipid accumulation [32].
However, the AMPK pathway involves hepatic lipophagy and hepatic lipid metabolism. The fatty acid translocase cluster of differentiation (CD36) is a multifunctional membrane protein promoting long-chain fatty acid uptake [33]. The current study revealed that CD36 expression in mice fed a high-fat diet is increased in the liver and autophagy is reduced. CD36 is involved in fatty acid oxidation by activating the adenosine monophosphate-activated protein kinase and regulating hepatic autophagy using the AMPK pathway [34].
Moreover, other proteins including ghrelin, TGF-
| Protein | Phosphorylation sites(s) | Stage of autophagy | Autophagy function |
|---|---|---|---|
| ULK1 | Ser467 (H, M, R) Ser555 (M, R)/Ser556 (H) Thr574 (M, R)/Thr575 (H) Ser637 (M, R)/Ser638 (H) Ser317 (M) | Autophagy initiation | Component of the ULK1 complex |
| Ser777 (M) | |||
| PIK3C3 | Thrl63 (H, M, R) Serl65 (H, M, R) | Autophagosome biogenesis | Component of the PIK3C3 complex |
| BECN1 | Ser91 (H, M, R) | Autophagosome biogenesis | Component of the PIK3C3 complex |
| Ser94 (H, M, R) | |||
| Thr388 (H, M, R) | |||
| RACK1 | Thr50 (H, M, R) | Autophagosome biogenesis | Promotes the assembly of PI3KC3 complex |
| PAQR3 | Thr32 (H, M) | Autophagosome biogenesis | Facilitates the formation of PI3KC3 complex |
| RAPTOR | Ser722 (H, M) | Regulation of autophagy | Negative regulator of autophagy |
| Ser792 (H, M) | |||
| mTOR | Thr2446 (H) | Regulation of autophagy | Negative regulator off autophagy |
| TSC2 | Serl342 (H, M, R) Thrl227 (H, R) | Regulation of autophagy | Negative regulator of mTOR |
| ATG9 | Ser76I (H, M, R) | Autophagosome elongation | Participates in the recruitment of lipids to the isolation membrane |
| ATG13 | Ser224 (M)/Ser225 (H) | Autophagy initiation | Component of the ULK1 complex |
A phosphatidylinositol 3-kinase-related kinase (PIKK) protein, an mTOR is a highly conserved serine/threonine (Ser/Thr) protein kinase. The mTOR exists as two protein complexes: mTORC1 and mTORC2. They behave as central controllers regulating cellular metabolism, growth, proliferation, survival, and autophagy processes [38]. mTOR inhibitors block the mTOR signaling pathway, leading to anti-inflammatory, anti-proliferative, autophagic, and apoptosis-inducing characteristics. Unlike the AMPK pathway, mTOR activation inhibits autophagy, and its inhibition activates autophagy. Among them, rapamycin can bind to the target of mTORC1 and is an mTOR inhibitor [39].
A recent study observed that Irbesartan (IRB) inhibits p-Akt and p-mTOR by
activating PPAR-
The mitochondrial fusion protein 2 (Mfn2) induces autophagy through the PI3K/AKT/mTOR signaling pathway. Mfn2 increases the expression of LC3-II, Atg5, and Bcl-2 in liver cells and down-regulates the expression of p62 and Bax [42]. Rapamycin Mfn2 significantly inhibits the expression of p-PI3K, p-Akt, and p-mTOR in liver cells, thereby inducing lipophagy [43].
In addition, the rate-limiting enzyme of peroxisomal
Additionally, branched-chain amino acids (BCAAs) in the liver activate mTOR, enhance hepatocyte apoptosis, block hepatic free fatty acids (FFA)/TG transformation, inhibit lipid-induced autophagy, and increase hepatocyte lipotoxicity and susceptibility to FFA [46]. BCAA inhibits autophagy, and obstacles the self-repairing mechanism that is protective against lipotoxicity, thereby aggravating liver injury [47].
There is evidence that ezetimibe induces autophagy even under hyperactivated mTOR conditions through the AMPK-TFEB pathway. A study indicated that ezetimibe reduces hepatic steatosis, inflammation, and fibrosis by enhancing autophagy through the PRKA activation and nuclear translocation of TFEB [48].
Vacuolar protein sorting 34 (Vps34) is inside a class III PI3Kinase (PIK3C3) among mammals. Phosphatidylinositol (PI) synthesizes phosphatidylinositol 3-phosphate (PI3P), which is essential for the extension of autophagy vesicles and the recruitment of ATG proteins to autophagy vesicles. Vps34 is activated by binding to Vps15 and binds to Beclin-1 to develop the Vps34-Vps15-Beclin1 complex [49].
A recent study indicated that Vps34 can be acetylated using acetyltransferase
p300. In contrast, p300-mediated acetylation can inhibit the activity of Vps34.
Acetylation reduces the affinity between Vps34 and the substrate PI and hinders
the formation of the Vps34-Beclin1 core complex [50]. The inactivation of p300
induces Vps34 deacetylation, PI3P production, and autophagy even in AMPK
Although the amino acid sequences of Beclin 1 and Atg6/vps30 differ significantly (24.4% sequence identity and 39.1% sequence similarity), Beclin 1 is a mammalian homolog of the yeast Atg6/vps30. Beclin 1 regulates autophagy in mammalian cells like yeast by forming complexes using different proteins [52].
The content of Beclin-1 in cells determines the degree of autophagy, and it is a marker protein for autophagy formation [53]. Beclin-1 is phosphorylated by Atg1 and is the whole scaffold of the PI3K complex to enhance the localization of autophagy proteins to autophagy vesicles [54]. It selectively participates in different stages of autophagy when the PI3K complex binds to other regulatory proteins. These include the formation of autophagy vesicles by binding to Atg14 and the maturation and transport of autophagy vesicles by binding to the UV radiation resistance-associated gene (UVRAG) [55].
Liver cells develop lipophagy under various stress conditions, such as hunger or hyperlipidemic nutrition (fatty acid deficiency or excess of lipids) [1]. The process of lipophagy involves subcellular structures, including LD, autophagosomes, and lysosomes, dynamic changes in the membrane structure and vesicle transport, and synergism with lipid intake and hydrolysis [9]. Hormones, natural compounds, and metal ions can control lipophagy. There are several relatively specific molecules involved in lipophagy in cells.
Sirtuin type1 (SIRT1) is a member of the family of silent information regulators and it is a nicotinamide adenine dinucleotide NAD+-dependent histone/protein deacetylase. The deacetylation of proteins and histones upregulates or downregulates gene transcription and protein function [56]. A previous study showed that the regulation of the deacetylation of SIRT1 can induce autophagy and decrease lipid accumulation in the liver [57].
Berberine stimulates SIRT1 deacetylation and induces autophagy in an autophagy protein 5-dependent manner. It also promotes gene expression and circulation of fibroblast growth factor 21 (FGF21) and ketone bodies within mouse liver in a SIRT1-dependent way. Autophagy and FGF21 activation in the liver regulate lipid storage as well as the utilization and whole-body energy metabolism [58].
Both resveratrol and caloric restriction upregulate the SIRT1-autophagy pathway to reduce hepatic steatosis in rats fed with a high-fat diet. Autophagy is later induced by the SIRT1-FOXO3 signal transduction pathway [59]. A recent study observed that the natural polyphenol resveratrol enhances the levels of cAMP, SIRT1, phosphorylated protein kinase A (pPRKA), phosphorylated AMP-activated protein kinase (pAMPK), and SIRT1 activity in HepG2 cells. Moreover, it can induce autophagy through the cAMP-PRKA-AMPK-SIRT1 signal pathway, whereas autophagy inhibition markedly abolishes resveratrol-mediated hepatic steatosis improvement [60].
Caloric restriction and resveratrol in animal studies can affect the sirtuin system. A randomized trial on healthy people revealed that the reduction of calories and supplementation with resveratrol significantly increases SIRT1 plasma levels [60]. Resveratrol increases the expression of SIRT1 mRNA in the liver of mice fed with a high-fat diet and controls the number and function of human adipocytes in a SIRT1-dependent manner [59].
However, SIRT1 also down-regulates the sterol regulatory element-binding protein (SREBP) homologs during fasting, inhibiting lipid synthesis and storage. SREBPs are steroid regulatory element binding proteins and transcription factors. A previous study revealed that SIRT1 can directly deacetylate SREBP [61]. Therefore, the modulation of SIRT1 activity may affect SREBP ubiquitination, protein stability, and target gene expression. SREBPs undergo cleavage-induced activation due to the low sterol levels in the cell, thus promoting the transcription of enzymes critical to sterol biosynthesis [62].
TGs in the liver are sequestered into the LDs, which are the main lipid storage organelles. LDs are home to proteins, the most abundant of which are the perilipins (PLINs) [7].
It has been observed that the overexpression of PLIN2 protects small LD from macroautophagy/autophagy, and PLIN2 deficiency enhances autophagy and depletes liver TG [63]. When mice are hungry, the degradation of PLIN2 and PLIN3 in hepatocytes is enhanced using autophagy. In contrast, the increase of adipose triglyceride lipase (ATGL) and LC3 is observed on the surface of LD [64]. In addition, PLIN1 can colocalized with the selective autophagy receptor p62, mediating the recognition of LD through the autophagy initiation membrane [65].
Neutral lipids in mouse liver and cultured hepatocytes are mobilized through the sequestration of perilipin 2-coated LDs in autophagosomes. This involves that LDs are delivered to the lysosomes, where triacylglycerols are hydrolyzed by lysosomal lipases [66].
The research identified that unphosphorylated perilipin 1 prevents Rab7 from docking to LDs and inhibits lipophagy. Moreover, Rab7 is recruited to lysosomes through conformational changes in perilipin 1, which are induced by phosphorylation with the association of LD with lysosomes [67].
ATGL is closely associated with lipophagy. Although ATGL is an essential molecule in direct lipid decomposition, it mediates lipid decomposition synergistically through lipophagy [68]. When lipophagy is inhibited, the increase in lipid decomposition due to ATGL overexpression is also inhibited. It is speculated that ATGL may decrease the size of LDs through lipolysis and develop the conditions for lipophagy [69].
In addition, ATGL is a selective autophagy receptor and mediates the specific recognition of LD by LC3 on the autophagy membrane through the LC3-interacting region (LIR) [70].
Autophagy stimulates lipophagy by the co-localization of ATGL and LD. Similarly, autophagy activation increases the co-localization of ATGL and LD [71]. Moreover, the correlation between ATGL and LD in autophagy-deficient cells significantly decreased, indicating that the localization ability of the lipase on LD is deficient [72]. The decrease in lipolytic activity of ATGL in autophagy-deficient cells cannot be attributed to the change in its association with LD. This is because when ATGL is reduced in hepatocytes, its binding to the autophagy inhibitor G0/G1 switch gene 2 (G0S2) decreases [70]. Moreover, its binding to the autophagy activation gene marker 58 (CGI-58) increases. These phenomena indicate that the relationship between ATGL and liver LD affects lipophagy activation [72].
Peroxisome proliferator-activated receptor-
PPAR
A study revealed that zinc is an effective promoter of fat phagocytosis. The
administration of zinc significantly reduces lipid accumulation in hepatocytes.
It enhanced the release of free fatty acids, which is associated with elevated
fatty acid oxidation and inhibition of fat formation, followed by autophagy
activation. Zn
High-fat diet significantly induces hepatic steatosis in rats. In contrast,
pioglitazone reverses hepatic steatosis due to a high-fat diet. Pioglitazone
combined with a high-fat diet dramatically decreases the content of serum insulin
and liver TG [78]. Pioglitazone significantly increases the expression of
lipolysis-related proteins (ATGL, HSL),
Some researchers also identified a nuclear receptor, farnesoid X receptor (FXR),
which has an opposite effect than that exerted by PPAR. PPAR
The Rab GTPase protein family is a crucial regulator of intracellular vesicular trafficking. The Rab family members regulate a series of molecular events by cycles between active GTP-states and inactive GDP-states [80]. The Rab protein family is vital in LD metabolism. Approximately 30 Rab family proteins have been identified on the LD surface [81]. Among them, Rab7 protein is widely involved in the regulation of the autophagosome maturation and intracellular transport. Hepatocyte starvation activates Rab7 on the surface of LDs and promotes lysosomal transport to LDs [82]. Rab7 expression in the liver of rats subjected to alcohol liquid diet is also significantly reduced. These results indicate that Rab7 regulates lipid metabolism through lipophagy [83]. Rab10 is a member of the Rab family of small GTPase located on the LD surface. Activated Rab10 in starved hepatocytes is co-located on the LD surface with the autophagy proteins Lc3 and Atg16. In addition, Rab25 and Rab32 can be involved in the molecular mechanism of lipophagy [84]. However, the involvement of other members of the RAB family in regulating lipophagy requires further exploration.
It has been observed that specific neurons within the central nervous system (CNS) can remotely control lipophagy in the liver [85]. Lysosomal-associated membrane protein 3 (LAMP3) is overexpressed, activating Akt and upregulating the expression of the lipogenase FASN and SCD-1 in HepG2 cells. Additionally, the increased TG content induced by LAMP3 overexpression is attenuated by the treatment with a PI3K/Akt pathway inhibitor. These effects can regulate hepatic lipid metabolism by activating the PI3K/Akt signaling pathway [86].
Oleic acid in unsaturated fatty acids can enhance the formation of triglyceride-rich LD while inducing autophagy. Palmitic acid in saturated fatty acids is rarely converted into triglyceride-rich LD, thereby inhibiting autophagy and inducing apoptosis [87]. However, palmitic acid can induce autophagy through the protein kinase C-mediated signal transduction pathway. Moreover, palmitic acid can phosphorylate the autophagy-promoting factor Hsp27 and enhance its induced autophagy [88].
TFEB is a basic-helix-loop-helix-leucine zipper (bHLH-Zip) transcription factor
in the MiT family. Many lysosomal genes are regulated by the single transcription
factor TFEB, such as lipid metabolism-related genes PGC-1
Many other factors affect autophagy, including calcium channel blockers, phosphatidylinositol-5-phosphate 4-kinase, and thyroid hormones [91, 92]. In addition, diets rich in polyunsaturated fatty acids (PUFA), excessive exercise, and a certain degree of radiation directly or indirectly regulate lipophagy [93].
Autophagy and lipid metabolism can influence each other and are crucial in maintaining the homeostasis of lipid metabolism [4]. Lipophagy can produce free fatty acids and remove excess lipids to maintain the balance of lipid metabolism along with the physiological function of hepatocytes [10]. In addition, lipid metabolism feedback induces autophagy to start or stop various abnormal lipid metabolisms, liver injury repair, and other pathological changes, thereby regulating lipid metabolism [94].
Lipophagy is a pathway of lipid catabolism in the liver. The inhibition of autophagy increases the content of intracellular TG and the number and size of LD [95]. In a situation of continuous starvation, liver autophagosomes prioritize the phagocytosis of lipids to provide energy, and lipophagy is activated when lipids accumulate excessively, inhibiting the excessive accumulation of fat inside cells [96]. The selectivity of autophagosomes in hepatocytes gradually increased with the extension of time during in vitro and in vivo experiments to form lipid-based selective autophagy, regardless of starvation or oleic acid incubation [97].
Hepatocyte in vitro experiments revealed that lipopolysaccharide-induced autophagy can enhance LD degradation and inhibit lipid accumulation in mouse liver [12]. In addition to the classical degradation pathway of LD by cellular solute lipase, LD is also isolated in autophagosomes. Autophagosomes selectively uptake LD and regulate the LD isolation membrane curvature through autophagy-associated proteins. The autophagic degradation of LD is also involved in the formation of LD themselves [98]. Lysosomal enzymes decompose the LD components after the fusion of autophagosomes and lysosomes [99]. In addition, experiments have revealed that cold-induced central autophagy activates liposomes and cellular solute lipase; thus, the inhibition of autophagy can prevent lipid utilization [100].
In contrast to mammalian cells, microlipophagy has been documented in Saccharomyces cerevisiae, in which the lipid droplet is trafficked to the vacuole by microtubules and not by Atg proteins [101]. Despite this, the latter is still necessary to catalyze the interactions between the lipid droplet and the vacuole, as evidenced by the formation of aggregates of larger droplets in their absence. Vac8 and Trs85 are special adaptor proteins used in microlipophagy to selectively dissolve lipids. Atg11, a component of other specialized autophagic machinery (pexophagy and Cvt pathway), also increases the efficiency of macrolipophagy [102].
A study observed that autophagy mediates hepatic lipid degradation and is involved in hepatic lipid anabolism to maintain lipid homeostasis. The main components of very low-density lipoprotein (VLDL) are TGs synthesized by hepatocytes using sugars and fatty acids (from lipid mobilization or residual chylous particles), and apolipoproteins APOB100, APOAI and APOE synthesized by hepatocytes plus a small amount of phospholipids, cholesterol and their esters [103]. However, a significant proportion (~70%) of the secreted VLDL is generated through the intracellular breakdown of TG, and autophagy activation can increase VLDL production. In contrast, the inhibition of autophagy causes a decrease in VLDL production [104].
In addition to lipid catabolism, lipophagy is also vital in lipogenesis, lipid synthesis, and lipid droplet biogenesis. The autophagy machinery participates in lipid droplet formation in hepatocytes and cardiomyocytes. A research found that the LC3 conjugation system is involved in lipid droplet formation [104]. Genetical ablation of Atg7 highly suppresses the cytosolic accumulation of LDs in hepatocytes and cardiac myocytes under both 12 and 24 h of starvation [105]. The researchers speculated that lipophagy enhances lipid droplet formation [104].
Lipophagy prevents fat accumulation by degrading intracellular LD, which are closely associated with various pathways of lipophagy. However, the excessive activation of autophagy can induce apoptosis and aggravate the disease in the later stage of the fatty liver [106]. Therefore, the proper regulation of lipophagy is more conducive to maintaining normal lipid metabolism. There are many ways to regulate hepatocyte lipid metabolism. One keyway is to increase lipid deposition in the liver by controlling lipophagy. However, the related mechanism is not precise and requires further research [107].
SCD1 is a crucial enzyme for controlling lipid metabolism. Sodium palmitate-treated hepatocytes had an increased SCD1 expression, AMPK inactivation, and lipophagy deficiency [108]. The inhibition of SCD1 expression in hepatocytes can enhance AMPK activity and lipophagy and reduce lipid deposition. SCD1 overexpression leads to a significant decrease in AMPK activity and lipophagy, and the lipid deposition decreased inside the hepatocytes [27]. It has been established that SCD1 controls lipophagy through AMPK and affects the lipid metabolism of primary mouse hepatocytes. Additionally, CAY10566 (a specific inhibitor of SCD1) significantly decreases hepatic steatosis and hepatic lipid droplet accumulation and promotes AMPK activity and lipophagy [109].
The average size of LD tripled by the ATGL gene knockout in AML12 cells cultured in vitro. Numerous small LDs accumulate in lysosomal acid lipase (LAL) knockout AML12 cells [110]. After ATGL+LAL knockout in the AML12 cells, the average size of LD increases by two times compared to ATGL knockout, but as opposite of LAL knockout, small LD significantly reduces [111]. These phenomena establish the synergistic effect between lipase lipolysis and lipophagy. However, the molecular mechanism between the interaction is unclear, which can be associated with the size of LD. Lipolysis may be a “tandem” process. Large LDs synthesize free fatty acids by lipase lipolysis, and lipophagocytosis occurs after esterification to synthesize smaller LD [97].
Studies on autophagy indicated that lipid metabolism can be regulated, and some external stimuli can enhance lipid metabolism in cells and lipophagy [112]. However, it is unclear whether lipid metabolism in adipocytes is promoted by the activation of lipophagy, and further research is required.
In recent years, lipophagy research expanded our understanding of autophagy and lipid homeostasis, improving the understanding of their related mechanisms. It also provides a novel way to target and treat various metabolic disorders, including obesity, fatty liver, and atherosclerosis [113].
However, there are still many problems with hepatic lipophagy. Different ATG gene knockout effects on hepatic lipid metabolism vary significantly among animals and cell models. It is difficult to determine their real effect on the lipophagy pathway [114]. Moreover, a study revealed that autophagy is involved in lipid degradation and can maintain the dynamic balance of lipids by controlling cholesterol intake [115]. In addition, different types of cells have various lipid swallowing functions, and liver cell-specific lipophagy molecules are not well identified.
Therefore, a deeper study of hepatic lipophagy might provide a deeper understanding of the molecular mechanism with the continuous development of biomedical technology. It is believed that an in-depth study of fat bacteriophages can provide new and better methods for treating liver diseases.
SF contributed to write and edit the manuscript. SF, ZS, XJ, LLi and YW collected the literature and the required information to write the review. SF, ZS, XJ, LLi and YW helped in formatting the manuscript. JL and BZ critically reviewed the manuscript. All authors contributed to the editorial changes in the manuscript. CW, LLin and YW contributed to the literature collecting and the required information. All authors read and approved the final version of the manuscript.
Not applicable.
This work was supported by the Key Special Projects of the National Key Research and Development Plan (2021YFA1301302), the Belt and Road Initiative and China-Africa Science and Technology Cooperation Project of the Department of Science and Technology of the Jiangxi Province (20202BDH80007), the National Natural Science Foundation of China (32200606), and the Natural Science Foundation of Top Talent of SZTU (GDRC202118). The authors declare no conflicts of interest.
This research was financially supported by the Key Special Projects of the National Key Research and Development Plan (2021YFA1301302), the Belt and Road Initiative and China-Africa Science and Technology Cooperation Project of the Department of Science and Technology of the Jiangxi Province (20202BDH80007), the National Natural Science Foundation of China (32200606), and the Natural Science Foundation of Top Talent of SZTU (GDRC202118).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.