
Academic Editors: Fabio Moda and Giorgio Giaccone
Background: Platelets (thrombocytes) are small anuclear cells that play
an important role in blood clotting. They are activated and dysfunctional in
brain disorders, such as Alzheimer’s disease (AD) and depression. Platelets
express the amyloid-precursor protein (APP) and release beta-amyloid
The life expectancy of humans has markedly increased over the last 100 years. As
age is the key risk factor for Alzheimer’s disease (AD), the number of patients
suffering from AD and mixed forms of dementia will dramatically increase over the
next 50 years. It is expected that there will be about 80 million AD
patients worldwide in
2050. Sporadic AD is a progressive chronic
neurodegenerative disorder (at least 97% of all cases are
non-genetic) and is characterized by two major
pathologies in the brain: the
Platelets are of particular interest in AD research because there is clear
evidence that they become activated and dysfunctional in AD patients [11, 12, 13]. In
an AD mouse model, we recently showed that platelets are early signs in vessels
before A
Tau is a microtubule-associated protein and physiologically stabilizes and
regulates axonal transport [25, 26, 27, 28]. However, the physiological roles of tau seem
to be more complex and by far not fully explored. Tau has more than 40 possible
phosphorylation sites and the tau phosphorylation (pTau) is regulated by several
kinases and phosphatases. It is extremely important to identify these critical
phosphorylation sites in the tau protein, as they are either therapeutic or
diagnostic targets [26, 27, 28, 29]. Tau and phospho-tau-181 are well-established
biomarkers in cerebrospinal fluid to diagnose AD, with levels of
Cognitively healthy subjects and patients suffering from AD, MCI or depression were recruited at the Landeskrankenhaus Hall/Tirol, Austria as reported in detail in several previous studies [19, 20]. The following groups were included in this study: healthy controls (group 1, n = 93), MCI patients (group 2, n = 111), AD patients (group 3, n = 233) and depressive patients without marked cognitive impairment (group 4, n = 76). All four groups were assessed by the same diagnostic procedure. Diagnosis of AD was established by a structured routine process including clinical assessment, extensive neuropsychological tests (including MMSE) and neuroimaging (magnetic resonance imaging, MRI) to exclude other brain pathologies. For each subject, the neuropsychological examination was conducted between 9:00 and 11:00 in the morning. Probable AD was diagnosed according to the current NINCDS-ADRDA (National Institute of Neurological and Communicative Disorders and Stroke and the AD and Related Disorders Association) criteria and confirmed for all participating patients. MCI was diagnosed according to previously published criteria [33]. The geriatric depression scale (GDS) was applied to each participant. A general blood examination was part of the routine diagnostic procedure. Exclusion criteria for healthy subjects, MCI and AD patients included (1) another primary neurological or mental disorder, (2) any kind of metabolic decompensation or any sign of peripheral inflammation (e.g., rheumatic disease), (3) long-term alcohol or drug abuse, (4) or any current, clinically significant cardiovascular disease. The study was approved by the ethics committee of Medical University of Innsbruck. All subjects and/or their caregivers enrolled in the study gave their informed consent.
Human platelets were isolated as previously described [20]. In short, 10 mL
blood was collected in ethylenediaminetetraacetate (EDTA) tubes during normal
clinical routine, centrifuged at 250
FACS analysis was performed as described in detail by us [34]. Immediately after
isolation, 2
Western blot analysis was performed for tau and phospho-tau396 as described by
us in detail [35]. Platelet extracts (80
Collected platelets of 15 patients were pooled in a tube, homogenized by
sonication and total RNA was extracted with QIAGEN RNeasy Mini Kit (Qiagen,
Germany) according to manufacturer’s protocol. Total RNA concentrations were
determined using Nanoquant Infinite M200 (Tecan, Switzerland). RNA integrity was
checked in random samples with Fragment Analyzer (Advanced Analytical
Technologies). In vitro reverse transcription was performed from 250 ng
of total RNA in 20
Platelet tau was characterized using our novel WANGEL (Western Blot-native
Agarose gel elution) model developed by us recently (Humpel, 2022 submitted).
Native Western Blots were performed as described above but directly after the gel
run, different molecular weight bands were dissected with a scalpel with a size
between 80 and 0 kDa related to the color molecular weight marker. For the
elution, 2.5 mL sample vials (PE, 14 mm, Roth 5863.1) were used and 1 mL 4%
agarose was pipetted at the bottom of the tubes and cooled. Then, the cut Western
Blot bands were carefully placed on top of the hardened agarose. Next 1 mL 1%
handwarm agarose was pipetted onto the extracted bands and cooled to harden. In
the next step, the bottom of the vial was cut, and the tube closed with a
Millicell culture insert (3
Tau was analyzed by immunoprecipitation and mass spectrometry similar as
reported earlier in detail for NT-proBNP [36, 37]. Briefly, a full length 2N4R
tau standard (1000 pg/mL) or a CSF sample (1024 pg/mL) or a pooled platelet
extract from 15 human patients was immunoprecipitated with a biotin-conjugated
anti-Tau monoclonal antibody (20
In the present study, 513 subjects were included, which were grouped in healthy
controls (group 1, n = 93), MCI patients (group 2, n = 111), AD patients (group
3, n = 233) and patients with depression (group 4, n = 76) (Table 1). The age of
the patients was 72–75 years, and only AD patients were slightly older (80.9
| Analyte | Control | MCI | AD | Depression |
| n (before pooling) | 93 | 111 | 233 | 76 |
| Age [years] | 74.0 |
75.2 |
80.9 |
72.2 |
| Male/% | 35/37.6 | 39/35.1 | 87/37.3 | 17/22.3 |
| MMSE | 29.4 |
27.6 |
21.1 |
28.3 |
| GDS | 4.3 |
5.1 |
4.0 |
8.2 |
| Pool of 15 (n) | 6 | 7 | 15 | 5 |
| Protein [mg/mL] | 35.0 |
42.9 |
45.8 |
35.0 |
| Tau [pg/mg] | 818 |
765 |
673 |
658 |
| pT181 [pg/mg] | 3.2 |
3.0 |
3.3 |
4.0 |
| Platelets were isolated from healthy controls or patients with mild cognitive
impairment (MCI) or Alzheimer’s disease (AD) or with depression. Values are given
as mean | ||||
Isolated human platelets were characterized for CD62P compared to a negative IgG control by FACS (Fig. 1A–C). The Lumipulse assay shows that platelets from healthy controls contain tau (approx. 820 pg/mg protein) and pTau-181 (approx. 3 pg/mg protein) (Table 1). Tau levels were significantly reduced in AD and depression but not in MCI (Table 1). Phospho-tau-181 levels were slightly increased in depression, but not in AD or MCI (Table 1).
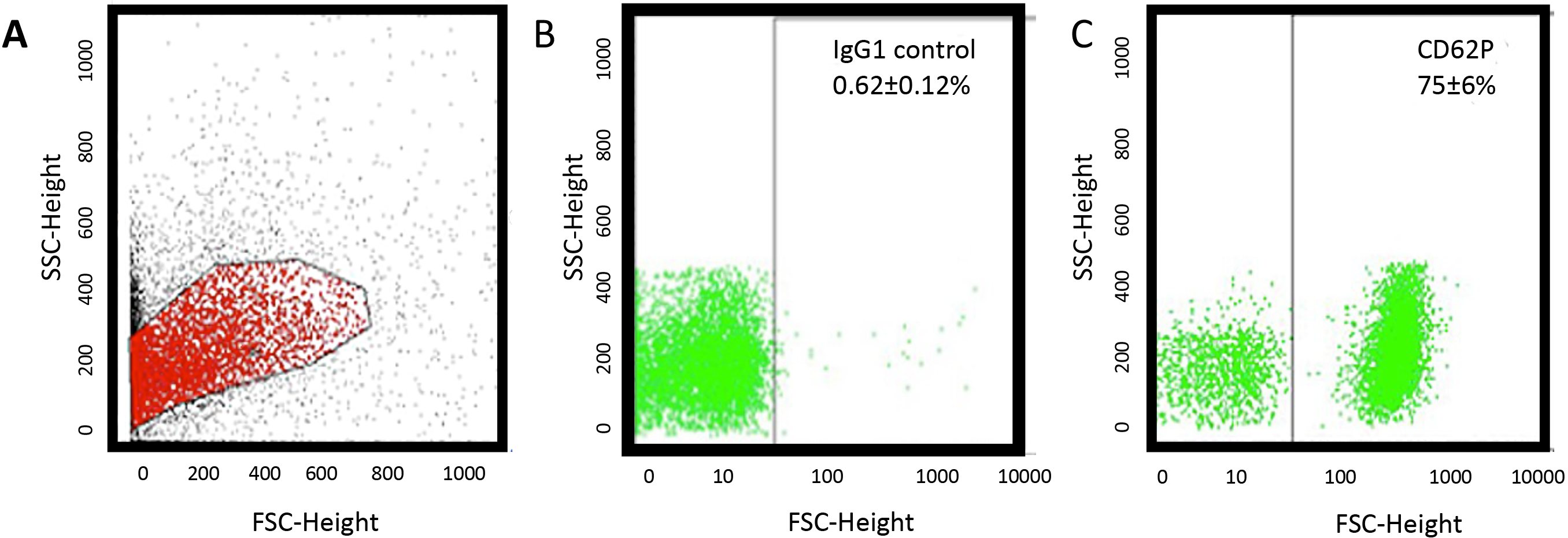 Fig. 1.
Fig. 1.Flow cytometry (FACS) analysis. (A) A representative sample of human platelets isolated from a healthy control shows a common population of cells in forward and sideward scatter and (C) markedly expressed CD62P (B) compared to an IgG control.
Western blot data shows a full-length tau standard with a size of approx. 60 kDa (Fig. 2) using the tau-5 antibody. No staining for platelet tau was found with the tau-5 antibody (Fig. 2). Using the HT-7 antibody, 2 clear bands were visible at approx. 45 and 30 kDa size, while a signal for phospho-tau-396 (approx. 50 kDa) was present in all samples (Fig. 2). A loading control GAPDH displayed a positive signal at 37 kDa (Fig. 2).
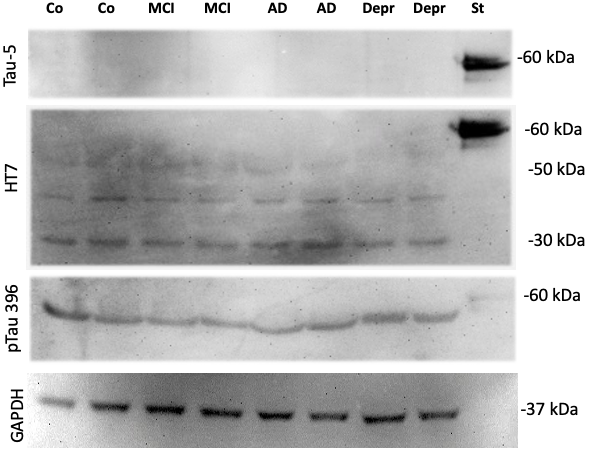 Fig. 2.
Fig. 2.Western blot of platelet extracts. Human platelets were
isolated, characterized by FACS and 15 samples per group pooled, extracted and 80
Platelets are anuclear cells, however, there are reports that mRNAs can be
detected in platelets by qRT-PCR. RNA was isolated from human platelets using the
Quiagen minikit and we achieved 1.1
Using our novel WANGEL system, we show that full-length tau migrates to the cathode and elutes from the agarose gel as a 50–60 kDa protein (Fig. 3). However, when the platelet extract (a pool of 32 lanes) was eluted from the WANGEL system, several large and small molecular weight species could be detected, but the majority of the tau+ detection was seen as a 20–30 kDa fragment (Fig. 3).
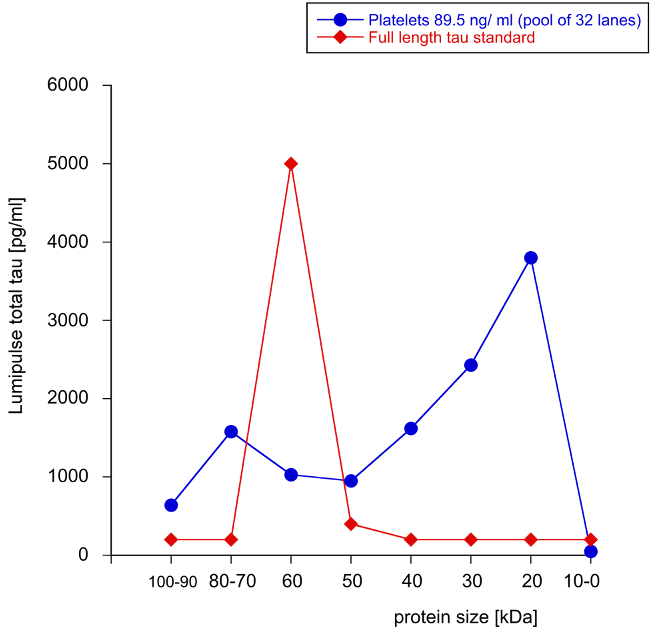 Fig. 3.
Fig. 3.Elution of tau from Western Blots linked to agarose gels. Tau protein (100 ng/lane) is separated on a Western Blot gel, then bands with between 0–100 kDa are cut, and tau eluted from agarose gels via the cathode. Note that tau elutes as a 60 kDa protein from Western-agarose gels (red triangles). In order to charcterize tau from platelet, an extract (89.5 ng/mL) was seperated on Western Blots, the lanes cut out and pooled from 32 lanes and analyzed by Lumipulse. Note that many large and small tau fragments were detected, but the majority of tau had a size of 20–30 kDa (blue circles). Values are given as pg/mL total tau measured by Lumipulse assay.
Applying an immunoprecipitation method with the HT7 antibody on the 2N4R full-length tau standard, 21 peptides could be identified by nanoLC ESI-MS/MS that covered 57% of the amino acid sequence of tau441. In a further experiment with cerebrospinal fluid of an AD patient (1024 pg/mL total tau), four tau peptides were identified with high confidence (data not shown). We used this method to investigate the different human platelet samples. Only a single peptide could be identified in a platelet extract pooled from 15 human patients of the same group (1577 pg/mg tau) with the HT7 antibody. The peptide with the sequence SGYSSPGSPGTPGSR corresponds to amino acid residues 203–217 of tau441, with an m/z = 697.32059 Da, charge state = 2, cross-correlation score (XCorr) = 3.74. The MS/MS spectrum and the assigned y- and b-series ions are shown in Fig. 4. The same peptide was found in the tau standard and in cerebrospinal fluid, respectively (data not shown). Tau could not be detected in all other platelet samples. Applying the tau-5 antibody instead of the HT7 antibody, no tau could be identified, neither in the standard sample nor in CSF or platelets.
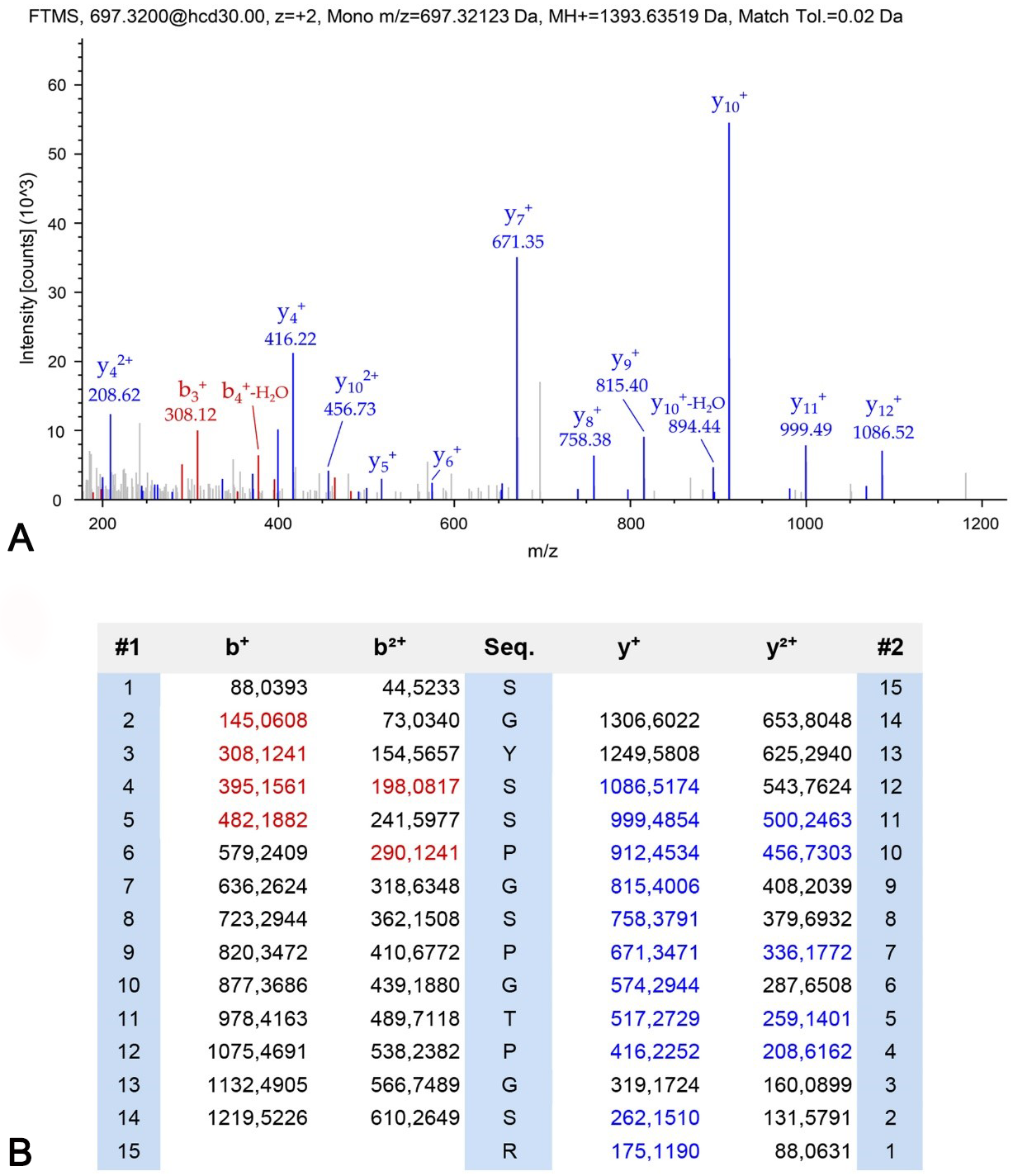 Fig. 4.
Fig. 4.Identification of a peptide from tau protein in platelets by mass spectrometry. Immunoprecipitation was performed to enrich tau from pooled platelets. Enriched proteins were analyzed with mass spectrometry and one unique peptide corresponding to amino acid residues 203–217 of tau441 was identified. (A) Shown is the MS/MS spectrum of peptide SGYSSPGSPGTPGSR (B) and the assigned y- and b-series ions.
In the present study, we show that tau protein is expressed in human platelets. It occurs especially as a fragmented form. We demonstrate that tau protein levels in platelets of AD and depressive patients are significantly changed and reduced tau levels could distinguish AD patients and depressed people from healthy individuals.
Platelets are about 3
Platelets modulate as mediators multiple cellular processes including angiogenesis and inflammation and are involved in a variety of pathologic conditions including AD. It is well known that both inflammatory and vascular processes accompany AD pathology and hence, it seems likely that platelets actively contribute to the progression of AD [9]. In murine models we have shown that platelets are first signs in AD as they concentrate in brain vessels and can also damage brain vessels and thus contribute to the AD pathology [15, 16]. Moreover, platelets contain proteins such as amyloid-precursor protein (APP) and produce beta-amyloid peptides, mainly the shorter 40 amino acid form [10, 17, 18, 19, 20, 40]. It is assumed that platelets use the beta-amyloid(40) peptide to clot the damaged vessel. Regarding the role of tau in platelets and AD, there is a strong mismatch in the literature. Although there are several reports that platelets contain tau (especially C-terminal tau), their role and structure is completely unknown [21, 22, 23, 24]. In the present study, we indeed verify that tau is found in platelets in a fragmented form and slightly decreased in late AD, however, we cannot link tau to any functional process.
Many studies now indicate that platelets in depressed people have a different level of activation [41, 42, 43, 44, 45]. Platelets contain a high concentration of serotonin, which is stored in vesicles and released by stimulation. Since serotonin plays a major role in depression, it could be that serotonin, which comes from platelets, plays a role in the pathogenesis of depression. In a previous study, we used platelets from depressive patients as an additional control, but also because AD and MCI patients complain from depressive episodes. Thus, a bio-profiling of platelets in severely depressed patients treated with medication was performed. We detected that the serotonin levels in blood platelets and different immunomodulatory compounds were significantly altered in these patients [46]. Interestingly, platelets not only contribute to the progression of depression, but also play a role in stress. In two previous papers we reported on differential changes in platelets reactivity induced by acute physical stress compared to persistent mental stress and also showed that stress enhances pro-inflammatory platelets activity [47, 48]. This present study shows that tau protein levels are decreased in depressed people followed by a slight enhanced phospho-tau-181 activation. Future studies are needed to further investigate this finding especially how and what the relationship is between serotonin and tau protein.
In order to characterize tau better, we performed a qRT-PCR, a Western Blot, a WANGEL detection and mass spectrometry. Platelets are anuclear cells; however, there are reports that mRNAs can be detected in platelets [49]. Although it seemed very unrealistic to detect tau mRNA in platelets, we performed a qRT-PCR. Our data supported our suggestion, that no tau mRNA was detectable in human platelets, but only in 1 sample out of 6 a very low tau expression was found by qRT-PCR, which could also be an artefact; this again shows that tau in platelets is expressed at very low concentrations. Thus, in order to catch tau in platelets we pooled up to 15 samples to concentrate tau. Using Western blots we did not see a full length tau protein (of approx. 60 kDa), but rather some smaller tau fragments of 40 and 30 kDa or smaller using the HT7 antibody. Only with an antibody recognizing the phospho-tau 396 form a large protein was seen, where we cannot really determine the specificity. In order to characterize the tau protein better, we performed a WANGEL analysis followed by the high sensitive Lumipulse assay. Here, a pool of 32 Western Blot lanes was extracted into agarose gels and eluted and analyzed and again we found that only very small tau fragments of 20–30 kDa were detectable. And finally applying immunoprecipitation and nanoLC MS, only a single peptide of tau was identified in one of the pooled platelet samples, while it was undetectable in all other platelet samples investigated. A comparison of different sample preparation protocols for the analysis of human platelets showed huge differences in the obtained protein concentrations [50]. A more efficient and replicable method may increase protein isolation and tau concentration, respectively. The fact that tau could not be detected clearly in platelets by mass spectrometry again suggests that it is of low abundance and/or fragmented, which results in a decreased binding to the HT7 antibody.
Taken together, our data highly suggest that tau is indeed present in human
platelets, but at very low levels and only in a highly fragmented small (20–40
kDa) form. This was surprising to see but this result fully agrees with a prior
study [51]. They report that multiple N-terminal and mid-domain fragments of tau
were detected in pooled cerebrospinal fluid (CSF) with apparent sizes ranging
from 20 kDa to approx. 40 kDa. The pattern of tau fragments in AD and control
samples were similar. They also did not detect full-length tau and C-terminal
fragments in their CSF samples. In order to concentrate the CSF, they used a
RP-HPLC followed by a Western Blot and also used the same HT7 antibody (as we
did) and found tau in fractions 3–7 representing small (20–40 kDa) tau
fragments [51]. This is partly surprising, as tau is a well-established biomarker
in CSF for AD with a pathological concentration of
Due to a new generation of ultrasensitive immunoassays, phosphorylated tau
epitopes are detectable in blood plasma. The currently best validated epitopes
are phospho-tau-181 and phospho-tau-127. Plasma phosphorylated tau-181, for
example shows 1.5 to 3.5 times higher concentrations in AD patients compared to
controls [52]. The recently published plasma-phospho-tau-231 is also a promising
new biomarker for incipient AD [53]. Furthermore, plasma phospho-tau-181
identified individuals who were A
The question now arises, if platelet tau may become a good biomarker for
dementia or depression. The answer is definitely no. As we have shown tau and
phospho-tau-181 are expressed in platelets at very very low concentrations. This
limits our tools to quantify tau. In order to overcome such a methodological
problem, more sensitive assays must be used. We thus highly suggest to use SIMOA
(single molecule array technology) to ultimately enhance the sensitivity for low
abundance biomarkers up to the single molecule level. It will be highly
interesting to use this technique, which is, however, very expensive and not
useful for routine diagnosis. On the other hand, our data show that the
differences between AD/depression and healthy controls are very low
(1.3
Definitely, this study has several limits. (1) The changes of tau in the brain of people suffering from severe neurocognitive disorder are well studied. However, in platelets, a functional role of tau is still largely unexplored. More work in humans, or in mouse models including tau transgenic will be necessary. (2) In platelets, the concentration of tau protein is very low. Therefore, future studies are necessary to examine whether it is possible to detect tau levels reliably at a specified level of confidence. The reliable reproducibility of the results are the prerequisite that the change in tau levels could establish as a future biomarker. More work is necessary to link tau to other vascular-related diseases. (3) As mentioned, tau exists as a small fragmented protein. This is really surprising, but it confirms work in human CSF and also questions why tau is so highly fragmented [51]. This fragmentation, however, does not limit the use as a biomarker in CSF for AD, as this method is well established internationally. It however opens the question, why tau is fragmented, and if is this only due to degradation and metabolism or has fragmented tau also a physiological role in the brain, e.g., to enter the nucleus. (4) In this regard, we suggest that full-length tau can be very instable and is metabolized to smaller fragmented peptides. This high fragmentation also limits our approach to detect tau by mass spectrometry.
Taken together we show that tau is expressed in human platelets, but exists mainly as a very highly small fragmented form. We show that tau in platelets is reduced in AD and depression and may differentiate them from healthy controls. We show that tau is very low in platelets and display a high heterogeneity. Thus, we conclude that tau in platelets may have a prominent function, but will not serve as a novel biomarker.
CH designed the study, wrote the manuscript, and acquired funding. DSK performed the Western Blots. JM collected blood and diagnosed patients. BS performed the MS analysis. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
The study was performed according to local ethical approval protocol no. AN2015-0159 351/4.7 405/5.5 (4484a). Informed consent was obtained from all subjects enrolled in the study. The study was in accordance with the guidelines of the ethical commission of the Medical University Innsbruck.
We thank Karin Albrecht for excellent technical help.
This research received no external funding.
The authors declare no conflict of interest.