




 , Da Young Lee 1, Ju Hyeon Lim 3, Won Keun Oh 4, Jun Tae Park 5, Sang Chul Park 6, Kyung A Cho 1,3,*
, Da Young Lee 1, Ju Hyeon Lim 3, Won Keun Oh 4, Jun Tae Park 5, Sang Chul Park 6, Kyung A Cho 1,3,*1 Department of Biochemistry, Chonnam National University Medical School, 58128 Jeonnam-do, Republic of Korea
2 Research Institute of Pharmaceutical Sciences, College of Pharmacy, Chonnam National University, 61186 Gwangju, Republic of Korea
3 Research center, Medispan Co., Ltd. 13486 Gyeonggi-do, Republic of Korea
4 Korea Bioactive Natural Material Bank, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, 08826 Seoul, Republic of Korea
5 Division of Life Sciences, College of Life Sciences and Bioengineering, Incheon National University, 22012 Incheon, Republic of Korea
6 The Future Life & Society Research Center, Advanced Institute of Aging Science, Chonnam National University, 61469 Gwangju, Republic of Korea
Academic Editor: Amancio Carnero Moya
Abstract
Background: Cancer is a representative geriatric disease closely
related to senescent cells and cell aging in tissues. Senescent cells that
surround cancer tissues reduce the effects of various cancer treatments and
induce cancer recurrence through senescence-associated secretory phenotype (SASP)
secretion. Thus, for good therapeutic effect, candidate drugs should be selective
for both cancer and senescent cells. In this study, we investigated the selective
effect of piperine as a potential senostatic agent as well as an anticancer drug.
Methods: The effect of piperine on cytotoxicity and cell proliferation
was tested by lactate dehydrogenase (LDH) or water-soluble tetrazolium salt (WST)
assay. The levels of p16
Keywords
- senescence
- senostatic
- anticancer
- senescence-associated secretory phenotype
- piperine
- human diploid fibroblasts
Senescent cells secrete a senescence-associated secretory phenotype (SASP) that leads to chronic inflammation, playing a crucial role in age-related functional decline and senile diseases [1]. Thus, the development of senotherapeutic interventions to remove senescent cells (senolytic drugs) or to modulate the SASP (senostatic drugs) could extend a person’s healthspan or treat various diseases [2]. Several senolytics target the prosurvival pathway, such as kinase inhibitors (e.g., dasatinib), flavonoids (e.g., quercetin and fisetin), BCL-2/BCL-xL inhibitors (e.g., navitoclax), and BCL-xL inhibitors (e.g., A1331852 and A1155463) [3].
Cancer is a representative geriatric disease and is closely related to senescent cells constituting tissues. Cellular senescence itself suppresses cancer development despite the accumulation of various genetic mutations, but the SASP secreted from senescent cells promotes the development of surrounding cancer cells [4]. Therefore, to develop effective therapeutic agents to remove cancer, it is crucial to identify candidate substances that can selectively act only on cancer cells. Additionally, SASP secreted from senescent cells that surround cancer tissues reduce the effects of various cancer treatments and induce cancer recurrence through SASP secretion [5, 6, 7]. Therefore, increasing attention is being paid to the development of cancer therapeutics using senotherapies that remove senescent cells or suppress SASP secretion. Senolytics, such as quercetin, navitoclax, and fisetin, are being studied as potential cancer treatments in nonclinical or initial clinical trials [8, 9, 10]. However, for good therapeutic effect, candidate drugs should be selective for both cancer and senescent cells.
Piperine is a bioactive phenolic component that has been isolated from plants of the Piper species, such as black pepper (Piper nigrum) and long pepper (Piper longum) [11], and has attracted attention as a dietary phytochemical and medicine [12]. Various pharmacological properties of piperine have been suggested, including antioxidant activity [13], anti-inflammatory activity [14] and biological enhancement [15]. Piperine also exerts a chemopreventive effect [16] and causes cellular toxicity in cancer cells by inducing various effector proteins involved in apoptosis [17, 18, 19]. Piperine was reported to suppress tumor development and metastasis in mouse models [17]. In cancer cells, piperine triggers both cell cycle arrest by activating p21 and apoptosis by activating caspase [20]. Interestingly, a combination therapeutic model of piperine with curcumin, a yellow pigment in the Indian spice turmeric (Curcuma longa), demonstrated neurotrophic and neuroprotective effects in a D-galactose-induced brain aging model [21], preventing the progress of aging induced by D-galactose as well as reversing hippocampal memory function due to antioxidant activity [22]. Curcumin is a well-known, promising antiaging intervention that is easy to add to one’s diet. Curcumin was reported to induce an extended lifespan in various models, including fruit flies, nematodes, and mice [23, 24, 25, 26]. While curcumin has demonstrated a direct antiaging effect, the antiaging effect of piperine has only been attributed to its antioxidant effect on brain aging in combination with curcumin. Like piperine, piperlongumine is a natural product found in various Piper species, and its analogs have also been suggested as senolytic agents through activation of the caspase pathway in senescent cells [27].
We previously screened single natural compounds that acted differently on cancer cells compared with premature senescent cells. Most substances showed similar effects on cancer and senescent cells, but piperine induced toxicity in cancer cells only. This study investigates the selective effect of piperine as a potential senostatic agent as well as an anticancer drug.
Piperine was kindly provided by Prof. WK Oh (Seoul National University, Korea)
and purchased (Merck, NY, USA). CT26 (mouse colon carcinoma, CRL-2638), T98G
(human glioblastoma, CRL-1690), A431 (human skin carcinoma, CRL-1555), MCF7
(human breast adenocarcinoma, HTB-22), HepG2 (human hepatocellular carcinoma,
HB-8065), and HeLa (human cervix adenocarcinoma, CCL-2) cell lines were purchased
from American Type Culture Collection (Manassas, VA, USA). All cancer cell lines,
except MCF7, were cultured in Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin
(P/S) at 37
We investigated the selectivity of piperine by observing its effect on the
growth of cancer and normal cells. Cancer and normal cells were cultured in
complete medium at 37
Cytotoxicity was assessed using an LDH assay kit (DG-LDH500; DoGen Bio, Seoul,
Korea) according to the manufacturer’s protocol. Briefly, cancer cells (2
To count the viable cells, we seeded various cancer cell lines (2
Cell proliferation assay was assessed using an EZ-Cytox water-soluble
tetrazolium salt (WST) cell proliferation assay kit (EZ-3000; DoGen Bio)
according to the manufacturer’s protocol. Briefly, S-HDFs (8
SA-
We used MATLAB software (MathWorks Inc., Natick, MA, USA) to quantify the
SA-
Western blot analysis was performed as previously described [28]. Briefly, total
proteins were extracted from the cells using radioimmunoprecipitation assay
buffer (Biosesang, Seongnam, Korea) containing Protease Inhibitor Cocktail
(Sigma-Aldrich, St Louis, MO, USA) and Phosphatase Inhibitor Cocktail I and II
(Sigma-Aldrich). The protein samples were separated using sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and then transferred to polyvinylidene
fluoride membranes (Millipore, Burlington, MA, USA). The membranes were incubated
with primary antibodies at 4
We used tetramethylrhodamine (TMRM) (I34361; Invitrogen) to analyze MMP and
dihydroethidium (DHE) (D23107; Molecular Probes, Eugene, OR, USA) to quantify the
levels of cellular ROS. Briefly, S-HDFs were treated with piperine (70
S-HDFs were treated three times with piperine (70
Statistical analysis was performed using GraphPad Prism software (V 8.0)
(GraphPad Software, San Diego, CA, USA). Data were presented as the mean
To confirm the effect of piperine on cancer cells, we treated various cancer
cell lines with piperine (70
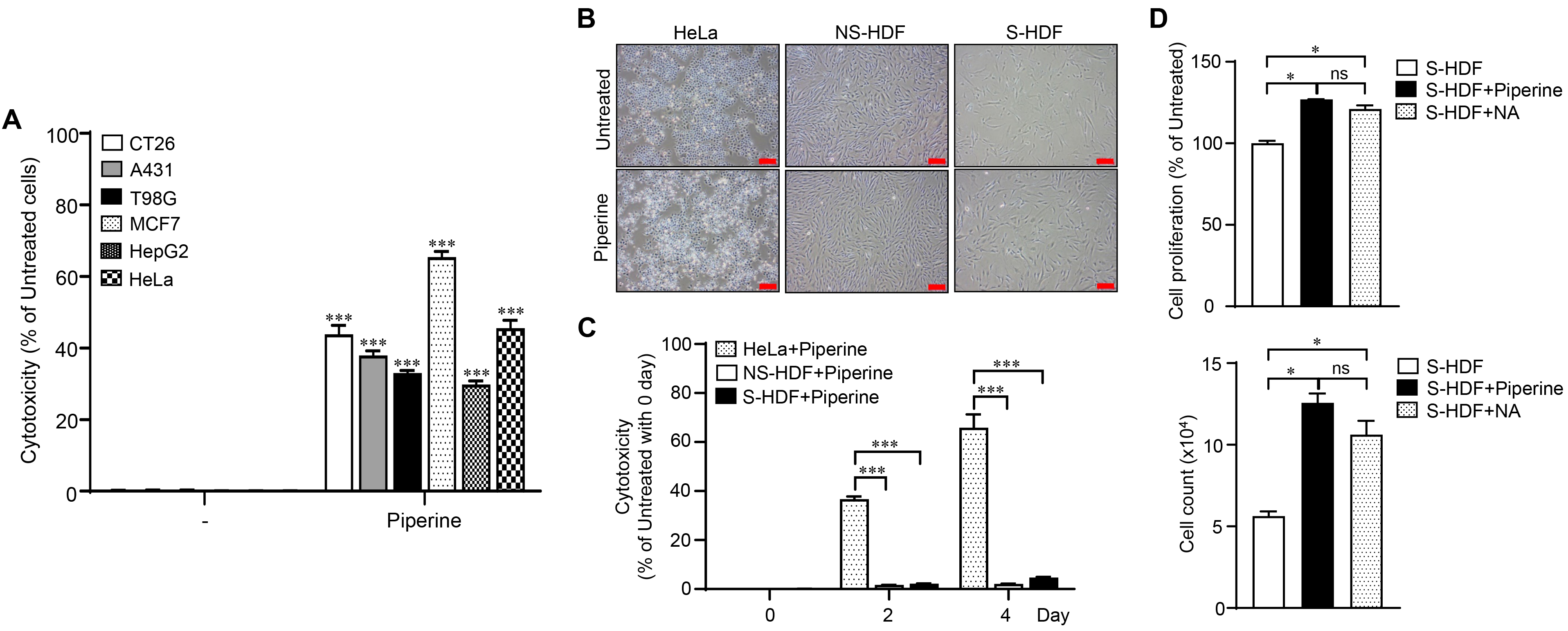 Fig. 1.
Fig. 1.Selective inhibition of cancer cell proliferation by piperine.
Various cancer cells were treated with piperine (70
We extended the piperine treatment period to three times every 2 days to observe
the senomorphic effects of piperine on senescent cells (Fig. 2A). Cytotoxicity
was not observed in the S-HDFs after the cells had been treated with piperine
three times (Fig. 2B). The effect of piperine treatment was compared with that of
nicotinamide (NA), which induces senescent cell proliferation [29], to confirm
the effect of piperine treatment on S-HDF growth. Then, we performed a WST cell
proliferation assay and counted the number of viable cells. Interestingly,
piperine induced a higher rate of cell proliferation than NA in S-HDFs (Fig. 2C,D). The expression of p16
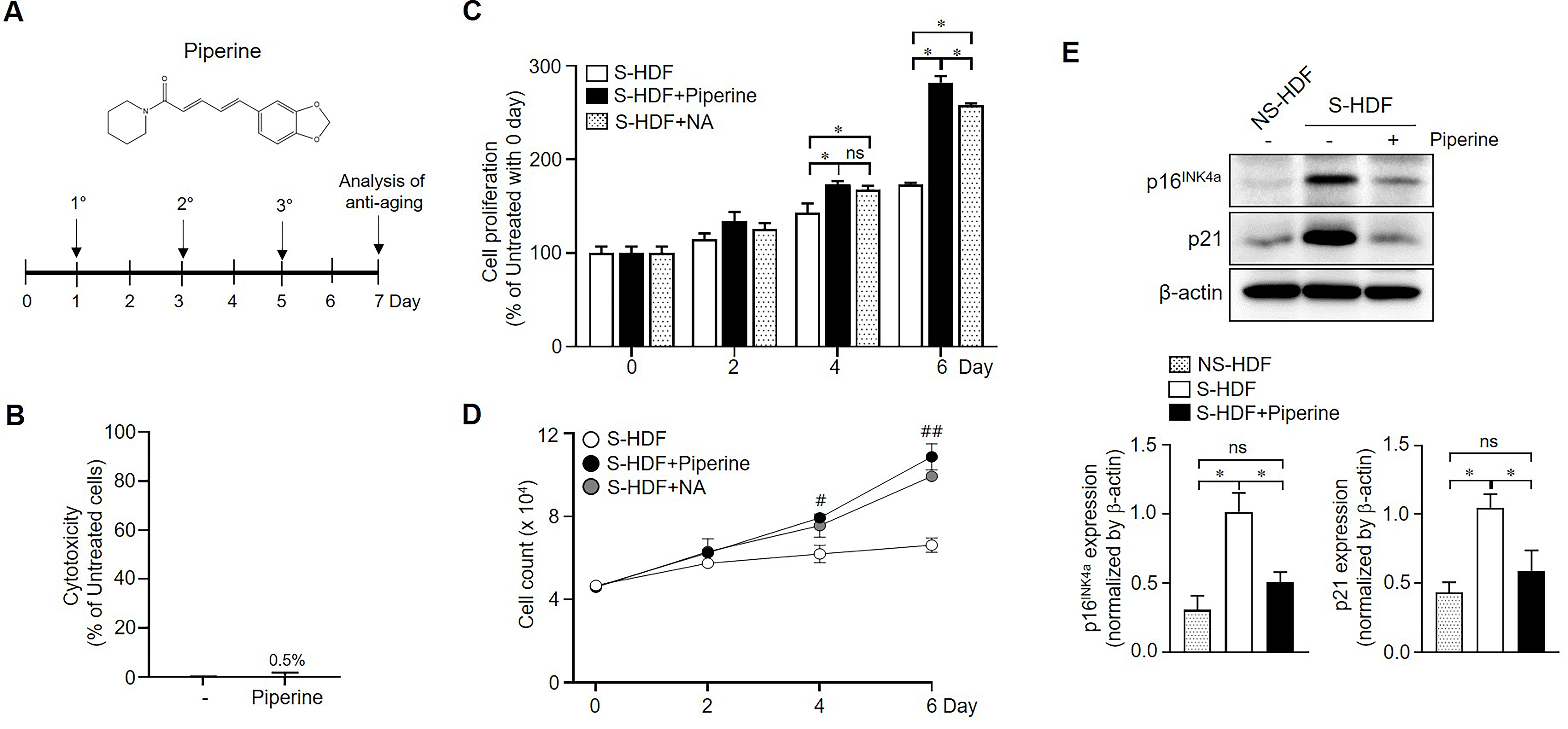 Fig. 2.
Fig. 2.Effects of piperine on the proliferation of senescent cells.
(A) The chemical structural of piperine (top panel) and the experimental scheme
of piperine treatment in senescent cells (bottom panel). S-HDFs were treated
three times with piperine (70
Mitogen-activated protein kinases (MAPKs), including Erk1/2, p38, and JNK, have
been implicated in senescence phenotypes such as growth arrest [30, 31],
apoptosis resistance [32], and the SASP secretion [33, 34]. We examined whether
piperine regulates MAPK pathways in S-HDFs in an effort to elucidate the
mechanism of cell division S-HDFs following piperine treatment. Although p38
phosphorylation was unaffected in piperine-treated S-HDFs, Erk1/2 and JNK
phosphorylation were remarkably increased (Fig. 3A). We also investigated the
involvement of various signaling pathways, such as the 5’ adenosine
monophosphate-activated protein kinase pathway, mammalian target of rapamycin
(mTOR) pathway, and autophagy, in piperine-treated S-HDFs. Piperine treatment did
not affect signaling in S-HDFs (Fig. 3A, data not shown). We also investigated
these signaling pathways in HeLa cells. Piperine treatment increased the
phosphorylation of JNK, p38, and mTOR but not Erk1/2 (Fig. 3B). These results
suggest that the signaling mechanism of piperine in senescent cells differs from
that in cancer cells and imply that piperine activates Erk1/2 and JNK signaling
in senescent cells, leading to the reduction of cell cycle inhibitors
p16
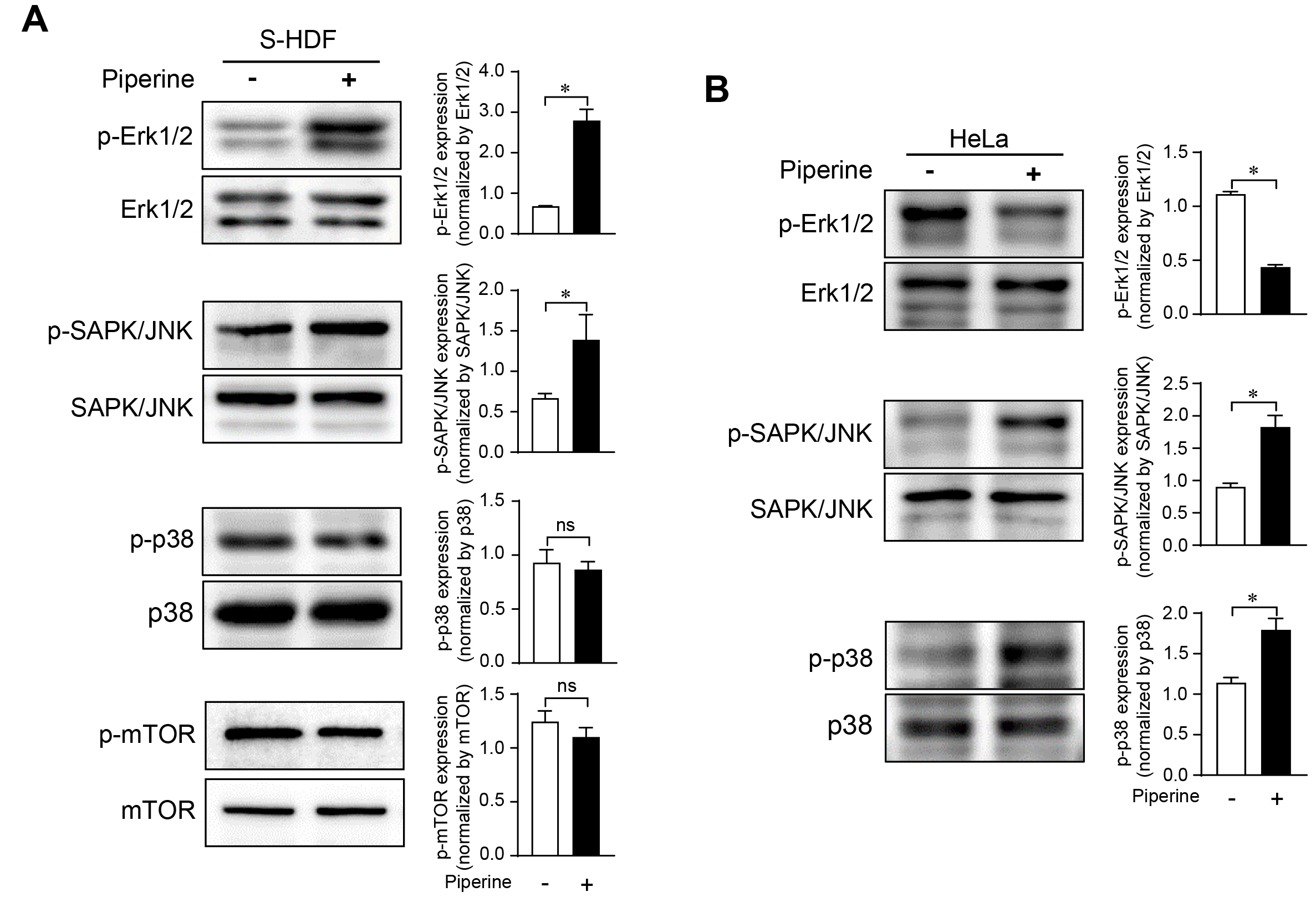 Fig. 3.
Fig. 3.Differential regulation of signaling in senescent cells and
cancer cells following piperine treatment. S-HDFs (A) and HeLa cells (B) were
treated with piperine (70
To further determine whether piperine rescues cellular senescence phenotypes, we
examined the SA-
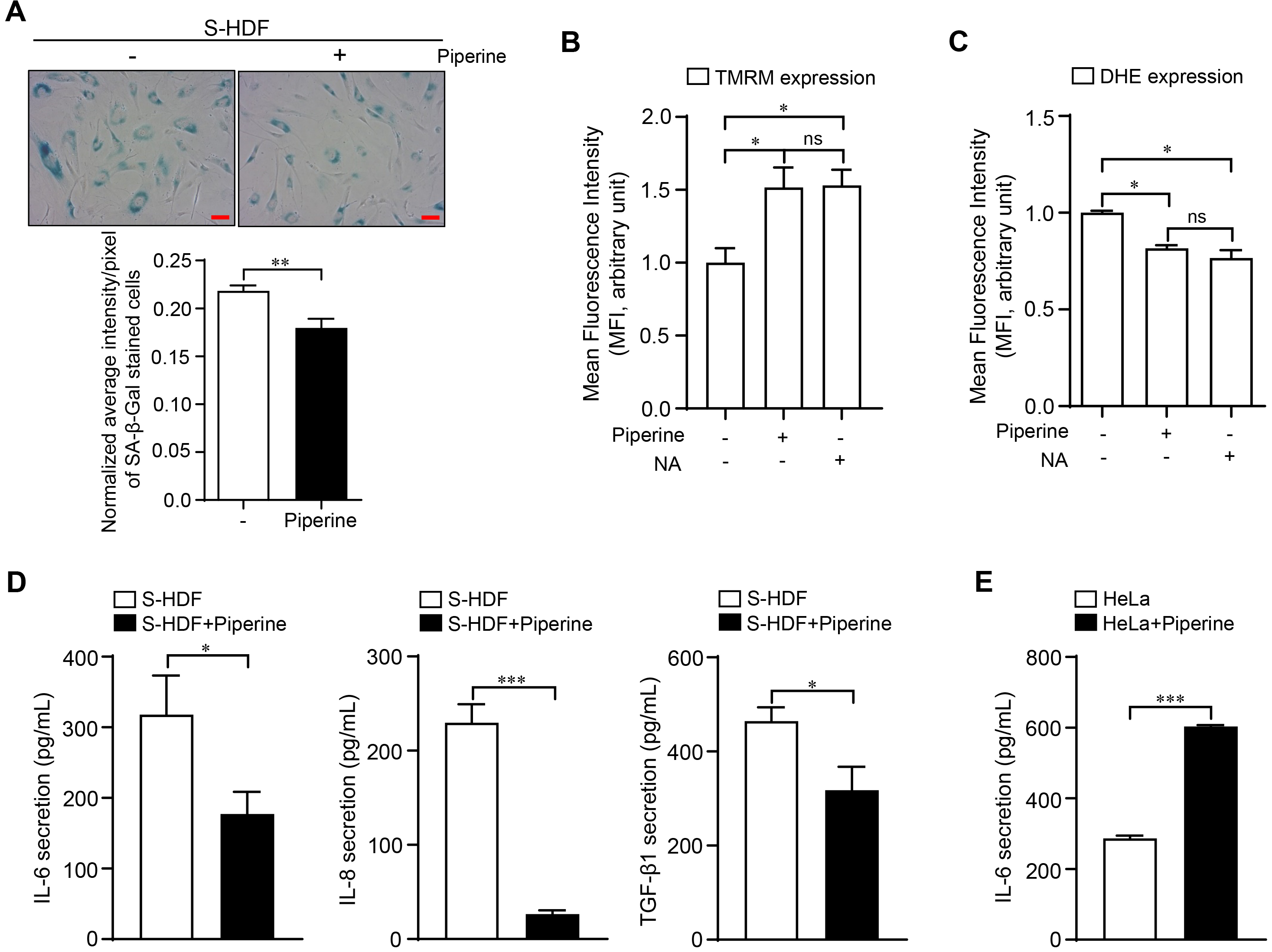 Fig. 4.
Fig. 4.Rejuvenation effects of piperine on senescent phenotypes.
S-HDFs were treated three times with piperine (70
Cellular senescence is not only closely related to the occurrence and promotion
of cancer but also affects its treatment and recurrence. Although chemotherapy
kills cancer cells, it also provides an environment in which cancer can recur by
inducing senescence of the surrounding cells [47, 48]. When
cyclophosphamide was administered in an animal model with myc-induced lymphoma,
cancer cell senescence was induced by p16
Piperine inhibits the proliferation and survival of various cancer cell lines by regulating the cell cycle and activating apoptosis-related signaling within cells [51]. This compound directly modifies functions involved in the activity of various enzymes and transcription factors involved in cancer cell invasion, metastasis, and angiogenesis.
Similar to previous reports, piperine induced high cytotoxicity in various
cancer cell lines was not toxic to normal and senescent cells (Fig. 1).
Additionally, piperine stimulated cell division, decreased SA-ꞵ-gal activity,
recovered MMP, and reduced ROS generation in senescent cells, similar to the
effect of NA, which has been reported to restore the function of senescent cells
[29] (Figs. 2,4). We found that piperine differently affected cancer cells and
senescent cells due to the different responses of intracellular signaling
pathways. In senescent cells, piperine promoted Erk1/2 phosphorylation, which is
involved in cell growth, whereas in HeLa cells, a cancer cell line, Erk1/2
phosphorylation was decreased and JNK and p38 phosphorylation were increased
(Fig. 3). Interestingly, piperine induced different responses in senescent and
cancer cells, not only in cell signaling but also in SASP secretion. Piperine
increased SASP secretion in cancer cells while significantly decreasing the
secretion of three SASP factors (IL-6, IL-8, and TGF-
From recent studies, the specific removal effects of senolytics on senescent cancer cells have been demonstrated. The specific inhibitor of the BCL-2 family (ABT263) successfully remove a range of senescent cancer cells and in vivo study, ABT263 suppresses cancer recurrence and metastasis by eliminating chemotherapy-induced senescent cells [52]. However, dasatinib+quercetin, another senolytic cocktail drug, did not kill senescent hepatocellular carcinoma (HCC) cells and reduce the growth of HCC [53]. Senostatics is also effective cancer therapy by synergistic effects. Metformin leads reduction of prostate cancer cells cultured with media from metformin-treated senescent cells by suppressing SASP [52].
It is difficult to predict the various side effects of cancer treatment substances because most cell models or animal models show the death of cancer cells but do not prove the effect on surrounding normal cells. Piperine showed the effect of inducing cancer cell-specific toxicity that does not affect normal cells, and further showed the effect of restoring the function of senescent cells that may exist around cancer cells. Therefore, we propose piperine as an effective cancer treatment that can simultaneously induce senostatic effects and the removal of cancer cells, not as an adjuvant to the existing senostatics for cancer treatment.
KAC and WKO designed the research study. JSL, DYL and JHL performed the research. JTP and SCP advised experimental design. KAC and SCP analyzed the data. KAC and JSL wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
We thank Young Sam Lee at DGIST for the kind discussion of the results.
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. NRF-2017R1E1A2A02081815 and NRF-2017R1E1A1A01074674; K.A.C.) and by the Basic Science Research Program, through the NRF funded by the Ministry of Education (grant number NRF-2018R1D1A1B07051207; J.S.L).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/j.fbl2704137.