
†These authors contributed equally.
Academic Editor: Graham Pawelec
Background: Peripheral alterations of mitochondrial DNA copy number (mtDNAcn) in obesity and associated co-morbidities have been previously shown. Furthermore, the possibility that methylation could occur in the mtDNA (in particular in the displacement loop, D-Loop) and regulate its functions has been raised. However, limited data about mtDNA methylation in adipose tissue are currently available. Since a strict crosstalk between the nucleus and mitochondria exists, especially in terms of the one-carbon cycle (that supports methylation reactions in the cell), we investigated methylation in selected areas of the mitochondrial and nuclear DNA and their expression in visceral adipose tissue (VAT) samples of patients with severe obesity. Methods: VAT biopsies were collected from surgery patients to isolate DNA and RNA. Gene expression and mtDNAcn were assessed through qPCR. DNA methylation in both nuclear and mitochondrial areas were determined through bisulfite pyrosequencing. Results: Methylation levels of the mtDNA were only marginally associated with the obesity degree (higher D-Loop methylation in severe obesity) and were not correlated with mtDNAcn. A significant correlation between D-Loop methylation and LINE-1 methylation was observed in VAT samples, and this was independent from the obesity degree. A progressive reduction of mtDNAcn and increase in NRF1 expression levels were measured in VAT in severe obesity. NRF1 expression was directly correlated with PPARG and MTHFR expression levels, while mtDNAcn was associated to TFAM expression. The correlation between mtDNAcn and TFAM expression was affected by the obesity status. Conclusions: This evidence supports the hypothesis that mtDNA alterations occur in obesity and a complex dynamic correlation between mitochondrial and nuclear DNA methylation exists, highlighting the need for further investigations.
Visceral adipose tissue (VAT) is a hormonally active component of total body fat, with unique biochemical features affecting both physiological and pathological processes in the human body. Abnormally high deposition of VAT is known as visceral obesity. This body composition phenotype is associated with several complex disorders, in particular metabolic syndrome, diabetes and cardiovascular disease [1, 2]. Given the complex multifactorial origin of this condition, a special interest is directed towards epigenetic mechanisms that might contribute to shift healthy to pathological VAT phenotypes in response to environmental triggers [3, 4, 5].
Among cellular organelles that play a major role in regulating the functionality of VAT there are mitochondria [6]. Mitochondria are involved in many key cellular processes such as apoptosis, calcium homeostasis, reactive oxygen species production, and most importantly, adenosine triphosphate (ATP) generation by oxidative phosphorylation (OXPHOS). Mitochondrial dysfunction results in detrimental effects on adipocyte differentiation, lipid metabolism, insulin sensitivity, oxidative capacity, and thermogenesis; indeed, it has been strongly correlated with metabolic diseases pathogenesis [6]. Mitochondria have their own genome, the mitochondrial DNA (mtDNA); in humans, it is a double-stranded, closed-circular molecule of 16569 base pairs, present in each cell in multiple copies, whose number (mtDNAcn) can vary according to the cell type and health status [7]. One strand of the mtDNA is referred to as the heavy strand, and the other strand is referred to as the light strand; each one has its own promoter, a light-strand promoter (LSP) and a heavy-strand promoter (HSP), regulating mitochondrial gene’s transcription [8]. mtDNA does not contain introns and encodes just 13 polypeptides which are subunits of the respiratory complexes involved in the OXPHOS. Interestingly, most of them are located into the heavy strand, except for the ND6 gene, which is located in the light strand [9, 10]. The promoters for transcription (LSP and HSP) and the origin of replication of the H strand (OH) are located in a specific area of the mtDNA, the displacement loop (D-loop) which is a noncoding control region [10], both involved in mtDNA transcription and replication [11, 12]. As a result, this region is the site of interaction for the nuclear-encoded transcription and replication factors that translocate to the mitochondrion to mediate mitochondrial genomic activities. Interestingly it has been recently shown that mtDNA can be methylated [13, 14, 15], especially in the D-loop area, and in a strand-specific manner [16, 17], thus potentially differentially affecting mtDNA functions [18]. Variation of mtDNA methylation have been reported in particular due to environmental exposures [15, 19, 20, 21, 22, 23, 24], including also diet, making this mark a potential intermediary agent acting at the interface between the environment and molecular pathways’ activation. Although promising, mtDNA methylation and its impact on replication and transcription are still a discussed topic [25].
Not only mitochondrial genes, but also several nuclear genes regulate
mitochondrial functions as well as the epigenetic homeostasis [26]. Indeed, genes
expressed in mitochondria work in concert with those expressed in the nucleus to
regulate OXPHOS [27]. Nuclear-encoded genes that are important for adipose tissue
metabolism and mitochondrial functions include mitochondrial transcription factor
A (TFAM), that encodes for the protein primary driving the mtDNA
packaging generating nucleoids [28, 29], peroxisome proliferator-activated
receptor
Recent evidence associated the 1C metabolism to body composition and epigenetic features in obesity [24, 37]; moreover, a reduction of mtDNAcn in circulating blood has been detected as a common feature in both individuals with obesity [24] and patients with cardiovascular diseases [38].
Since epigenetics is tissue-specific [39], the aim of this study was to evaluate
if these alterations, previously measured at systemic level, are also typical of
dysfunctional VAT in severe obesity (BMI
VAT biopsies from omental depot were obtained from 53 subjects with a wide BMI range, spanning from 22.8 to 53.5, undergoing bariatric and non-bariatric (i.e., cholecystectomy) surgery at the General Hospital Azienda Ospedaliera Universitaria (AOU Ospedali Riuniti; Ancona, Italy). Samples from VAT were collected, immediately snap frozen in liquid nitrogen, and stored at –80 °C. The study protocol was approved by the Ethics Committee of Ospedali Riuniti (Ancona, Italy). The information collected included anthropometric data (BMI) and medical history. Exclusion criteria were the presence of infectious or autoimmune diseases, cancer and neurodegenerative disorders.
DNA was isolated from adipose tissue using QIAamp Fast DNA Tissue Kit (Qiagen, Milano, Italy) according to the manufacturer’s instructions. The amount of input genomic DNA was quantified by Qubit dsDNA BR Assay Kit (Applied BioSystems, MA, USA). DNA was stored at –80 °C until use. Total RNA was isolated using the Norgen Biotek Kit (Thorold, ON, Canada), according to the manufacturer’s instructions. A first step with Trizol (ThermoFisher Scientific, MA, USA) and chloroform (Sigma-Aldrich, MO, USA) was performed to remove lipid content. RNA was stored at –80 °C until use. RNA amount was determined by spectrophotometric quantification with Nanodrop ONE (NanoDrop Technologies, Wilmington, DE, USA).
Total RNA was reverse-transcribed using High-Capacity cDNA RT Kit with RNase
Inhibitor (Applied BioSystems, MA, USA) in a total volume of 20
As previously published [38, 40], relative mtDNAcn quantification was performed
by real-time PCR (CFX96; Biorad, CA, USA) considering nDNA as a normalizer
(mtLEU_fw; 5′-CACCCAAGAACAGGGTTTGT-3
Bisulfite pyrosequencing was performed to evaluate DNA methylation levels in
both nuclear and mitochondrial sequences. To avoid inaccurate bisulfite
conversion due to the circular structure of the mtDNA, 600 ng of DNA were digested
with BamHI (New England Biolabs, MA, USA). Digestion with BamHI was performed in
order to linearize the mtDNA, thus reducing the risk of false positive
methylation results [41]. Then, DNA was converted with sodium bisulfite using the
EZ-96 DNA methylation Gold kit (Zymo Research, CA, USA) according to the
manufacturer’s instructions. Primers used to amplify the areas of interest (both
nuclear and mitochondrial) are shown in Supplementary Table 1B.
Mitochondrial DNA methylation was assessed in two areas of the D-loop, one
located in proximity of the mtDNA replication initiation site (Oh) in the heavy
strand (heavy chain D-Loop region, HDLR), the other in proximity of the
transcription initiation site in the light-strand promoter (light chain D-Loop
region, LDLR), as previously described and validated by Vos et al. [16].
The D-loop region is highly relevant to mitochondrial gene transcription, and is
one of the few regions on mtDNA that is not included in the nuclear DNA as a
NUMTs [42]. For this reason, analysis of methylation in this area does not
require previous isolation of mtDNA from nuclear DNA. Concerning nuclear genes,
we studied promoters of MTHFR, PHGDH and PPARG, whose
methylation has been previously associated to obesity [35, 36, 43, 44]. For
MTHFR, a sequence in the 5
Statistical analysis was performed by using SPSS (IBM SPSS Statistics for
Windows, Version 24.0, USA) and R version 3.5.3 (R Core Team, Austria). The
Shapiro-Wilk test was used for the analysis of the normality of data
distribution. Parameters with skewed distributions were appropriately
log-transformed before parametric analyses. Pearson’s correlation was measured to
test parametric correlations between continuous variables. Multiple linear
regression was applied to test correlations between continuous variables
adjusting the model for covariates (age, sex and diagnosis of diabetes).
Principal Component Analysis (PCA) with Varimax rotation was used as a technique
for dimension reduction in multivariate analysis. Samples with missing data were
excluded from the analysis; no imputation was performed. A p value
The analysed population was composed of 53 subjects (25% males; 75% female)
with a mean age of 51
The mtDNAcn was inversely correlated to BMI (Pearson’s correlation = –0.344;
p = 0.011) (Fig. 1A). This association was even more significant after
adjusting the analysis for confounding factors (age, sex and diagnosis of
diabetes) (
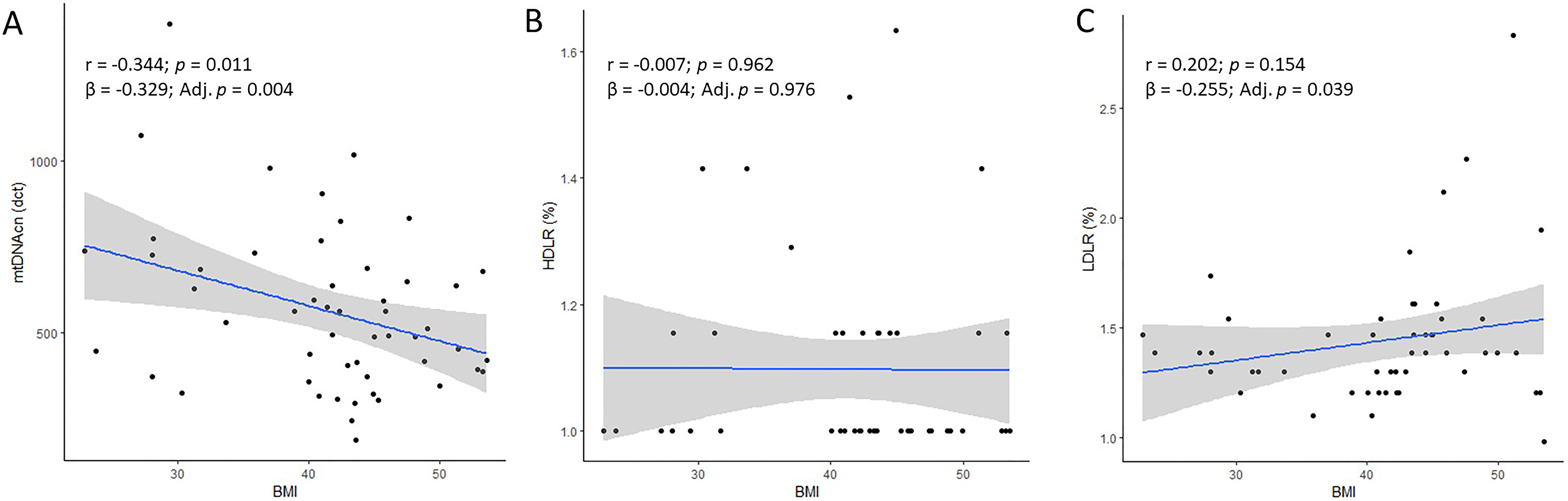 Fig. 1.
Fig. 1.Correlations between BMI and mitochondrial DNA copy number and methylation. Scatter plots showing unadjusted Pearson’s correlation between BMI and mtDNAcn (A), % of DNA methylation in the HDLR (B) or in the LDLR (C).
DNA methylation levels measured in the D-loop area of the mitochondrial DNA were
different between HDLR and LDLR. In particular, methylation levels measured in
the HDLR were low (mean% = 1.22
A correlation matrix to explore the relationship between the expression pattern
of genes involved in mitochondrial functions, mtDNAcn and obesity degree was
computed in the whole population. A cluster of correlated expression levels
including genes that regulate mitochondrial biogenesis
(PGC1
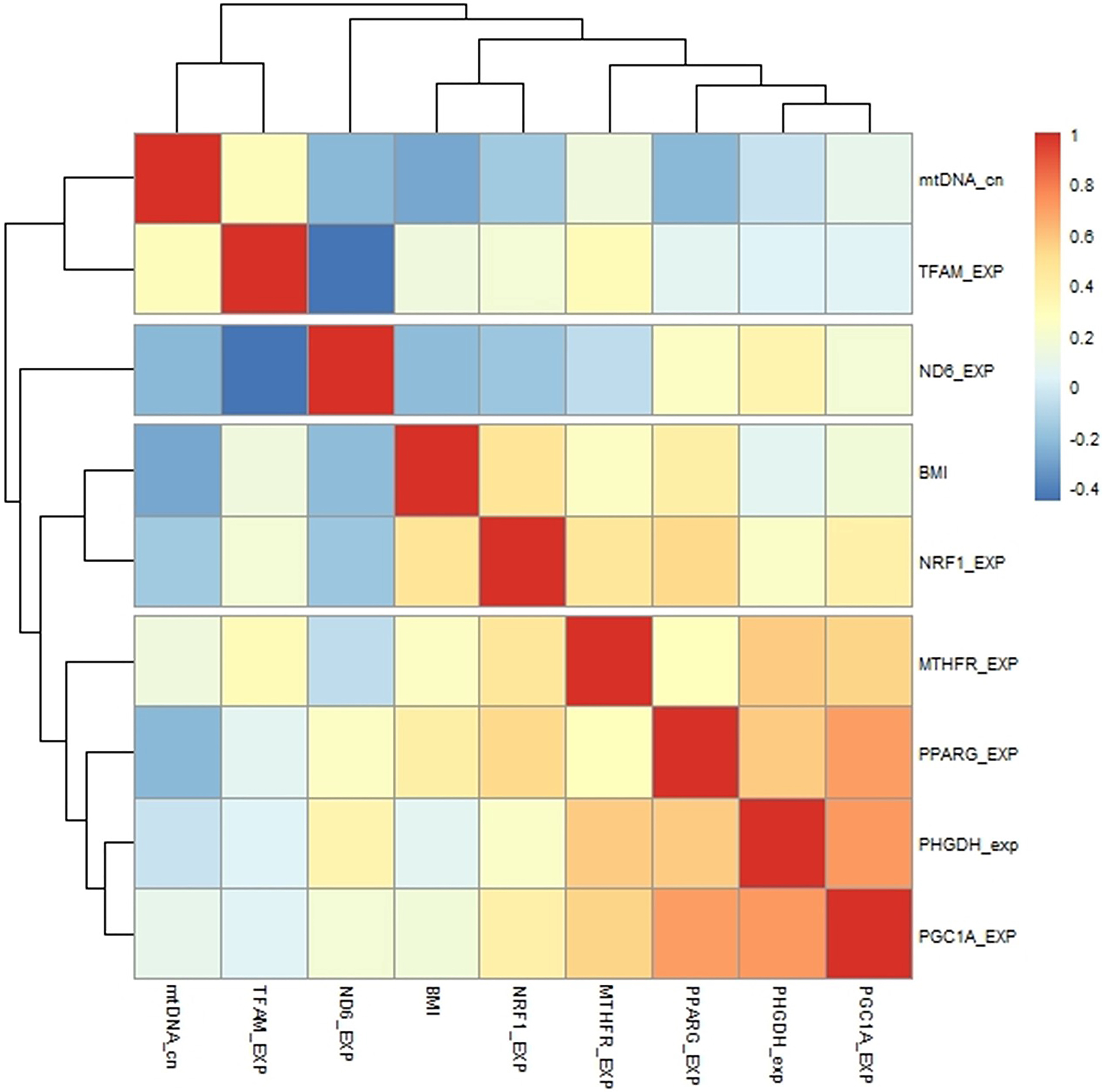 Fig. 2.
Fig. 2.Heatmap of expression of genes involved in mitochondrial and 1C metabolism. This heatmap shows correlations (Pearson’s correlations; no adjustments) between expression of mitochondrial genes, genes regulating the 1C cycle and BMI.
Given the presence of multiple co-correlations among these variables (Bartlett’s
Test of Sphericity, p = 7.5
BMI was significantly correlated to PC3 (Spearman’s Rho = 0.524; p =
7.3
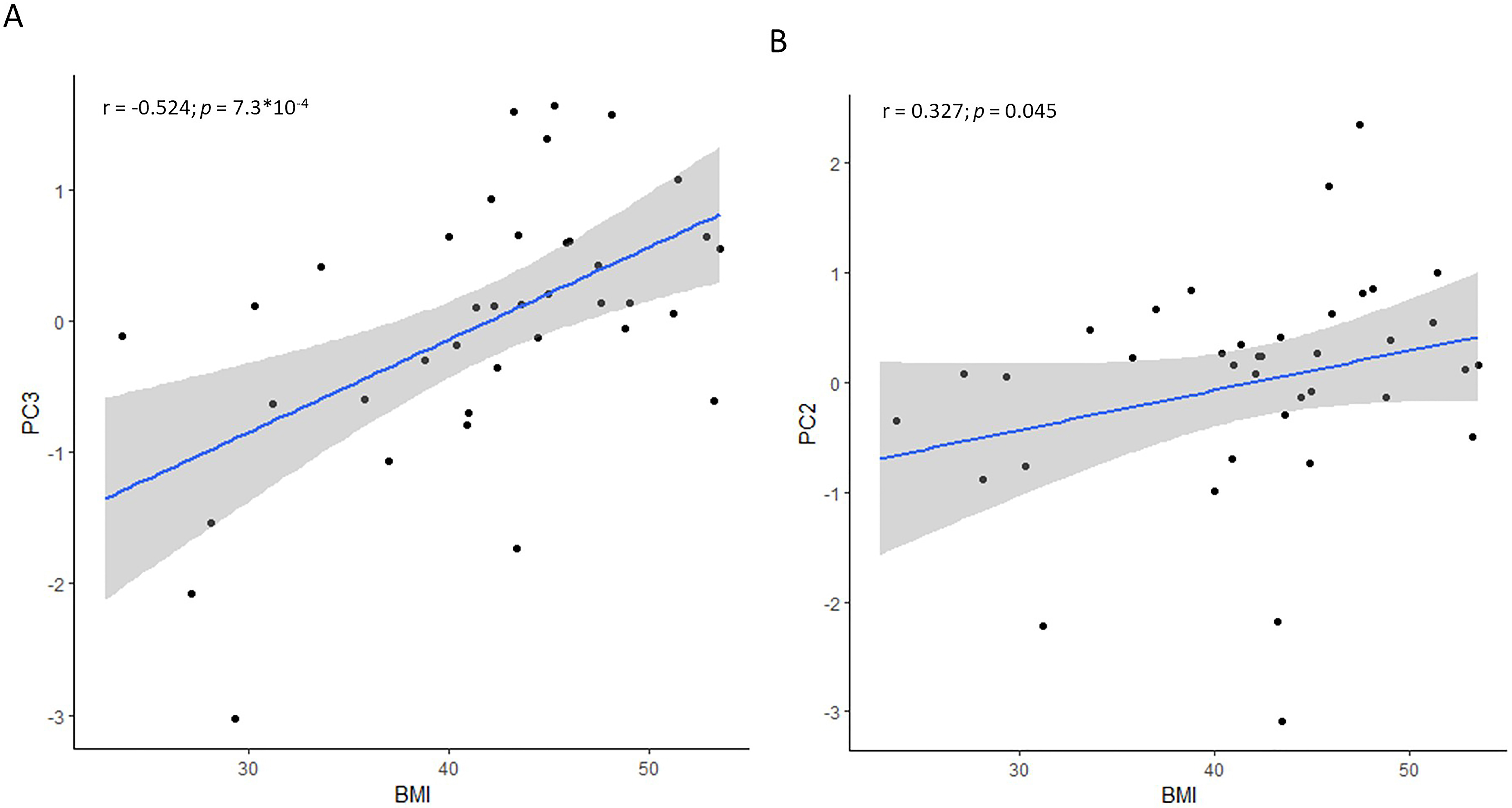 Fig. 3.
Fig. 3.Correlation between BMI and PC describing mitochondrial and 1CC gene expression. Correlations between BMI and PC3 (A) and PC2 (B) resulting from the PCA.
Despite the genes involved in the 1C cycle (mostly represented by PC1) were not
directly associated to BMI, MTHFR and PPARG expression levels
were strongly correlated to NRF1 (Supplementary Table 2A).
Moreover, adjusting for age, sex and diabetes, PC1 was positively correlated to
the global levels of D-Loop methylation (HDLR + LDLR) (
PCA results suggest that individuals with high BMI had lower mtDNAcn (PC3), but
higher TFAM expression (PC2) values. This was confirmed by a linear regression
model including both these parameters, showing that TFAM expression
(
Since methylation of MTHFR promoter has been previously associated to
obesity [24], associations between methylation levels and MTHFR
expression and obesity degree were tested in VAT samples. No significant
correlations between methylation in the analysed area and MTHFR
expression (r = –0.243; p = 0.111) or BMI (p = 0.384) were
detected. No significant association with PPARG expression (r = –0.130;
p = 0.400) or with BMI (
Finally, since ND6 is the unique mitochondrial gene located in the light strand of the mtDNA, the correlation between its expression level and LDLR methylation was tested. No significant correlations were measured (r = –0.069; p = 0.668) in these samples. Plots are shown in Supplementary Fig. 3.
Surprisingly, mtDNAcn was nominally negatively correlated with methylation levels of MTHFR and PHGDH nuclear genes (Fig. 4), despite it was not associated with their expression (Supplementary Table 2B). These correlations were independent from the BMI.
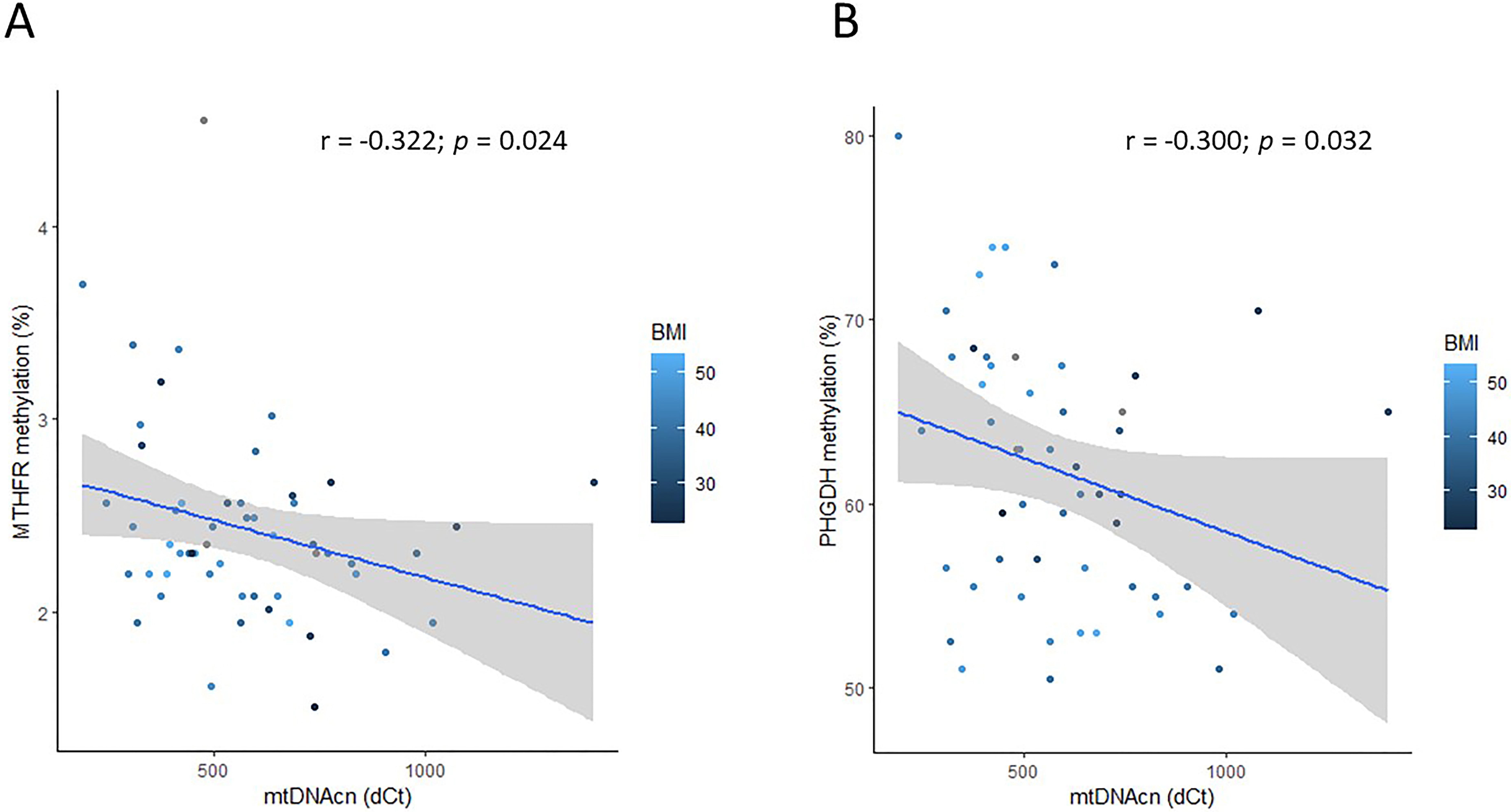 Fig. 4.
Fig. 4.Correlation between mtDNAcn and methylation in nuclear genes involved in 1CC. MtDNAcn nominally correlates with methylation level at MTHFR (A) PHGDH (B) promoters.
Given the potential alteration of the overall cellular methylation pattern in
obesity, methylation levels at the LINE-1, a surrogate marker of global nuclear
DNA methylation, were measured in the VAT samples. Adjusting the analysis for age
and sex, no direct associations between LINE-1 methylation and BMI (
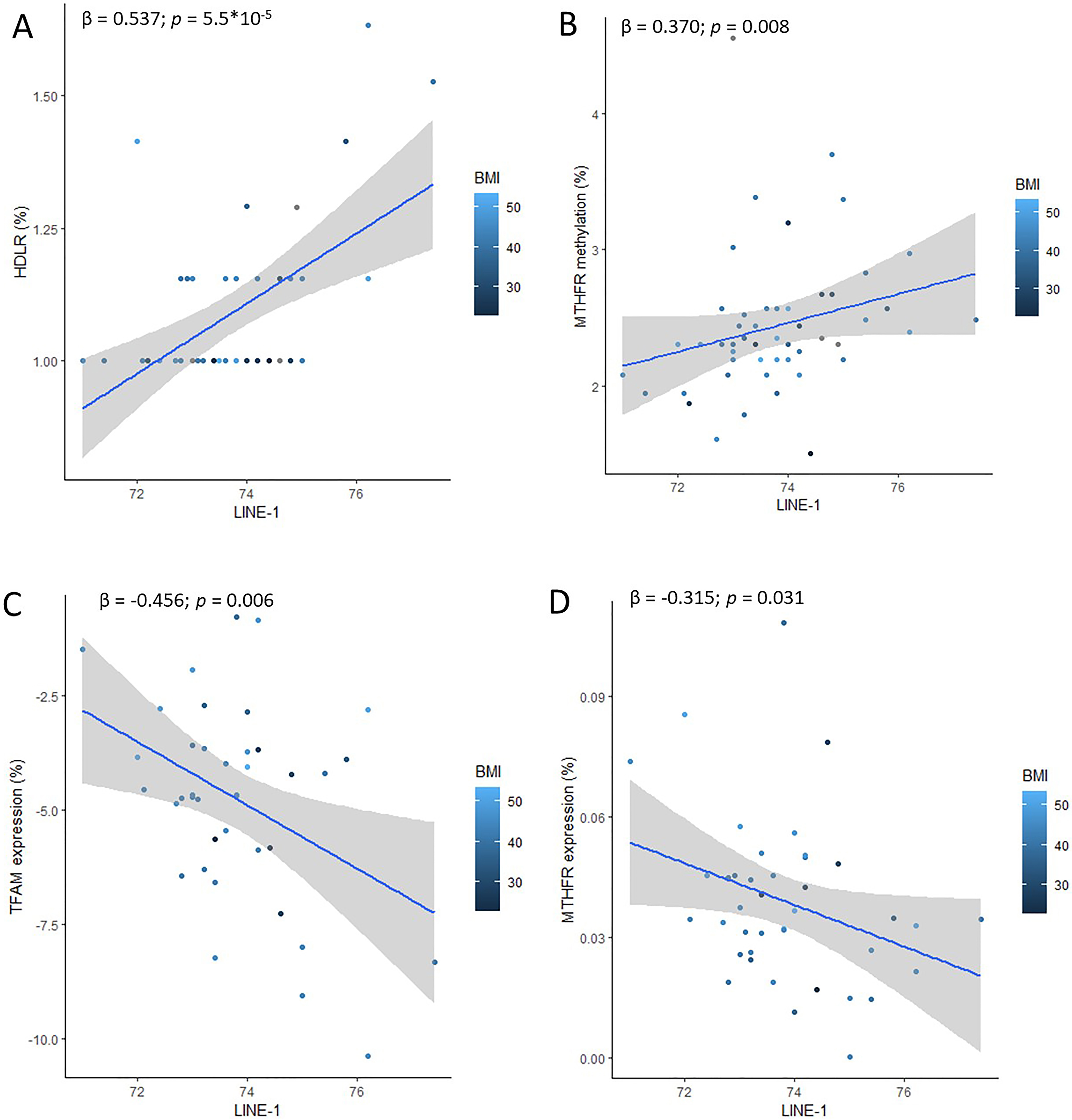 Fig. 5.
Fig. 5.Associations between global DNA methylation measured in LINE-1 and methylation in specific areas of the genome involved in the regulation of mitochondrial dynamics. Associations between LINE-1 methylation levels and HDLR methylation (A), MTHFR methylation (B), TFAM expression (C) and MTHFR expression (D). All the associations are independent from BMI.
Peripheral changes in mtDNAcn have been associated with obesity complications such as cardiovascular disease and diabetes [38, 56, 57]. Moreover, lower mtDNAcn has also been associated with frailty and all-cause mortality [58]. Cells with reduced mtDNAcn show reduced expression of vital complex proteins, altered cellular morphology and lower respiratory enzyme activity [59].
Previous studies have shown that obesity is associated with mtDNAcn changes in peripheral blood [24, 60, 61]. However, contrasting evidence about the association between obesity and mtDNAcn measured in adipose tissue was reported. Lindinger and colleagues showed that omental adipose tissue in obese subjects had a decreased mtDNAcn, which was not correlated with basal metabolic rate and fat oxidation rate [62]. On the contrary, recent investigations did not find changes in mtDNAcn in omental adipose tissue of obese people with respect to controls, while the diagnosis of diabetes was associated to an increase of this parameter [63], suggesting that diabetes rather than obesity might have driven the previously reported association. Litvinova and collaborators have recently shown that mtDNAcn in various fat depots was higher in obese patients with type 2 diabetes than in obese patients without diabetes or in the control subjects and it was related to the levels of leptin and proinflammatory cytokines [64]. In our study, adjusting the analysis for age, sex and diagnoses of diabetes, we found that mtDNAcn in VAT decreased linearly with BMI. This result was coherent with data collected in subcutaneous adipose tissue (SAT) by Fischer et al. [65] and Heinonen et al. [66], that showed a decrease of mtDNAcn in isolated subcutaneous adipocytes of subjects with obesity. According to these findings, a down-regulation of mtDNA-encoded proteins has been measured in VAT of subjects with obesity and associated with aging progression [67]. Coherent findings from animal models showed that a high-fat diet was able to reduce adipose tissue mitochondrial proteins, triggering glucose intolerance in rats [68].
In addition, methylation of the mtDNA has been recently proposed as a potential
regulator of mitochondrial functions, despite this is still a controversial topic
[21, 25, 69]. Alterations of mtDNA methylation patterns have been measured in
obesity analysing peripheral samples [22, 23, 70, 71, 72], but no previous studies
investigated this parameter in the omental adipose tissue of people with severe
obesity. In our population, methylation in the LDLR was mildly correlated with
the BMI adjusting the analysis for sex, age and diagnosis of diabetes. This
suggests that increased methylation levels in this area might be implicated in
dysfunctional mitochondria, that are typical of people with obesity. However,
despite significant, this association is driven by a few individuals with extreme
phenotype (BMI
Interestingly, HDLR (but not LDLR) methylation was strongly correlated to LINE-1 methylation level (an estimator of global level of DNA methylation in the nucleus also associated to genome stability and to the adiposity status [73]), suggesting a correlation between mitochondrial and nuclear dynamics also from the epigenetic point of view. Indeed, previous studies highlighted a connection between mtDNA and epigenetic regulations in the nucleus [69, 74]. For example, cells harbouring distinct mitochondrial polymorphisms showed differences in nuclear DNA methylation [75, 76]. Recent evidence suggests that also nuclear DNA methylation might contribute to regulate mtDNA methylation. For example, some nuclear encoded proteins involved in mitochondrial function (e.g., PGC1a and NRF1) can up-regulate or down-regulate, respectively, the levels of intra-mitochondrial DNMT1 [77]. However, this is the first study reporting a correlation between methylation at mitochondrial DNA and nuclear LINE-1 sites (which do not encode for specific proteins working in the mitochondrion). It has been recently described that LINE-1 demethylation and consequential retrotransposons activation can be triggered by mitochondrial stress [78], thus representing an additional link between mitochondrial dysfunctions and the nuclear epigenome to investigate. Moreover, in our study LINE-1 methylation was also highly correlated with TFAM expression, which is involved in the regulation of mtDNA copy number, stability, replication and transcription [55, 79]. Remarkably, it has been recently shown that D-loop methylation has the potential to impact TFAM-DNA recognition [18], thus possibly regulating the TFAM-dependent activities in vitro. Since TFAM expression is also correlated to global methylation levels in the nuclear DNA (LINE-1), we can speculate that epigenetic regulations might operate both at mitochondrial (where TFAM operates) and nuclear level (where TFAM is encoded) to regulate the nucleoid functions. These findings suggest further investigations to corroborate this hypothesis. Remarkably, low mtDNAcn and high TFAM expression were independently associated with the obesity degree in the VAT samples analysed in this study. Interestingly, the correlation between mtDNAcn and TFAM was perturbed by obesity, suggesting a deregulation of the ratio between mtDNAcn and the proteins around which it is wrapped (TFAM) to assemble nucleoids. Despite it has been previously demonstrated that mtDNAcn regulation is directly dependent on the TFAM protein levels [55], recently it has emerged that also the ratio of TFAM-to-mtDNAcn is critical for regulating the normal mtDNA expression and is associated with nucleoid replication and transcription both in vitro [54] and in vivo [80]. In mice, a moderate increase in TFAM protein levels still resulted in a normal TFAM-to-mtDNA ratio and unaltered mtDNA expression. However, very high TFAM levels lead to deficient OXPHOS and early postnatal lethality [80]. The authors suggested that TFAM can act as a repressor of mtDNA expression, and this effect can be counterbalanced by tissue-specific expression of regulatory factors. Despite we could not quantify TFAM protein in our samples, results suggest that the direct correlation existing between TFAM expression and mtDNAcn is lost in subjects with severe obesity, which display high TFAM expression without increasing their mtDNAcn.
PCA analysis confirmed that TFAM and ND6 expression (PC2) were correlated to BMI. Also, lower mtDNAcn and higher NRF1 expression (PC1) were identified as markers of severe obesity in our VAT samples. Of note, NRF1 expression was correlated with genes regulating the one-carbon cycle (MTHFR, PHGDH), despite they were not directly associated to BMI. No direct correlations between gene expression and DNA methylation in their respective promoters were detected.
Independently from the BMI, LINE-1 methylation was also associated to methylation in the MTHFR promoter and negatively correlated with MTHFR expression. This is coherent with previous findings suggesting that demethylation of MTHFR promoter might be a strategy to promote MTHFR transcription only in case of suppression of methylation pathways, such as in case of folate deficiency [24]. However, these associations were independent on the obesity condition in this sample. High methylation at MTHFR and PHGDH promoters’ level were nominally correlated to mtDNAcn. According to our findings, recent evidence has shown that changes in mtDNAcn can affect nuclear DNA methylation at specific loci and results in differential expression of specific genes with an impact on human health and disease via altered cell signalling [81]. A bidirectional control of the nuclear and mitochondrial genomes through modulation of DNA methylation to control mtDNA copy number and gene expression has been demonstrated also in tumours [82]. Moreover, adipose tissue mitochondrial dysfunction in obesity has been associated to a specific DNA methylation signature in adipose-derived stem cells [83]. Our findings corroborate the hypothesis of the existence of a crosstalk between the nuclear and the mitochondrial genome [26], also in terms of epigenetic regulations, even though the direction of this association is still to be clarified.
A limit of this study is the low number of samples, especially VAT from healthy normal weight individuals, whose adipose tissue biopsies from surgery were difficult to obtain. However, analysing extreme phenotypes (i.e., VAT from people having a BMI from 22 to 53) should increase the possibility to identify pathways that are normally subtly altered in mild obesity. Secondly, VAT is mostly but not only composed by adipocytes. It also contains pre-adipocytes, fibroblasts, immune cells and vascular cells, which are collectively known as the stromal vascular fraction [84]. The number and phenotype of these cells vary depending on the adipose tissue depot and is also different between obese and lean individuals [85]. Thus, cellular heterogeneity might blur differences in molecular marks which are cell specific such as epigenetic and gene expression changes. Also, we cannot exclude that methylation in other areas of both the nuclear and the mitochondrial genome might play a relevant role in regulation of gene expression in the instances where we did not identify a direct correlation with this epigenetic mark.
In addition, even though we reduced the risk of potential failure of bisulfite conversion due to three-dimensional structures of the mtDNA by linearizing it before the conversion, some other limitations must be recognized when applying bisulfite pyrosequencing to the analysis of mitochondrial DNA: (1) the assessment of methylation with this technical approach was limited to CpG sites, (2) it was not possible to distinguish between methylation and hydroxymethylation, (3) methylation levels in the mtDNA are typically low and near to the limit of detection of the instrument and (4) preferential amplification of low-methylated sequences might occur (in case methylation exists also out of the CpG, as it has been suggested in mtDNA [86]), thus leading to an underestimation of the methylation levels. Despite these limitations, bisulfite pyrosequencing remains the gold standard for targeted analyses of DNA methylation because of its high scalability, specificity and robustness of the results which are given as a percentage of the overall amplicons. Further investigations through Nanopore sequencing technology are planned to provide the missing information.
Finally, the observational design of this study reduces the possibility of mechanistic speculations; in particular, it is not possible to demonstrate the causality of the associations measured between molecular marks and obesity. Also, no information about dietary habits of these individuals were available. Thus, it was not possible to adjust results for different dietary patters. Nevertheless, the impact of diet on mtDNA copy number and methylation has been investigated in another study from our group [87].
Evidence from this study suggests that mtDNA can be methylated, especially in the light-strand promoter area (LDLR). According to previous findings [16], despite the low levels of methylation detected in the initiation of mtDNA replication site in the heavy chain (HDLR) do not correlate with mtDNAcn, a possible biological relevance is suggested by their association to global nuclear DNA methylation levels. Since mitochondrial dynamics are correlated to DNA methylation pathways also through the regulation of mtDNAcn, further studies on this topic, including a specific focus on epigenetic regulation of the mitochondrial DNA are warranted. Moreover, additional investigations are required to clarify the role of mtDNAcn in adipocytes, especially in association to the TFAM protein, since conflicting findings exist on its involvement on inflammatory processes [88, 89], and also taking into account the possibility of an involvement of mitochondrial epigenetics in regulating its functions.
The data that support the findings of this study are available in the methods and/or supplementary material of this article.
ATP, Adenosine triphosphate; D-Loop, Displacement Loop; HDLR, Heavy chain D-Loop
region; LDLR, Light chain promoter D-Loop Region; LINE-1, Long interspersed
nuclear element-1; MtDNAcn, Mitochondrial DNA copy number; MTHFR,
Methylenetetrahydrofolate reductase; NRF1, Nuclear respiratory factor 1; OXPHOS,
Oxidative phosphorylation; PGC1
LB Designed research, conducted epigenetic research, analysed data, performed statistical analysis, drafted and reviewed the manuscript. JP Designed research, collected samples, carried out gene expression analyses, review the manuscript. IP Carried out mtDNAcn assessment and epigenetic assays. EDM Collected samples and carried out gene expression analyses. GL, and MG—Resources. AG and RG Supervised the project, reviewed and edited the manuscript.
Not applicable.
We thank the School of Pharmacy of the University of Camerino for having co-founded the PyroMark Q24 Pyrosequencer (Qiagen, Germany).
This work was supported by grants from University of Camerino Institutional funds awarded to Rosita Gabbianelli (FPA300015) and by grants from Marche Polytechnic University awarded to Antonio Giordano.
The authors declare no conflict of interest. LB, AG and RG are serving as Guest Editors of this journal. We declare that LB, AG and RG had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to GP.