



1 ELKH-SZTE Neuroscience Research Group, Danube Neuroscience Research Laboratory, Eötvös Loránd Research Network, University of Szeged (ELKH-SZTE), Danube Neuroscience Research Laboratory, H-6725 Szeged, Hungary
2 Institute of Biophysics, Biological Research Centre, Eötvös Loránd Research Network (ELKH), H-6726 Szeged, Hungary
3 Department of Neurology and Cerebrovascular Diseases, Pándy Kálmán County Hospital, H-5700 Gyula, Hungary
4 Department of Medical Microbiology and Immunobiology, Albert Szent-Györgyi Medical School, University of Szeged, H-6725 Szeged, Hungary
5 Department of Medical Physics and Informatics, Albert Szent-Györgyi Medical School, University of Szeged, H-6720 Szeged, Hungary
6 Department of Neurology, Albert Szent-Györgyi Medical School, University of Szeged, H-6725 Szeged, Hungary
†These authors contributed equally.
Academic Editor: Graham Pawelec
Abstract
Background: Earlier studies reported alterations of the kynurenine (KYN) pathway of tryptophan (TRP) metabolism in Parkinson’s disease (PD). The first rate-limiting enzymes indoleamine 2,3-dioxygenase (IDO) and tryptophan dioxygenase were observed upregulated, resulting elevated KYN/TRP ratios in the serum and cerebrospinal fluid samples of patients with PD. More and more single nucleotide polymorphisms (SNPs) have been identified in a population of PD. However, little is known about the impact of genetic variations of the IDO on the pathogenesis of PD. Methods: SNP analysis of IDO1 was performed by allelic discrimination assay with fluorescently labelled TaqMan probes and a subgroup analysis was conducted according to the age of PD onset. The frame shifts variant rs34155785, intronic variant rs7820268, and promotor region variant rs9657182 SNPs of 105 PD patients without comorbidity were analyzed and compared to 129 healthy controls. Results: No significant correlation was found in three SNPs between PD patients and healthy controls. However, the subgroup analysis revealed that A alleles of rs7820268 SNP or rs9657182 SNP carriers contribute to later onset of PD than non-carriers. Conclusions: The study suggested that SNPs of IDO1 influenced the age onset of PD and genotyping of SNPs in certain alleles potentially serves as a risk biomarker of PD.
Keywords
- kynurenine
- tryptophan
- indoleamine 2
- 3-dioxygenase
- IDO
- Parkinson's diseases
- single nucleotide polymorphisms
- biomarker
- age
- onset
Parkinson’s disease (PD) is the second most prevalent chronic progressive neurodegenerative disease characterized by motor symptoms such as tremor, rigidity, and hypokinesia. The prevalence of PD is approximately 0.2% on average in the general population, but it is increasing with age up to 1.9% [1]. The disease affects primarily the elderly, imposing a serious burden on the aging societies. The non-motor symptoms may appear in early stages of the disease and even before the appearance of classical motor symptoms [2]. The comorbidity is common in PD patients [3, 4, 5, 6, 7, 8]. Relatively significant findings are autonomic dysfunction including arrhythmia, blood pressure irregularity, asymmetric sweating, and incontinence and psychobehavioral manifestations including dementia, depression, anxiety, paranoia, and psychosis [9, 10]. The occurrence of dysfunctions in autonomic modulation in PD patients mostly impacts on psychobehavioral responses, and usually overlaps with behavioral disorders [11, 12, 13, 14, 15]. The histopathological and clinical hallmarks of PD include the degeneration of dopaminergic neurons in the substantia nigra pars compacta, the presence of Lewy bodies, and the positive response to dopamine (DA) replacement therapy [16, 17].
The etiology of PD remains poorly understood. Genetic disposition, neurodevelopmental insuts, and environmental factors are considered to play an important role in the pathogenesis of PD [18]. Pathognomic findings are abnormal protein aggregation, elevated oxidative stress, mitochondrial dysfunction, increased glutamate excitotoxicity, alteration of immune response, disturbance of the kynurenine pathway (KP) of tryptophan (TRP) metabolism, and among others [19, 20, 21, 22, 23, 24, 25, 26, 27]. Altered levels and ratios of kynurenine (KYN) metabolites have been observed in neurologic and psychiatric diseases; furthermore, KYN metabolites have attracted a growing attention not only as potential biomarkers, but also as cross-species and environmenental indicators [10, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36]. Earlier studies revealed that the activities of indoleamine 2,3-dioxygenase (IDO) 1/TRP 2,3-dioxygenase (TDO) were upregulated in PD patients compared to controls, which was indicated by elevation of L-KYN/TRP ratios in the serum and in cerebrospinal fluid (CSF) samples of the patients [37]. The cascade of the KP produces several neuroactive metabolites such as 3-hydroxykynurenine (3-HK) quinolinic acid (QUIN), 3-hydroxyanthranillic acid (3-HAA), and kynurenic acid (KYNA) [38].
3-HK and 3-HAA generate reactive oxygen species (ROS) [16]. An elevation of 3-HK
levels was related to excitotoxic injury and is observed in patients with
neurodegenerative diseases [10]. The neurotoxic effects of 3-HK and 3-HAA involve
the generation of superoxide anion and hydrogen peroxide, which contribute to the
oxidative processes implicated in the pathophysiology of meningitis [39]. QUIN is
a free-radical metabolite. Interferon (IFN)-
KYNA is a broad-spectrum, competitive antagonist of all three ionotropic
excitatory glutamate receptors including
The indoleamine 2,3-dioxygenase IDO1 is one of the first rate-limiting enzymes that converts L-TRP to N-formyl KYN in TRP metabolism, which play a crucial role in governing concentrations of downward bioactive KYN metabolites [16] (Fig. 1). Activation of IDO1 and the KYN system promotes immunosuppressive effects by inhibition of Natural Killer cells, inhibition of T cell functions, and activation of the regulatory T cells [47]. The relationship between PD and the KP have been investigated in in vivo and in vitro studies; however, only limited data are available on the significance of genetic alterations of KP enzymes in PD patients [48, 49].
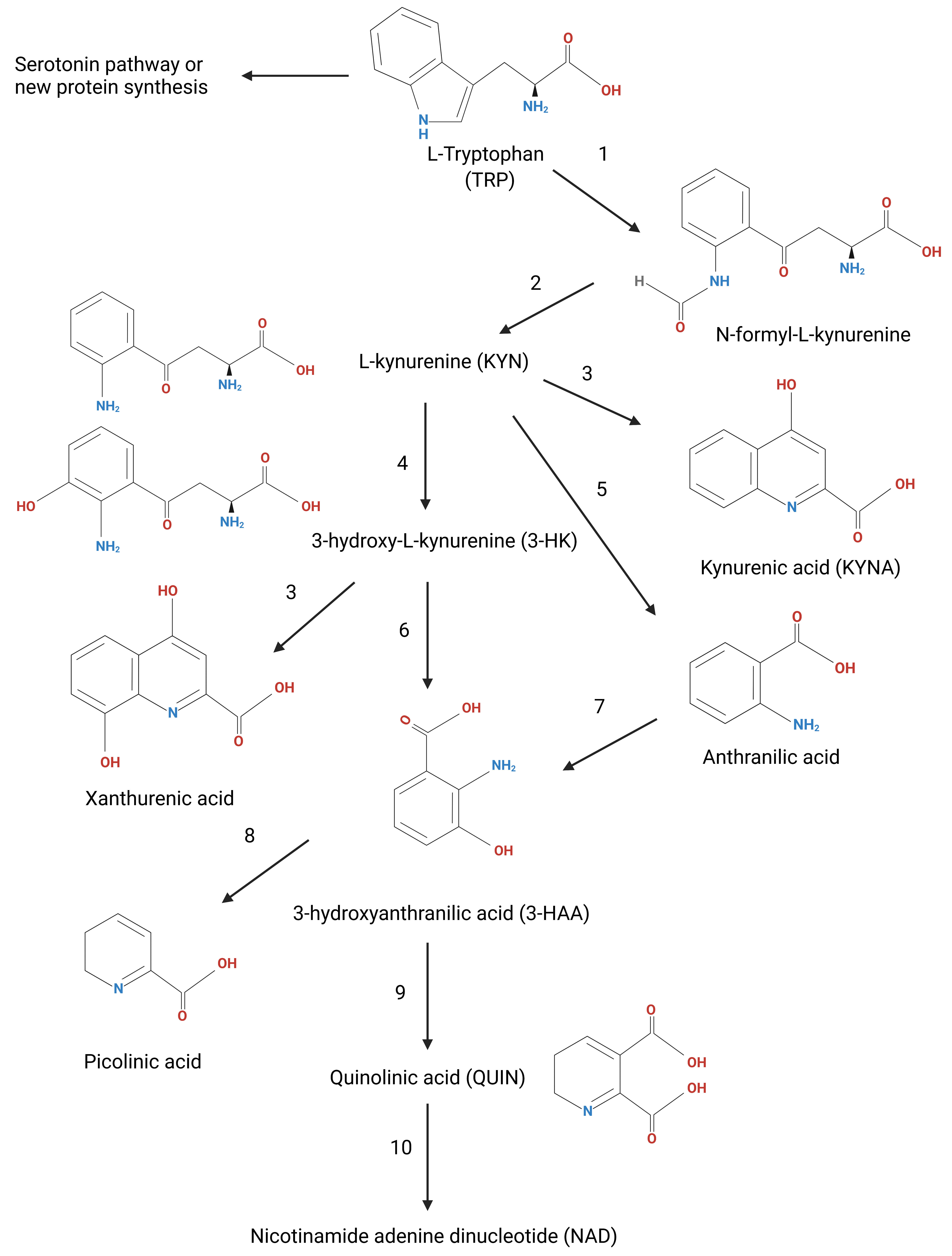 Fig. 1.
Fig. 1.The kynurenine pathway (KP). The KP is the degradation route of tryptophan (TRP) metabolism producing an end-product nicotinamide adenine dinucleotide (NAD). The indoleamine 2,3-dioxygenase (IDO) 1, IDO2, and the tryptophan 2,3-dioxygenase (TDO) (1) are the first rate-liming enzymes that convert the L-tryptophan (TRP) to N-formyl-L-kynurenine. N-formyl-L-kynurenine is converted by formamidase (2) to L-kynurenine (L-KYN). L-KYN is metabolized into various bioactive compounds: kynurenic acid, picolinic acid, 3-hydroxy-L-kynurenine (3-HK), and quinolinic acid. The following are main enzymes of the KP: 1: tryptophan dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO), 2: formamidase, 3: kynurenine aminotransferases (KATs), 4: kynurenine 3-monooxygenase (KMO), 5: kynureninase, 6: kynureninase, 7: non-specific hydroxylation, 8: 2-amino-3-carboxymuconate-semialdehyde decarboxylase (ACMSD), 9: 3-hydroxyanthranilic acid oxygenase, 10: quinolinic acid phosphoribosyltransferase.
In this study we performed single nucleotide polymorphisms (SNPs) analysis in
three loci of IDO1 in PD patients and healthy controls. The frameshift
mutation rs34155785 SNP causes a drastic change in the gene product. Both
intronic variant rs7820268 SNP, and promotor region variant rs9657182 SNP affect
the immune system. The T allelic variants of the rs7820268 showed impaired
CD8
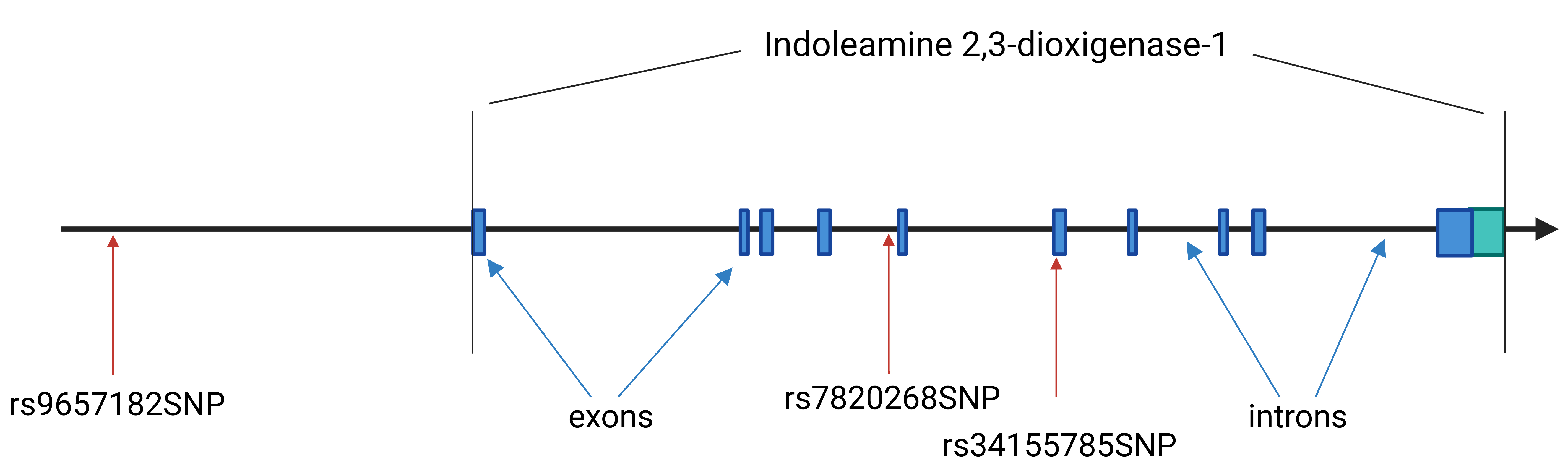 Fig. 2.
Fig. 2.Schematic of the indoleamine 2,3-dioxygenase (IDO) 1 gene. The IDO1 gene consist of 10 exons and 9 introns localized in 8p11.21 chromosome region. It has 9 transcript variants. The encoded protein is 403 amino acid long. The rs9657182 SNP is located 5’ upstream from the promoter region of the IDO1 gene. The rs7820268 SNP is localized in the fourth intron and the last rs34155785 SNP, which is a frame shift mutation, is located in the sixth exon.
The blood samples of 105 PD patients and 129 healthy controls were examined for
the study. All patients gave their informed consent in accordance with the
Declaration of Helsinki, and the study was approved by the Medical Research
Council Scientific and Research Ethics Committee (47066-3/2013/EKU (556/2013)).
The samples were collected at two sites (at the Department of Neurology, Faculty
of Medicine, University of Szeged, Hungary, and at the Department of Neurology
and Cerebrovascular Diseases, Pándy Kálmán County Hospital, Gyula,
Hungary). The patient and the control groups were age- and gender-matched (gender
ratio p = 0.908 by chi-square test; mean age difference p =
0.310 by Student’s t-test). For data analysis, the patient’s group was
divided into two subgroups based on the appearance of the first symptoms: the
early-onset (EOPD; disease onset
| Group | Male | Female | Age | Age at onset (Mean | |||
| Mean |
Median | Min | Max | ||||
| PD patients (n = 105) | 48 | 57 | 66.42 |
68 | 34 | 84 | 58.81 |
| Controls (n = 129) | 58 | 71 | 65.26 |
63 | 53 | 87 | - |
| Min, minimum age in the group; Max, maximum age in the group. | |||||||
Peripheral whole blood samples (stored at –80 °C) were subjected to genomic DNA isolation by the desalting method developed by Miller et al. [51]. The purified genomic DNA were stored at –20 °C at the biobank of the Department of Neurology, Faculty of Medicine, University of Szeged (biobank license: Regional Human Biomedical Research Ethics Committee: 135/2008).
The IDO1 genotypes were determined by allelic discrimination study with
TaqMan probes. Three SNP of the IDO1 gene were investigated. The
rs34155785 SNP is a frame shift mutation in the human genome (results a Phe (F)
For the rs34155785 SNP, the following primers were used for the amplification of the DNA forward primer: 5’- CTA AAC TTC TTG CCT TCC TTA TC-3’; reverse primer: 5’- AGA CGT ACT TTG ATT GCA GA-3’. The following probes were applied for allelic discrimination: wild type allele: 5’-Fam- GAC GTT TTG TTC TCA TTT CGT G-BHQ-1-3’; and C allele: 5’-Hex- GAC GTT TTG TTG CTC ATT TCG TG-BHQ-1-3’.
The rs7820268 SNP, the following primers were applied for the amplification of the G/A at chromosome 8: forward primer: 5’- TAA ATG TAA TGC CTA CTG AAG AA-3’; reverse primer: 5’- CCT TAT GAA AGC AGC CAT G-3’. The following probes were designed for allelic discrimination: G allele: 5’-Fam- GTA GCA TTC AAT CAA ATA GCA ACA AC-1-3’; and A allele: 5’-Hex- GTA GCA TTC AAT TAA ATA GCA ACA AC-1-3’.
The rs9657182 SNP, the following primers were used: forward primer: 5’- ATT GTT GTA GGT CAT AAA AGG AG-3’; reverse primer: 5’- TGA AGA CAC AAC ACT TAA GGA-3’. The following probes were applied for the separation of the alleles: G allele: 5’-Fam- CCA TCT TTA ACC ACG GCC A-BHQ-1-3’; A allele: 5’-Hex- CCA TCT TTA ACC ATG GCC A-1-3’.
The parameters for PCR amplifications were as follows: 95 °C for 3 min, followed by 44 cycles of 95 °C for 10 s, and then 59 °C for 50 s (rs34155785 SNP) or 57 °C for 50 s (rs7820268 SNP) or 55 °C for 50 s (rs9657182 SNP). The genotyping specific master mix from the PCR Biosystem (2x PCRBio Genotyping mix Lo-ROX) was used. The PCR experiments were performed with a Bio-Rad CFX96 C1000 real-time thermal cycler machine, and the data analysis was carried out with Bio-Rad CFX Manager version 1.6 (Applied Biosystems, University Park, IL, USA).
For the analysis of genotype frequencies and allele distributions SPSS software version 26.0 (IBM Corp., Chicago, IL, USA) and GraphPad Prism 6.01 (GraphPad Software, San Diego, CA, USA) was used. We applied the chi-square test for comparing the distributions of the examined genotypes and alleles, and the t-test for comparing the averages in the two groups. Odds ratio (OR) and 95% confidence interval (CI) was calculated for the SNP variants in the groups of early and late onset. A p value less than 0.05 was considered statistically significant.
We determined the frequencies of three polymorphisms of IDO1 gene in PD patients and matching healthy controls. The observed genotype frequencies of the PD and the control groups were in accordance with the Hardy-Weinberg equilibrium.
The genotype distribution of rs34155785 was 105 (100%) homozygous wild-types in the PD patient group, and 129 (100%) in the control group, we did not detect the insertion allele in either group. The allele frequencies were 210 (100%) wild-type alleles in the patient and 258 (100%) in the control group. This SNP variant is probably not associated with PD and do not affect the age at disease onset either, as no allelic differences could be observed between the two groups.
The genotype distribution of rs7820268 in the patient cohort was 56 GG, 38 GA and 11 AA, and in the control group was 59 GG, 63 GA and 7 AA. The allele frequencies were very similar, with 71.4% G allele and 28.6% A allele in the PD group and 70.2% G and 29.8% A in the controls (Table 2). This SNP variant is not significantly associated with the PD (genotype: p = 0.093, G allele: p = 0.149, A allele: p = 0.248), but it affects the age at disease onset (genotype: p = 0.042). The frequency of A allele was significantly higher in LOPD group than in EOPD group (p = 0.024, OR = 2.471 95% CI (1.117–5.467)) (Table 3).
| rs7820268 | |||||
| Genotype | PD patients | Controls | p value | ||
| GG | 56 (53.3%) | 59 (45.7%) | 4.744 (df = 2) | 0.098 | |
| AG | 38 (36.2%) | 63 (48.8%) | |||
| AA | 11 (10.5%) | 7 (5.4%) | |||
| Allele | |||||
| G | 150 (71.4%) | 181 (70.2%) | 0.091 (df = 1) | 0.763 | |
| A | 60 (28.6%) | 77 (29.8%) | |||
| Allele distribution | |||||
| G | Carriers | 94 (43.5%) | 122 (56.5%) | 2.079 (df = 1) | 0.149 |
| Non-carriers | 11 (61.1%) | 7 (38.9%) | |||
| A | Carriers | 49 (41.2%) | 70 (51.8%) | 1.337 (df = 1) | 0.248 |
| Non-carriers | 56 (48.7%) | 59 (51.3%) | |||
| rs9657182 | |||||
| Genotype | PD patients | Controls | p value | ||
| GG | 29 (27.6%) | 23 (17.8%) | 3.368 (df = 2) | 0.186 | |
| AG | 48 (45.7%) | 70 (54.3%) | |||
| AA | 28 (26.7%) | 36 (24.9%) | |||
| Allele | Patients | ||||
| G | 106 (50.5%) | 116 (49.5%) | 1.412 (df = 1) | 0.235 | |
| A | 104 (45%) | 142 (55%) | |||
| Allele distribution | |||||
| G | Carriers | 77 (45.3%) | 93 (54.7%) | 0.045 (df = 1) | 0.832 |
| Non-carriers | 28 (43.8%) | 36 (56.3%) | |||
| A | Carriers | 76 (41.83%) | 106 (58.2%) | 3.210 (df = 1) | 0.073 |
| Non-carriers | 29 (55.8%) | 23 (44.2%) | |||
| rs7820268 | ||||||
| Genotype | EOPD | LOPD | p value | OR (95% CI) | ||
| GG | 34 (64.2%) | 21 (42%) | 6.352 (df = 2) | 0.042 | - | |
| AG | 13 (24.5%) | 24 (48%) | ||||
| AA | 6 (11.3%) | 5 (10%) | ||||
| Allele distribution | ||||||
| G | Carriers | 47 (51.1%) | 45 (48.9%) | 0.047 (df = 1) | 0.828 | 1.149 (0.327–4.031) |
| Non-carriers | 6 (54.5%) | 5 (45.5%) | ||||
| A | Carriers | 19 (39.6%) | 29 (60.4%) | 5.073 (df = 1) | 0.024 | 2.471 (1.117–5.467) |
| Non-carriers | 34 (61.8%) | 21 (38.2%) | ||||
| rs9657182 | ||||||
| Genotype | EOPD | LOPD | p value | OR (95% CI) | ||
| GG | 19 (35.8%) | 9 (18%) | 4.795 (df = 2) | 0.091 | - | |
| AG | 23 (43.4%) | 24 (48%) | ||||
| AA | 11 (20.8%) | 17 (34%) | ||||
| Allele distribution | ||||||
| G | Carriers | 42 (56%) | 33 (44%) | 2.280 (df = 1) | 0.131 | 0.508 (0.210–1.232) |
| Non-carriers | 11 (39.3%) | 17 (60.7%) | ||||
| A | Carriers | 34 (45.3%) | 41 (54.7%) | 4.141 (df = 1) | 0.042 | 2.546 (1.020–6.351) |
| Non-carriers | 19 (67.9%) | 9 (32.1%) | ||||
| EOPD, Early onset | ||||||
The genotype distribution rs9657182 was 29 GG, 48 GA and 28 AA in the PD group, and 23 GG, 70 GA and 36 AA in the control group. The allele frequency results did not show remarkable differences, with 50.5% G allele in the PD group vs 45% G allele in the controls, and 49.5% A allele in the PD group vs 55% A allele in the control group (Table 2). This SNP variant is likewise not associated with PD (genotype: p = 0.186, G allele: p = 0.832, A allele: p = 0.073), but it may influence the age at disease onset (genotype: p = 0.091, G allele: p = 0.131, A allele: p = 0.042) (Table 3). Similarly, the A allele seems to be a factor affecting the age at onset of PD (Table 3), as carrying the allele associates with LOPD (54.7% vs 45.3%), and non-carrying associates with EOPD (67.9% vs 32.1%) (OR = 2.546, 95% CI (1.020–6.351)), p = 0.042 for AA + AG vs GG).
DA receptors, catechol-O-methyltransferase, monoamine oxidase B, NMDA receptors, adenosine A2A receptors, and cholinergic receptors are main targets of PD medication approved for clinical use. The drugs frequently cause serious side effects, become less effectual during treatment, and eventually lead to development of drug-resistant PD such as L-DOPA-resistant PD [52, 53]. Thus, a search for novel therapeutic targets is under extensive research. Intervention in the KP of TRP metabolism, adjacent biosystems, and the gut microbiota has been shed light on in search of novel biomarkers and drugs for neurodegenerative and psychiatric diseases [54, 55, 56, 57, 58, 59, 60, 61]. Glutamate and acetylcholine are main neurotransmitters responsible for cognition and behavior in which bioactive KYNs mediate neurotoxic, neuromodulatory, and immunological response in reaction to pathological insults [10, 54, 62].
The decreased concentrations of L-KYN and KYNA in the frontal cortex, putamen and SNpc, and elevated concentrations of 3-HK in the putamen and SNpc were observed in postmortem brain of PD patients [62, 63]. Clinical manifestations of dysregulation of kynurenine metabolism, including impairments in memory and learning (i.e., memory consolidation, poor planning, defects in set-shifting, impaired working memory, and executive dysfunction) are common in PD and correlate with a typical cognitive pattern due to prefrontal cortex dysfunction [64, 65, 66, 67, 68, 69, 70]. In addition to the central nervous system, peripheral samples showed abnormalities such as the increased activity of kynurenine aminotransferase (KAT) and elevated KYNA levels in red blood cells, which may be a protective response [71]. Natural product curcumin was reported to relieve pain and stress through the KYN metabolic pathway [72]. The KP is proposed to be a potential biomarker and target for treatment of Alzheimer’s diseases, schizophrenia, and depression [55, 72, 73, 74, 75, 76, 77]. Psychedelic psilocybin was reported to relieve depressive and anxiety symptoms of patients with terminal illness [78]. The action is considered at least partly through the KP. Furthermore, delivery of active agents to the brain through the blood-brain barrier is under extensive research [79]. Other in vitro and in vivo studies showed therapeutic opportunities through elevated levels of KYNA in PD [48, 80, 81].
However, in recent years growing body of evidence has accumulated on the importance of the KP in PD pathogenesis, only limited data are available on the genetic alterations of KP enzymes in PD yet. Until now, only two kynurenine enzymes, ACMSD and KMO genes polymorphisms have been investigated in PD [82, 83]. ACMSD play a key role in the KP, generating neuroactive metabolites which may play a crucial role in PD pathogenesis by inhibiting excitotoxicity and inflammation, therefore it could be a potential therapeutic target in PD [84]. Genome-wide association studies described that ACMSD polymorphisms are risk factors in PD [85, 86, 87], however its importance could not be confirmed in different populations [88, 89].
Previously, SNP analysis of KMO in PD and healthy controls were compared and a subgroup analysis was conducted according to age of onset. Four loci of SNPs did not reveal any significance between PD and healthy controls and between subgroups of age of onset [90].
In this study we compared the frequencies of three SNPs of IDO1 gene between PD patients and control individuals. Our results revealed that the investigated SNPs were not associated with PD, however, the rs7820268 and the rs9657182 SNPs showed significant associations with age of onset of the disease. The A allele of rs7820268 and rs9657182 SNPs associated with LOPD. It is well known that there are some genetic factors which show associations with age of onset in PD. Several mutations in PINK-1, parkin, LRRK2 and GBA genes are associated with EOPD. Our findings suggest that the A allele of these SNPs potentially delayed the development of PD [91, 92, 93, 94, 95].
The IDO1 gene is located on chromosome 8 spanning 14,900 nucleotides.
It encodes the 403 amino acid IDO1 enzyme. IDO1 plays a crucial role in the
production of immune and neuroactive KP metabolites, therefore its importance in
immunomodulation and inflammatory processes is expected [96]. Previously have
been described, that the minor allele of the rs7820268 SNP was shown to impair
CD8
PD is a complex multifactorial disease, underlying mechanisms causing the pathological conditions of PD is not fully understood yet. In addition to the age, which is the most important risk factor for neurodegenerative disease, genetic, epigenetic, environmental and lifestyle are initiation factors which play a role in the pathogenesis of PD [16, 90, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113]. The preclinical studies, and diagnostic precision of PD are under extensive research, applying in vitro, in vivo, and in silico methodologies [114, 115, 116, 117, 118, 119]. Until now, 21 PARK genes have been described in human genome as causative factors of the disease, furthermore, genetic variants of 26 loci have been shown to be important risk modifiers for PD. Beside genetic factors, there are more evidence that epigenetic mechanisms, like DNA methylation, histone modifications and dysregulation of non-coding RNAs (e.g., long non-coding RNA and microRNA) play a role in pathogenesis of PD [109, 120]. Several studies described that the gene expression of different genes is altered in PD, which may help predict the PD progression, and identify new targets for therapeutic intervention [121, 122, 123]. Moreover, it is evident that the KP are involved in the pathogenesis of PD. Our result, in line with data of others’, strengthen the involvement of IDO1 in the pathogenesis of PD. Furthermore, the influence of SNPs on the non-motor symptoms of PD is of particular interest. The gene variants of alpha-synuclein SNCA rs11931074 and glucosylceramidase beta 1 rs708606 have been reported to correlate with hyposomnia in PD [124, 125]. The direct involvement of KP metabolism in the non-motor symptoms of PD is to be explored.
This study had several limitations including its relatively small sample size, the small number of SNP loci, heterogeneity of the study subjects, and the collection of blood samples at a single center. Furthermore, enzyme activities of IDO1, IDO2, and TDO were not measured. Thus, it was not known if IDO2 and/or TDO compensated the polymorphic variants of IDO1. Prospective studies in larger patient number, with the larger number of SNP loci, and data with IDO2 and TDO activities are needed to validate our findings.
This study explored the potential role of IDO1 gene polymorphisms in PD. None of three IDO1 SNPs investigated in this study were significantly associated with PD; however, two IDO1 SNPs showed correlated with the age onset of PD, suggesting that the gene polymorphisms may not play a direct role in pathogenesis of PD, but may influence the disease onset probably as secondary risk factors. The further investigation is expected in search of roles of gene polymorphism in risk, onset, prognosis, progression of PD including SNPs, structural variants, and the disease-related pathways.
AMPA, 2-amino-3-carboxymuconate-semialdehyde decarboxylase; AMPA, alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; CSF, cerebrospinal fluid; DA, dopamine; 3-HAA, 3-hydroxyanthranillic acid; 3-HK, 3-hydroxykynurenine; IDO, indoleamine 2,3-dioxygenase; IFN, interferon; KAT, kynurenine aminotransferases; KMO, kynurenine 3-monooxygenase; KP, kynurenine pathway; KYNA, kynurenic acid; NMDA, N-methyl-D-aspartate; PD, Parkinson’s disease; QUIN, quinolinic acid; ROS, reactive oxygen species; SNP, single nucleotide polymorphisms; TDO, tryptopan 2,3-dioxygenase; TRP, tryptophan.
NT designed the research study. NT performed the research. KB analyzed the data. NT, RMT, KM, ZS, FS, KB, MT, PK and LV wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
All patients gave their informed consent in accordance with the Declaration of Helsinki, and the study was approved by the Medical Research Council Scientific and Research Ethics Committee (47066-3/2013/EKU (556/2013)).
The authors are sincerely grateful to Dr. Michael D. Lovelace and his co-workers for critical reading and their kind help.
This research was funded by the Economic Development and Innovation Operational Programme [GINOP 2.3.2-15-2016-00034; GINOP 2.3.2-15-2016-00048], National Research, Development and Innovation Office [NKFIH-1279-2/2020 TKP 2020], TUDFO/47138-1/2019-ITM, National Scientific Research Fund [OTKA138125] and ELKH-SZTE. Project no. TKP2021-EGA-32 has been implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-EGA funding scheme.section is required for all papers.
Given his role as Guest Editor, Masaru Tanaka had no involvement in the peer-review of this article and has no access to information regarding its peer-review. Full responsibility for the editorial process for this article was delegated to Graham Pawelec. The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.