1. Introduction
Diabetes mellitus (DM) is the most common metabolic disease, with an increasing
morbidity and mortality rates worldwide and causes serious hyperglycaemia and
several complications, such as nephropathy, retinopathy,
neuropathy, and cardiovascular disease [1]. Vascular
endothelial cell dysfunction (VECD) induced by long-term hyperglycaemia and other
diabetes-associated physiological changes in individuals with DM is a critical
initiating factor and most fundamental pathological change in diabetes; it is
also a key factor contributing to the development of diabetic complications [2].
VECD may result in impaired vasodilation and barrier functions of endothelial
cells, disturbances in proliferative capacities, impaired migratory and tube
formation properties, impaired angiogenic properties, an attenuation of synthetic
function, and a deterrence of white blood cell adhesion and
diapedesis [3]. High glucose have been shown to trigger the
shift of endothelial to mesenchymal transition (EndMT). During the EndMT,
endothelial cells lose their characteristic phenotype and acquire
mesenchymal features, which are characterized by the
development of invasive and migratory abilities as well as the expression of
extracellular matrix proteins [4]. The EndMT appears to
represent the key link in the interaction between inflammation
and endothelial dysfunction in diabetic complications [4]. Furthermore,
transforming growth factor receptor (TGF) signalling is
central to the EndMT, and the Ras homolog gene family member A
(RhoA)/Rho-associated coiled-coil containing kinases (ROCKs) pathway is also
involved in the EndMT [4, 5]. ROCKs were shown to be downstream effectors of RhoA
activation and play important roles in regulating several cellular functions
including proliferation, migration, and angiogenesis [6, 7]. Two similar isoforms
of ROCK have been identified: ROCK1 and ROCK2. RhoA/ROCK signalling is involved
in hyperglycaemia-induced injury [8]. ROCK deletion attenuates diabetes induced
vascular endothelial dysfunction by preventing increased
arginase activity and a reduction in NO production [9]. Human
endothelial cells exposed to hyperglycaemia exhibit increased ROCK activity, and
hyperglycaemia stimulates ROCK activity via protein kinase C (PKC) and oxidative
stress-dependent pathways. ROCK1 plays a predominant role in
hyperglycaemia-induced increases in ROCK activity [8].
Hydrogen sulfide (HS) is an endogenous gasotransmitter with multiple
functions in the cardiovascular system [10]. HS
generation mainly depends on three major enzymes: cystathionine -lyase
(CSE), cystathionine -synthase (CBS) and 3-mercaptopyruvate
sulfurtransferase (MST) [11]. CSE is primarily responsible for most of the
HS produced within the cardiovascular system [12]. Recent studies have
shown that HS plays a critical role in HG-induced endothelial injury, such
as migration dysfunction [13], mitochondrial dysfunction [12], apoptosis [14],
oxidative stress, and matrix protein accumulation [15]. Moreover, Ying et
al. [16] indicated that HS protects against the endoplasmic reticulum
stress-induced EndMT in subjects with cardiac fibrosis.
Dopamine receptors (DRs) are classified into D1-like receptors
(DR1), including D1 and D5, which stimulate adenylyl cyclases
(AC), or D2-like receptors (DR2), including D2, D3 and D4. The effect of DR2 is
the opposite of DR1 [17, 18]. DR1 couples to G to modulate
phospholipase C (PLC), thus leading to the generation of inositol triphosphate
(IP) and diacylglycerol (DAG). This activation results in the activation of
PKC by DAG and increased intracellular calcium concentrations ([Ca])
in response to IP. The increase of [Ca] in the cytoplasm
induces the activation of calcium/calmodulin-dependent PK II (CaMKII) [17, 18].
DRs are widely expressed in in the brain and in the periphery, including blood
vessels, and the heart [19]. The DRs expressed at the highest levels in the blood
vessels is DR1 [20]. DRs activation are involved in the occurrence and
development of myocardial ischemia-reperfusion injury [21], diabetes and obesity,
atherosclerosis, hypertension and other diseases [22].
Additionally, DR1 activation can inhibits the proliferation and migration of
vascular smooth muscle cells, thereby exerting anti-atherosclerotic effects [23].
HS can protects against endothelial cell dysfunction induced by high
glucose. Yang et al. [24] reported that an increase of [Ca]
activates CSE, which in turn promotes the production of endogenous HS in
endothelial cells and protects endothelial cells from damage [24]. Moreover, DR1
activation can increases [Ca]. Researchers have not clearly
determined whether DR1 activation functions by increasing [Ca] to
promote the production of endogenous HS in vascular endothelial cells is
unclear. Therefore, in the present study, we will explore this question and the
related mechanism and signalling pathway (RhoA/ROCK1).
2. Materials and methods
2.1 Materials and drugs
Sodium hydrogen sulfide (NaHS), PPG (a CSE inhibitor) and
7-Azido-4-Methylcoumarin (AzMC) were purchased from Sigma-Aldrich (St. Louis, MO,
USA). SKF38393 (a DR1 agonist) was obtained from Abcam (Cambridge, MA, USA).
Y-27632 (a ROCK inhibitor) was obtained from MedChemExpress (Shanghai, China).
The primary antibodies for anti-CSE, Cyclin D1, proliferating cell nuclear
antigen (PCNA), p21, collagen I (Col-1), collagen III (Col-3),
matrix metalloproteinase 9 (MMP-9), osteopontin (OPN) and -smooth
muscle actin (-SMA) were purchased from Proteintech (Wuhan, China). The
anti-p-RhoA, t-RhoA, p-ROCK1, t-ROCK1 were from Affinity Biosciences (Cincinnati,
OH, USA). The anti-DR1 antibody was from
GeneTex (Irvine, CA, USA). Horseradish peroxidase-conjugated goat anti-rabbit
IgG, goat anti-mouse IgG antibody, the Cell Counting Kit-8 (CCK-8) and
anti--actin were obtained from Boster Bio-engineering Limited Company
(Wuhan, China). The EdU Cell Proliferation Assay Kit were obtained from Ribobio
(Guangzhou, China). Fluo-4 AM were obtained from Beyotime Biotechnology
(Shanghai, China). Enhanced ECL Chemiluminescent Substrate Kit
was obtained from Yeasen Biotechnology (Shanghai, China). All other chemicals
were from Solarbio (Beijing, China) or Beyotime Biotechnology.
2.2 Cell culture and treatment
Human umbilical vein endothelial cells (HUVECs) were purchased from the Cell
Resource Database of Chinese Academy of Sciences (China) and cultured in
Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) in a
humidified 5% CO at 37 C. When the cells had reached 80% confluence, they
were passaged by digestion with 0.25% trypsin-EDTA at a ratio of 1:2. Cells
between the 4th to 8th passages were used in experiments.
For cell treatment, HUVECs were starved for 12 h in serum-free medium and then
pretreated with NaHS (100
M), or DR1 agonists SKF38393 (10 M), or CSE inhibitor PPG (10
M), or ROCK inhibitor Y-27632 (10 M) respectively for 30 min before
expose to HG (30 mM) or normal glucose (Control, 5.5 mM). Cells were then treated for 48 h. SKF38393 and
Y-27632 were dissolved in DMSO with a final concentration of less than 0.1%.
Osmotic control group (5.5 mM D-glucose + 24.5 mM D-mannitol) showed that osmotic
pressure did not affect parameters tested in HUVECs (data not shown).
2.3 Cell viability
Cell Counting Kit-8 (CCK-8) was used according to the manufacturer’s
instructions. HUVECs were seeded at a density of 1 10 cells/well
in 96-well plates, after different treatments, the cells were
washed with PBS, and incubated with 10 L CCK-8 in culture medium at 37 C
for 1 h. The resulting absorbance was measured at 450 nm on a spectrophotometer.
2.4 EdU proliferation assay
HUVECs proliferation was detected through the EdU Cell Proliferation Assay Kit
according to the manufacturer’s instructions. Briefly, HUVECs were seeded in
96-well plate, after different treatments, the cells were incubated with 10
M EdU for 12 h then fixed with 4% paraformaldehyde,
after that, the cells were stained with Apollo for 30 min and Hoechst 33342 for
30 min respectively, the number of cells was counted in six random fields of each
well and presented as the ratio of EdU positive cells to total cells using
fluorescence microscope (Olympus, IX71, Japan).
2.5 Wound healing assays
Wound healing assays were conducted as previously described with minor
modifications [25]. Briefly, the cells were seeded into 6 well plates and were
cultured up to sub-confluence and serum deprived for 12 h. Then, the confluent
monolayer was scratched with a 200 L sterile pipette tip and cells were
washed twice with PBS and then fresh FBS-free DMEM was added. Next, the cells
were given different treatments. Migration was followed by phase-contrast
microscopy (Nikon Eclipse TS100-F microscope coupled to a
digital sight Nikon DS-L3 camera) at different time points (0, 24 and 48 h) up to
wound healing closure. The initial and final wound sizes were measured using
Image J software (version: 1.53c, National Institutes of Health, Maryland, United
States) [26]. Relative migration rate (%) was calculated according to area of
migration.
2.6 Transwell migration assay
To assess endothelial cell migration, a transwell assay was
performed as previously described [27]. Briefly, after
different treatments, serum deprived HUVECs were added to the upper chambers of
the transwell (8.0 m pore size, Corning, NY, USA) and allowed to migrate
for 24 h. The number of migrated cells in three random fields was counted.
2.7 Measurements of HS levels
The HS levels in the HUVECs were measured as previously described [28].
Briefly, after different treatments, cells were incubated with 50 M
7-Azido-4-Methylcoumarin (AzMC) in PBS for 30 min, followed by washing of the
cells with PBS. Visualization of the fluorescence response of AzMC to HS in
HUVECs was carried out using fluorescence microscope (Olympus, IX71, Japan), and
semi-quantitative fluorescence value were measured using Image J.
2.8 Detection of intracellular calcium concentration
([Ca])
The [Ca] was detected with the Fluo-4 AM calcium probe as described
previously with minor modifications [29, 30]. Briefly, after different
treatments, the HUVECs were washed with Ca-free PBS and incubated in 5
M Fluo-4 AM for 30 min at 37 C. Cells were then washed three
times with Ca-free PBS and incubated for 30 min. Excitation was set at 488
nm, and emission was set at 530 nm. The fluorescence intensity of Fluo-4 AM was
determined using fluorescence microscope (Olympus, IX71, Japan).
2.9 Western blotting
Western blotting was performed as described previously [31].
Cell lysates were prepared in radioimmunoprecipitation assay (RIPA) buffer with
protease inhibitor PMSF. Protein concentrations were confirmed by BCA Protein
Assay Reagent. Equivalent amounts of protein samples were separated by 8–12%
SDS-PAGE, and the target proteins were transferred to PVDF membranes. Membranes
were blocked in 5% skim milk for 1 h at room temperature, followed by incubation
with primary antibodies at 4 C overnight, and then washed with TBST three times.
Subsequently, the membranes were incubated with corresponding HRP-conjugated
secondary antibody. The resulting immunoreactive bands were visualized with the
Enhanced ECL Chemiluminescent Substrate Kit according to the manufacturer’s
directions. -actin was used as the loading control. Protein detection
was performed using Image J. Adobe Photoshop (version: 22.0.0 Free Trial, Adobe Inc., San Jose, California, USA) was used to prepare image panels and
annotations.
2.10 Statistical analysis
Data are expressed as mean standard error of the mean (SEM). Data are
from at least three independent experiments. Data involving
more than two groups were analyzed using one-way ANOVA followed
by Fisher’s LSD post-hoc test (GraphPad Prism 8.02 Free Trial, GraphPad Software, San Diego, California, USA), with
p values 0.05 considered statistically significant.
3. Results
3.1 DR1 activation upregulates the CSE/HS pathway in
HG-induced HUVECs
We investigated the effects of HG on DR1 and the CSE/HS pathway in HUVECs
by assessing the expression of DR1 and CSE and HS
production in HG-treated HUVECs. The expression of DR1 and CSE and the endogenous
HS production rate were reduced in the HG group compared with the control
group. Compared with the HG, the DR1 agonist SKF38393 markedly increased DR1 and
CSE expression and endogenous HS generation, whereas NaHS (a HS
donor) only increased CSE expression and endogenous HS generation but did
not affect DR1 expression (Fig. 1). In the present study, our data showed that
SKF38393 upregulates DR1 expression, consistent with a previous study [32]. Based
on these results, HG-induced injury of HUVEC injury is related to the
downregulation of the DR1-CSE/HS pathway, and DR1 is an upstream regulatory
factor of the CSE/HS pathway.
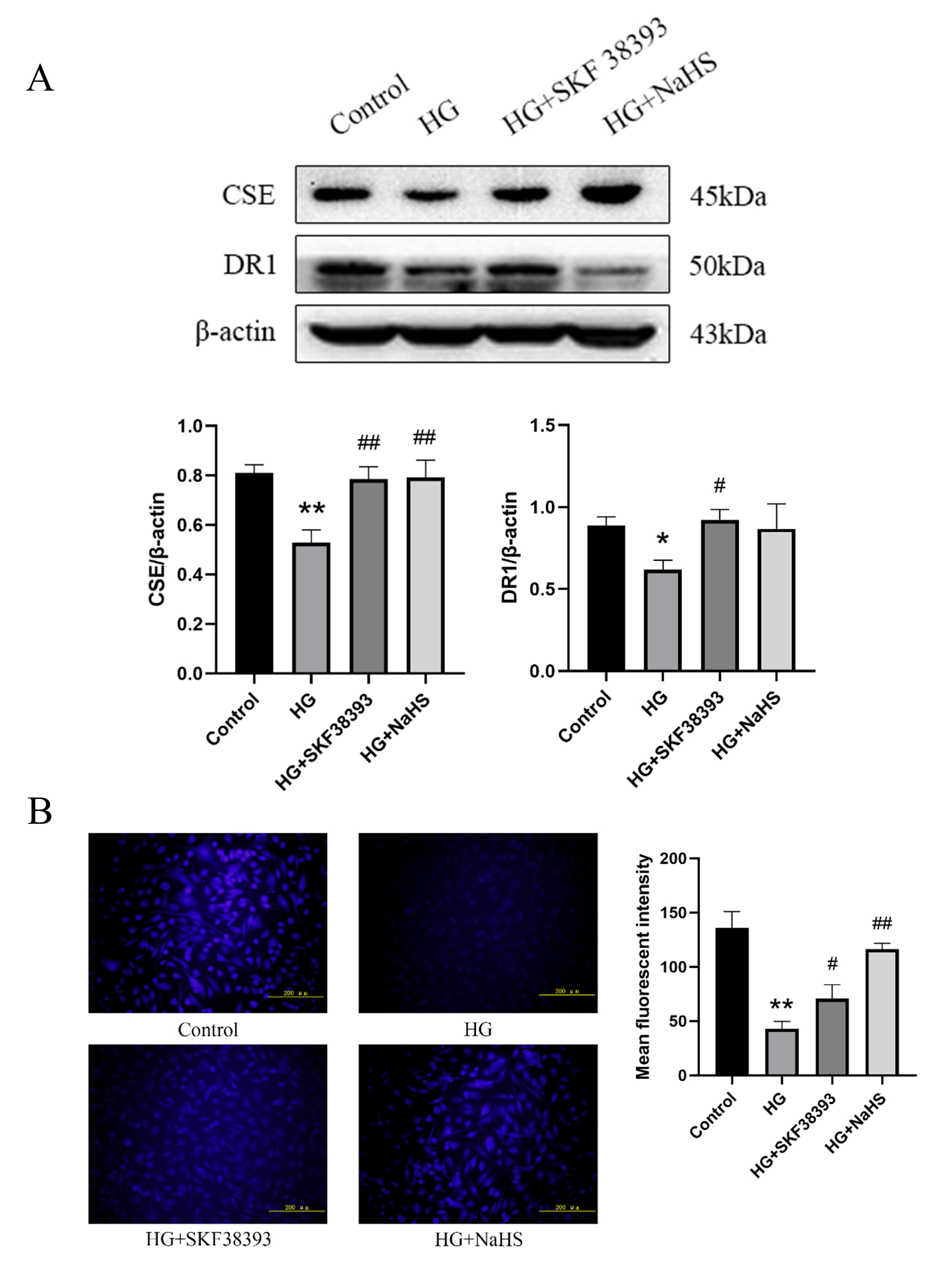 Fig. 1.
Fig. 1.
DR1 activation upregulates the CSE/HS pathway in
HG-induced HUVECs. (A) The expression DR1 and CSE was determined using western
blot (n = 5). The intensity of each band was quantified by densitometry, and data
was normalized to the -actin signal. (B) Fluorescence microscopy was
used to detect HS levels in the HUVECs (n = 4, magnification 200;
scale bar, 200 m). The results were expressed as the mean SEM. *
p 0.05 vs. control group; ** p 0.01 vs. control group;
# p 0.05 vs. HG group; ## p 0.01 vs. HG group.
3.2 DR1 activation upregulates the CSE/HS pathway by
increasing the intracellular calcium concentration in HG-induced HUVECs
Compared with the control group, the intracellular calcium
concentration ([Ca]) was not changed in the control + SKF38393 group
and was increased in the HG group. In addition, [Ca] was further
increased in the HG+SKF38393 group compared with the HG group (Fig. 2).
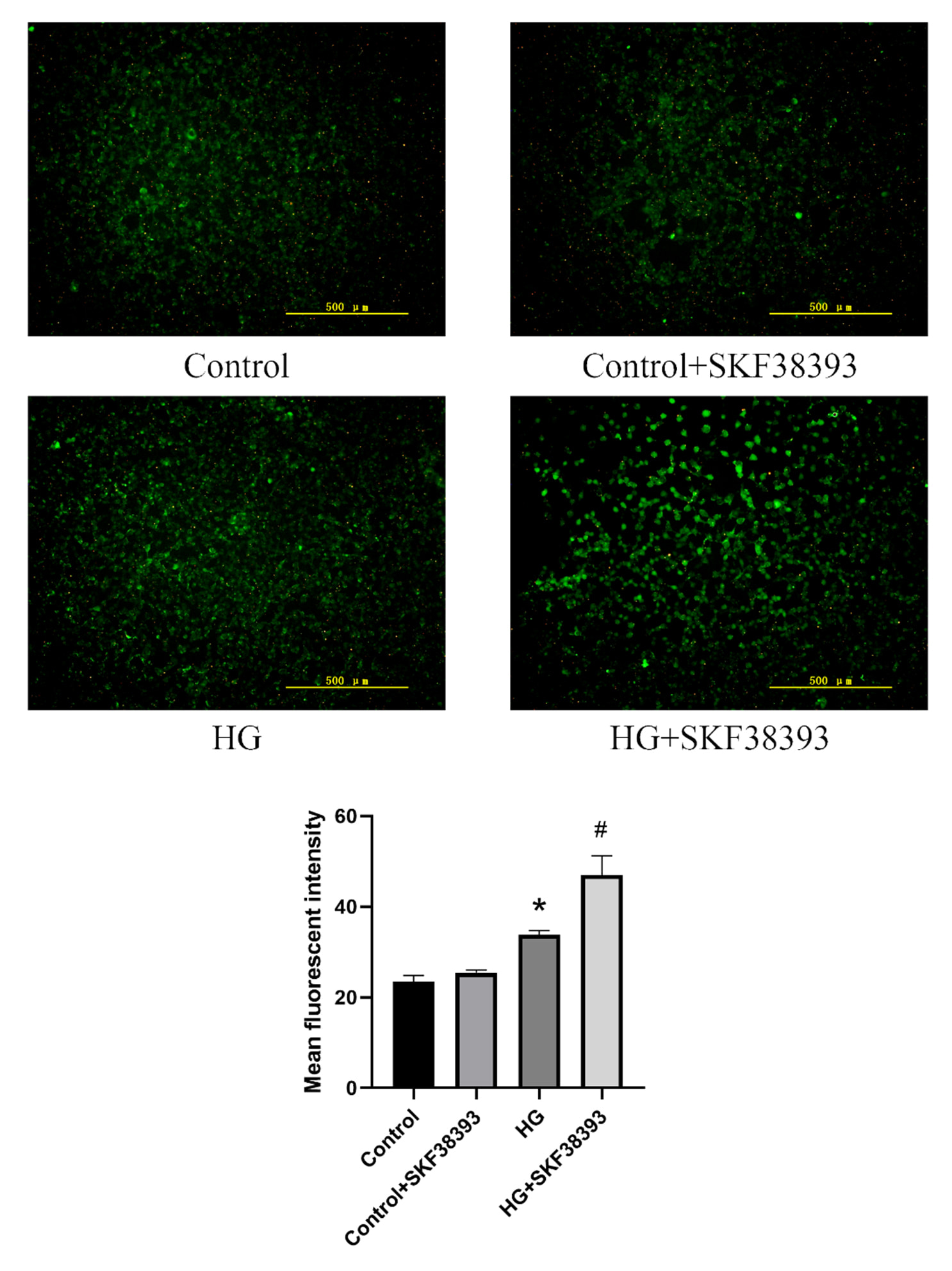 Fig. 2.
Fig. 2.
DR1 activation upregulates the CSE/HS pathway by
increasing intracellular Ca concentration in HG-induced HUVECs.
Fluorescence microscopy was used to detect intracellular Ca concentration
in the HUVECs (n = 3, magnification 100; scale bar, 500 m). The
results were expressed as the mean SEM. * p 0.05 vs. control
group; # p 0.05 vs. HG group.
3.3 DR1 activation increases the proliferation of HG-induced HUVECs
by activating the CSE/HS pathway
Compared with the control group, cell viability and proliferation and the
expression of PCNA and Cyclin D1 were decreased and the expression of
p21 was increased in the HG group. Compared with the HG group, cell
viability and proliferation and the expression of PCNA and Cyclin D1 were
significantly increased and the expression of p21 was obviously
decreased in the HG+SKF38393 and HG+NaHS groups. PPG blocked the effect of
SKF38393 on HG-induced HUVEC proliferation. The beneficial effect of SKF38393 was
similar to that of Y-27632 (a ROCK inhibitor) (Figs. 3,4). These results indicate
that DR1 activation promotes HUVEC proliferation by upregulating the CSE/HS
pathway.
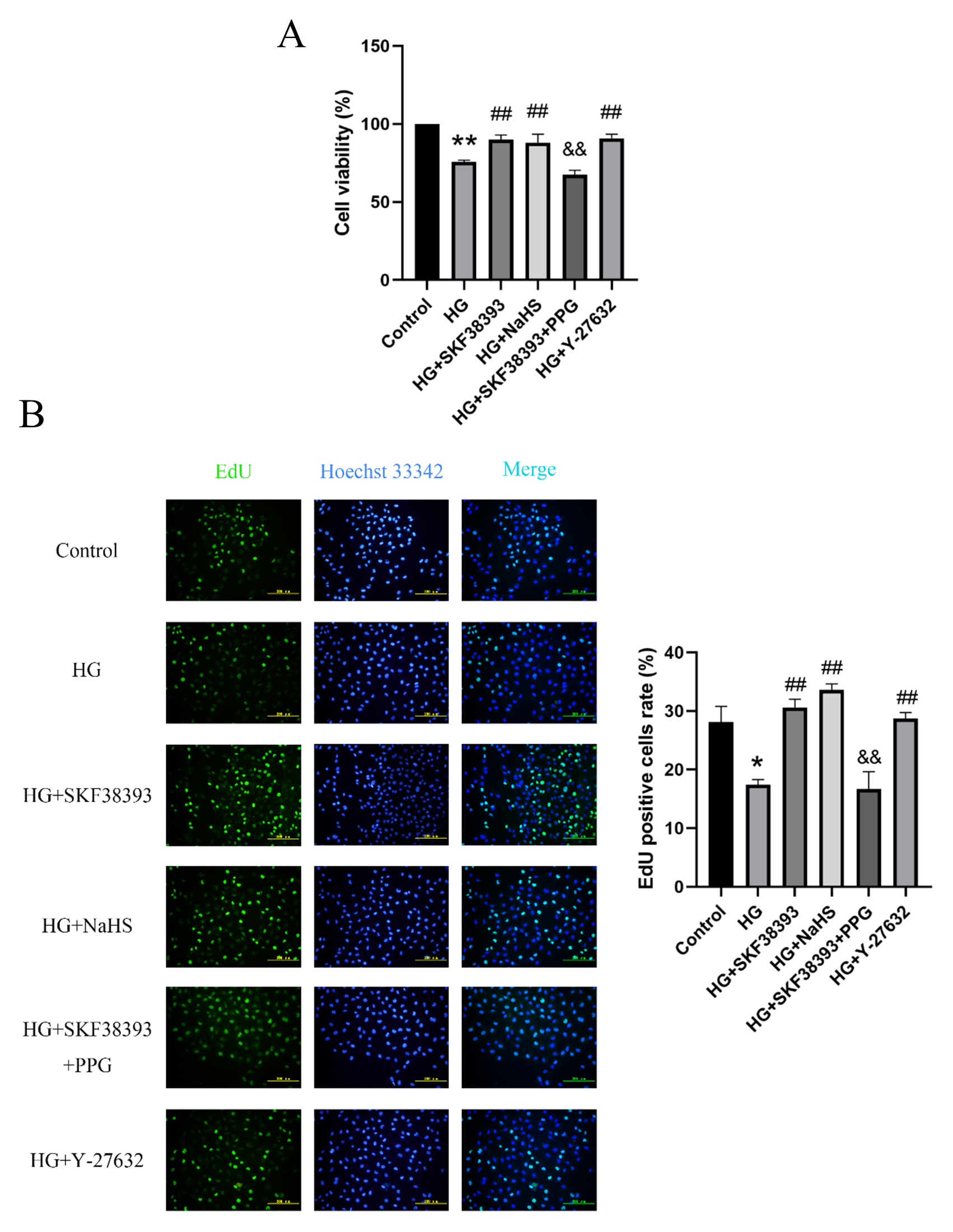 Fig. 3.
Fig. 3.
DR1 activation increases the proliferation of
HG-induced HUVECs by activating the CSE/HS pathway. (A) Cell viability was
detected by CCK-8 kit assay (n = 5). (B) Cell proliferation was detected by EdU
proliferation assay (n = 8, magnification 200; scale bar, 200
m). The results were expressed as the mean SEM. * p
0.05 vs. control group; ** p 0.01 vs. control group; # p
0.05 vs. HG group; ## p 0.01 vs. HG group; && p
0.01 vs. HG+SKF38393 group.
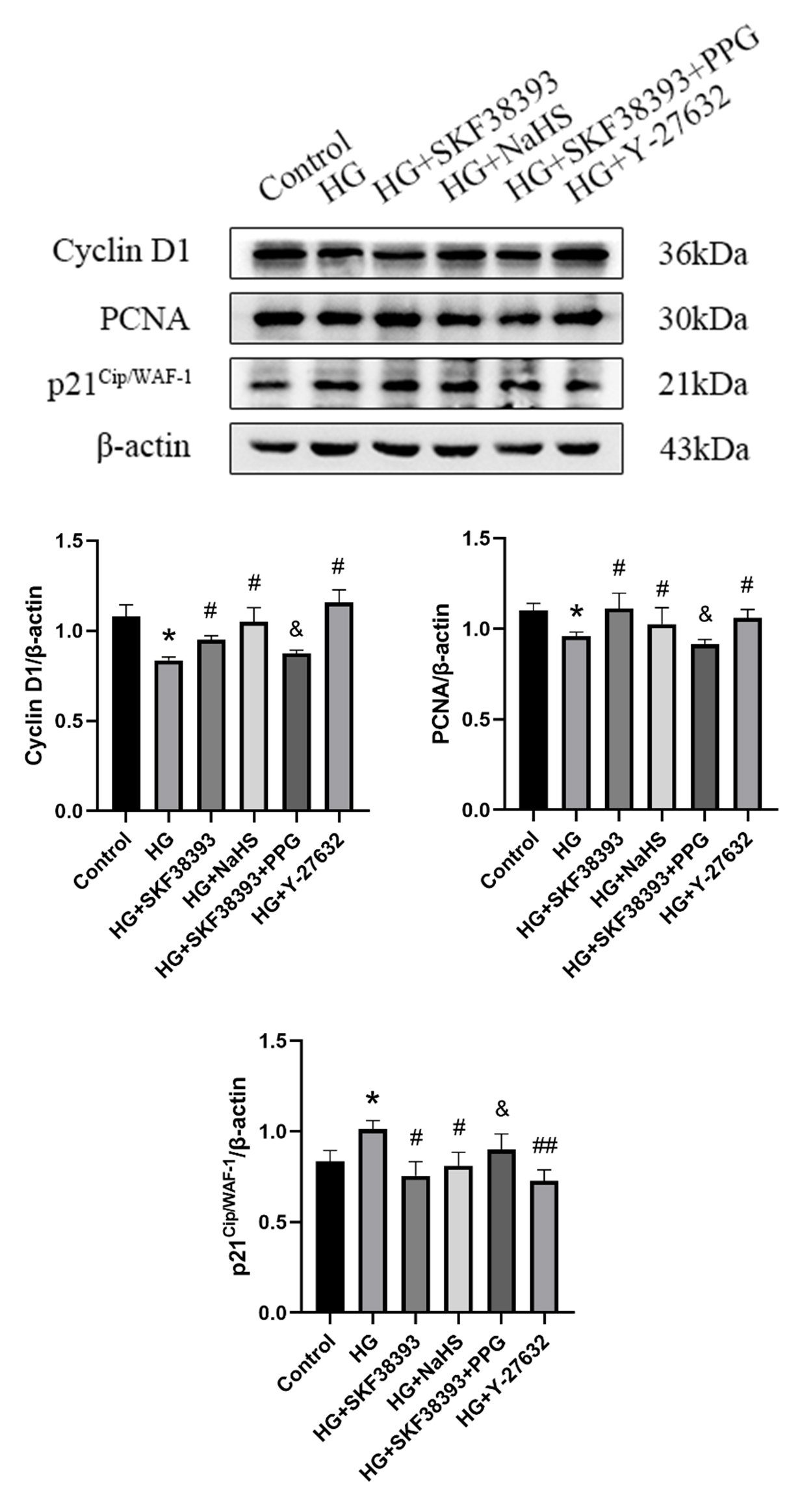 Fig. 4.
Fig. 4.
Effects of DR1 activation on cell proliferation
associated proteins by activating the CSE/HS pathway in HG-induced HUVECs.
Detection of Cyclin D1 (n = 4), PCNA (n = 3) and p21 (n = 3)
expression levels using western blot. The intensity of each band was quantified
by densitometry, and data was normalized to the -actin signal. The
results were expressed as the mean SEM. * p 0.05 vs. control
group; # p 0.05 vs. HG group; ## p 0.01 vs. HG
group; & p 0.05 vs. HG+SKF38393 group.
3.4 DR1 activation alleviates the EndMT and promotes the migration
of HG-induced HUVECs by activating the CSE/HS pathway
Our data showed that HG suppressed HUVEC migration at 24 h and
48 h, while the effect of HG was reversed by SKF38393 and NaHS.
PPG reversed the effect of SKF38393. The beneficial effect of
SKF38393 was similar to that of Y-27632 (Fig. 5).
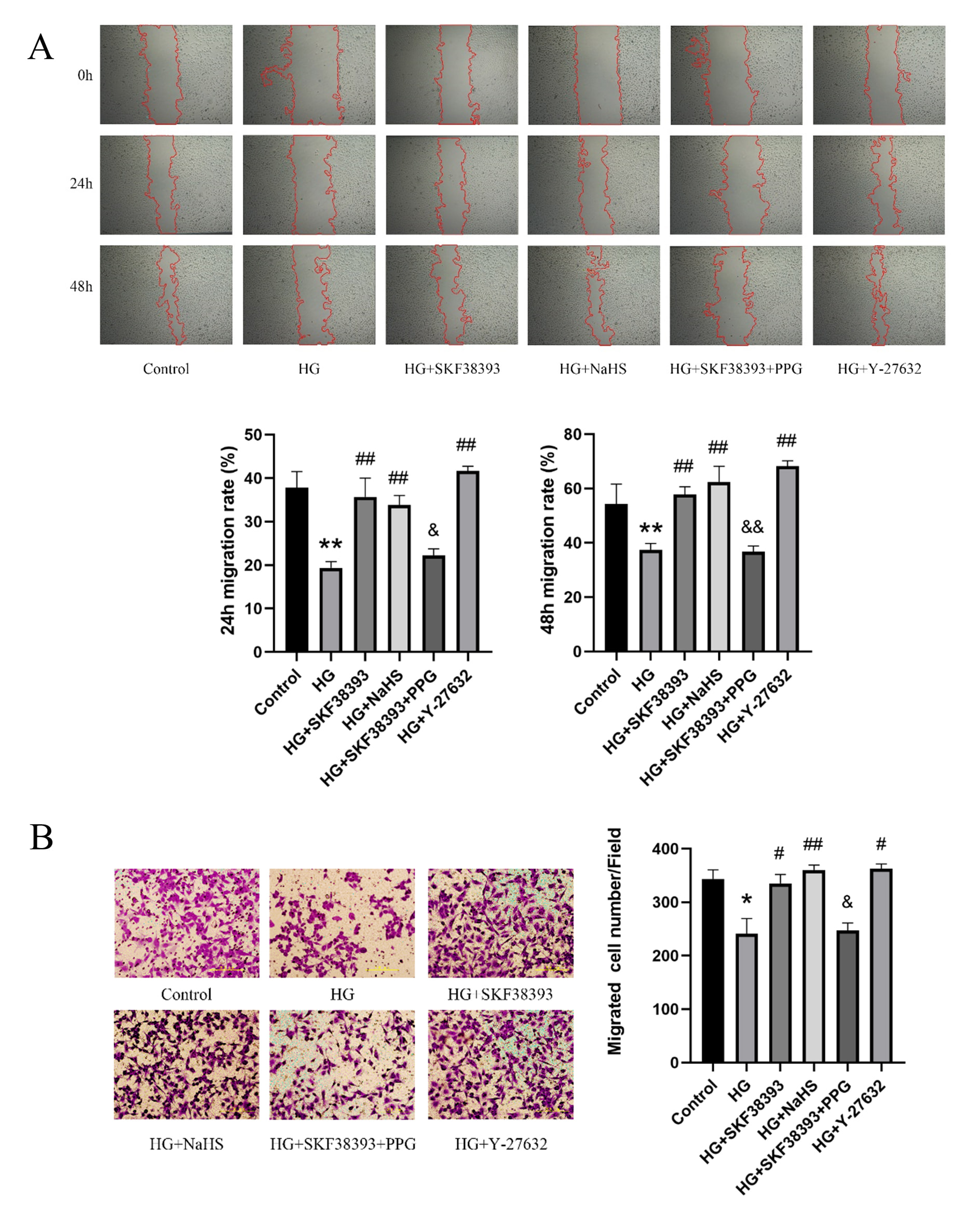 Fig. 5.
Fig. 5.
DR1 activation promotes migration by activating the
CSE/HS pathway in HG-induced HUVECs. (A) Cell migration was measured by
wound healing assays (n = 6). (B) Cell migration was tested via Transwell assay
(n = 3, magnification 200; scale bar, 200 m). The results were
expressed as the mean SEM. * p 0.05 vs. control group; **
p 0.01 vs. control group; # p 0.05 vs. HG group; ##
p 0.01 vs. HG group; & p 0.05 vs. HG+SKF38393 group;
&& p 0.01 vs. HG+SKF38393 group.
A previous study revealed that endothelial cells undergo the EndMT under HG
conditions [33]. We confirmed that the protective effects of the DR1-CSE/HS
pathway were associated with the regulation of EndMT by measuring the levels of
the EndMT markers Col-1, Col-3, MMP-9, OPN, and -SMA using Western
blotting [34]. HG increased the expression of Col-1, Col-3,
MMP-9, OPN and -SMA. However, all these changes were reversed by
SKF38393 and NaHS treatments. PPG abolished
the effect of SKF38393. The beneficial effect of SKF38393 was similar to that of
Y-27632 (Fig. 6). These results suggest that DR1 activation
inhibits the EndMT and promotes migration of HG-induced HUVECs by upregulating
the CSE/HS pathway.
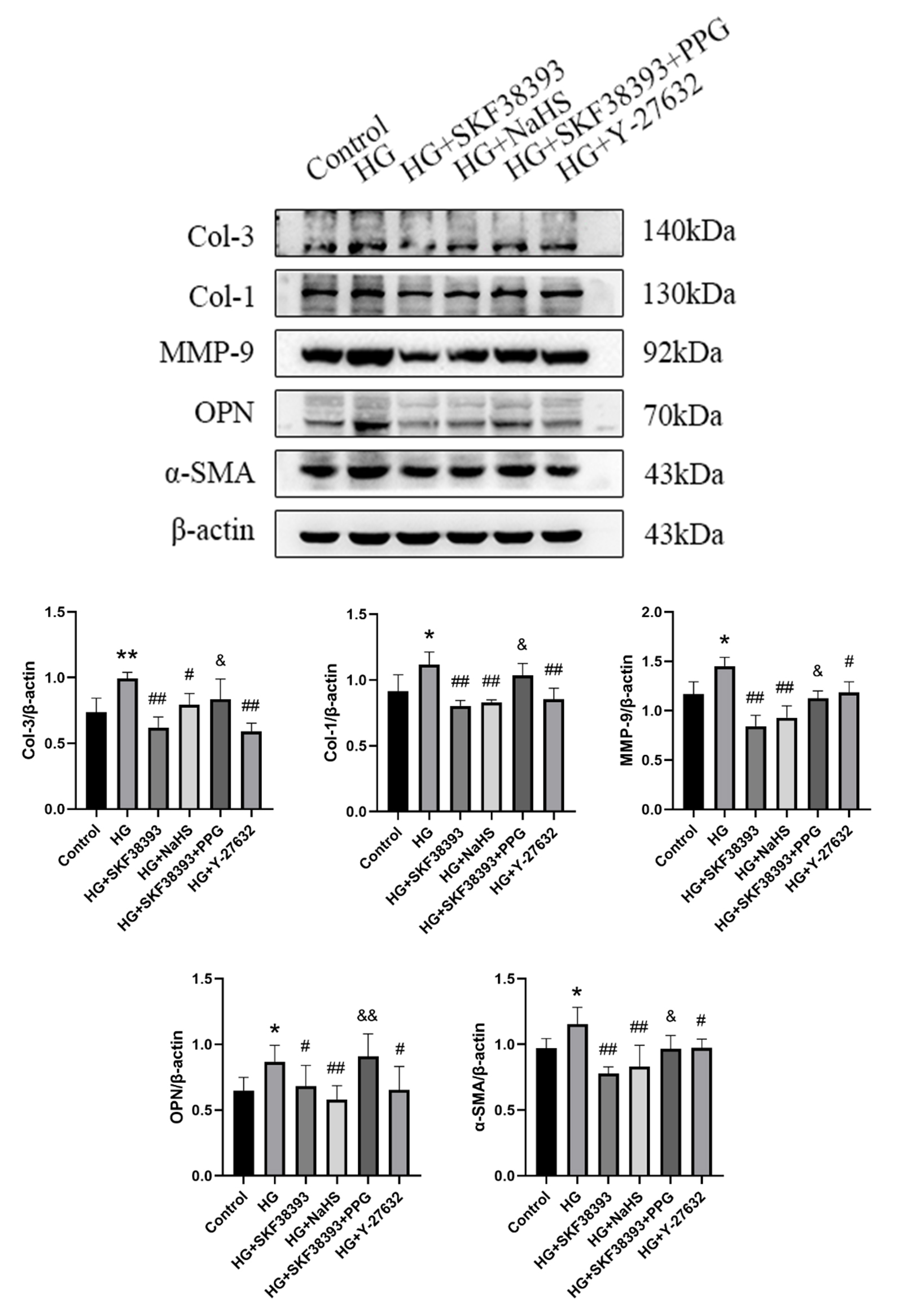 Fig. 6.
Fig. 6.
DR1 activation alleviates the EndMT of HG-induced
HUVECs by activating the CSE/HS pathway. Detection of Col-3 (n = 4), Col-1
(n = 4), MMP-9 (n = 3), OPN (n = 4) and -SMA (n = 3) expression levels
using western blot. The intensity of each band was quantified by densitometry,
and data was normalized to the -actin signal. The results were expressed
as the mean SEM. * p 0.05 vs. control group; ** p
0.01 vs. control group; # p 0.05 vs. HG group; ## p
0.01 vs. HG group; & p 0.05 vs. HG+SKF38393 group; &&
p 0.01 vs. HG+SKF38393 group.
3.5 Activation of the DR1-CSE/HS pathway
attenuates HG-induced HUVEC dysfunction by inhibiting the RhoA/ROCK1 pathway
A previous study indicated that RhoA/ROCK was activated under HG conditions
[35]. In the present study, we measured the levels of t-RhoA,
p-RhoA, t-ROCK1 and p-ROCK1 using Western blotting to determine the effects of
the RhoA/ROCK1 pathway on HG-induced HUVECs and whether the protective effects of
activation of the DR1-CSE/HS pathway were associated with the regulation of
the RhoA/ROCK1 signalling pathway. HG increased the levels of p-RhoA/t-RhoA and
p-ROCK1/t-ROCK1 compared with the control. The levels of p-RhoA/t-RhoA and
p-ROCK1/t-ROCK1 were markedly decreased in the HG+SKF38393, HG+NaHS and
HG+Y-27632 groups (compared with the HG group). PPG abolished the effect of
SKF38393 on the RhoA/ROCK1 pathway. The total levels of the RhoA and ROCK1
proteins remained unchanged after exposure to different stimuli (Fig. 7).
Therefore, activation of the DR1-CSE/HS pathway attenuates HG-induced HUVEC
dysfunction by inhibiting the RhoA/ROCK1 pathway.
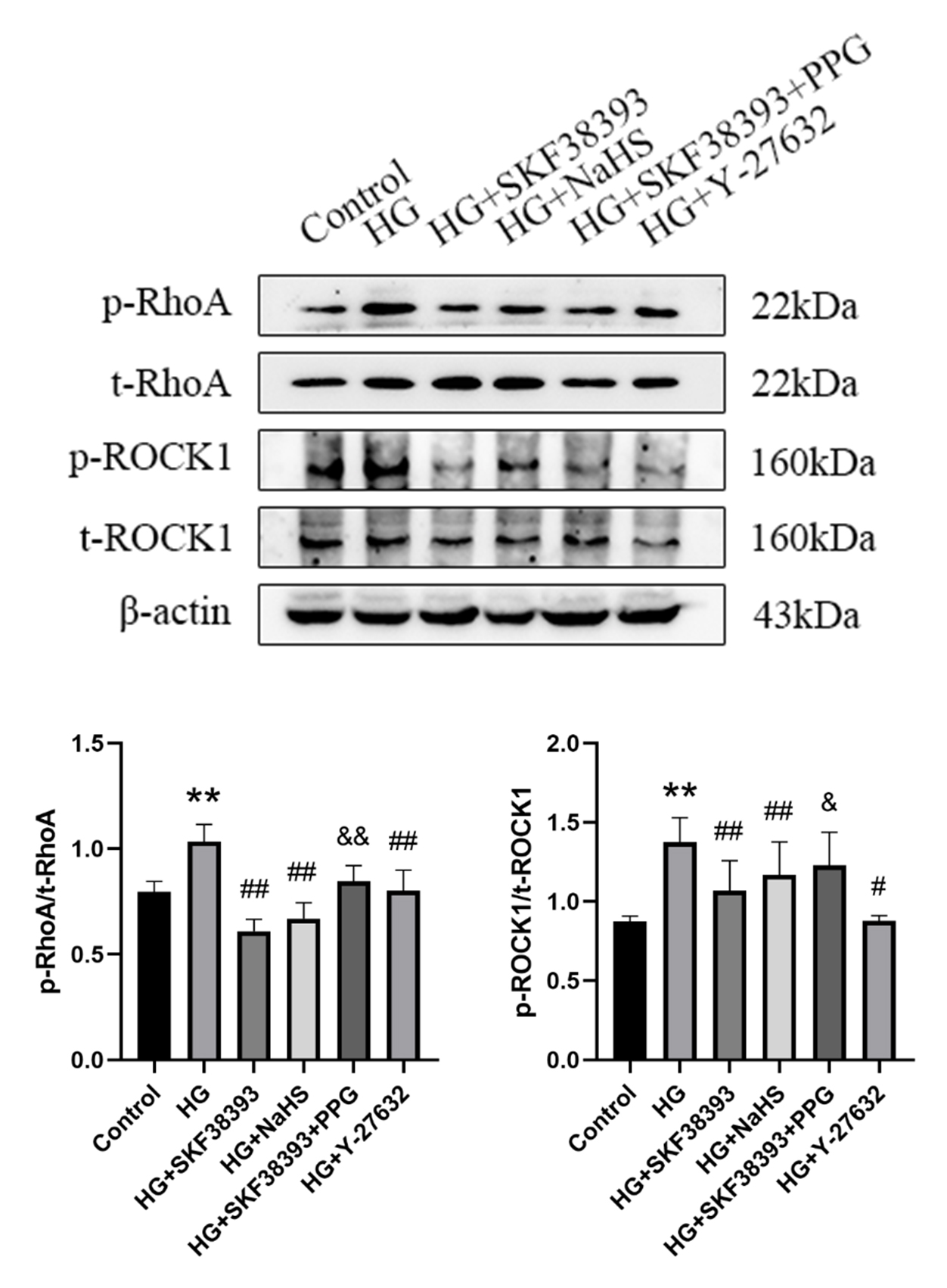 Fig. 7.
Fig. 7.
Activation of the DR1-CSE/HS pathway attenuates
HG-induced HUVEC dysfunction by inhibiting the RhoA/ROCK1 pathway. Analysis of
p-RhoA, RhoA, p-ROCK1 and ROCK1 levels using western blot (n = 4). The intensity
of each phosphorylated band was quantified by densitometry, and data was
normalized to the corresponding total band signal. The results were expressed as
the mean SEM. ** p 0.01 vs. control group; # p
0.05 vs. HG group; ## p 0.01 vs. HG group; & p 0.05
vs. HG+SKF38393 group; && p 0.01 vs. HG+SKF38393 group.
4. Discussion
Our findings provide new insights into the mechanisms of diabetes-induced
vascular endothelial cell dysfunction and the protective effect of DR1 on
regulating the CSE/HS pathway. Our results suggest that (i) DR1 expression
and the activity of the CSE/HS pathway are decreased in HG-induced vascular
endothelial cells. (ii) DR1 activation upregulates the CSE/HS pathway by
increasing [Ca]. (iii) DR1 activation protects endothelial cells
from HG-induced injury, which is related to the regulation of the CSE/HS
pathway and subsequent inhibition of the RhoA/ROCK1 pathway.
Hyperglycaemia causes vascular endothelial dysfunction, which is associated with
diabetic vascular complications [2], and the EndMT contributes to renal fibrosis
which frequently results in deleterious outcomes in patients
with diabetes [4]. These factors are in turn associated with the high
morbidity and mortality rates and an enormous
cost to global health care. HS exerts proangiogenic effects, such as
increased vascular endothelial cell proliferation and migration, microvessel
formation and wound and ulcer healing, both in vivo and in
vitro [36]. Many studies have indicated that the CSE/HS system is
downregulated under HG conditions, including
diabetic animal models and in vitro studies [37, 38]. According to
previous studies, stimulation of DR1 in dermal
fibroblasts restores vascular endothelial growth factor A
production, resulting in adequate angiogenesis and subsequent healing of
cutaneous wounds in diabetic mice [39]. However, the function
of DR1 in diabetic endothelial dysfunction has rarely been explored. The results
of the present study revealed that DR1 and CSE expression and HS production
were reduced in HG-treated HUVECs, while SKF38393 significantly
increased HS production and DR1 and CSE expression. In addition, NaHS only
increased CSE expression and HS production but had no effect on DR1
expression. Based on these results, HG-induced endothelial dysfunction is related
to a decrease in the activity of the DR1-CSE/HS pathway. DR1 activation
upregulates the CSE/HS pathway, and DR1 is an upstream regulatory factor of
the CSE/HS pathway.
How does DR1 regulate CSE/HS pathway? DR1 couples the PLC signalling
pathway that triggers intracellular calcium release [17, 18, 40]. A previous
study confirmed that DR1 activation promotes hypoxia/reoxygenation injury of
cardiomyocytes by increasing [Ca] [21]. Moreover, according to Yang
et al. [24], the increase in [Ca] and the activation of the
Ca-CaM complex activates CSE, which in turn stimulates endogenous HS
production in endothelial cells. Therefore, we detected the
[Ca] to further investigate whether DR1 exerts a protective effect
on HG-induced endothelial cell dysfunction by regulating [Ca], and
the results showed that HG induced [Ca] overload in HUVECs,
consistent with a previous study [41]. However, the CSE/HS pathway was
inhibited in HG. Furthermore, SKF38393 increased [Ca] and activated
the CSE/HS pathway under HG conditions. Thus, DR1 activation upregulates
CSE/HS by increasing [Ca] to a certain level.
Endothelial cell proliferation and migration are required to promote
angiogenesis, which are impaired under HG
conditions [13]. The endothelial to mesenchymal transition (EndMT) has been
identified as playing a vital role in the pathologic process of diabetic
fibrosis. Moreover, HG conditions have been shown to trigger the shift of the
endothelium towards the mesenchymal phenotype [4]. In this study, cyclin D1 and
PCNA protein expression levels were decreased, while
p21 protein levels were increased under HG conditions. In addition,
cell migration was inhibited under HG conditions. In addition, in the present
study, the increased protein levels of
mesenchymal markers Col-1, Col-3, MMP-9,
-SMA, and OPN indicated that HG participated in the occurrence of
EndMT, consistent with a previous study [33]. Furthermore,
HUVEC proliferation and migration were increased after SKF38393 and NaHS
treatments. Moreover, DR1 and CSE/HS activation mitigate mesenchymal marker
expression, thus alleviating the EndMT induced by HG. Our findings suggest that
DR1 activation attenuates HG-induced proliferation and migration dysfunction and
the EndMT by upregulating the CSE/HS pathway in HUVECs.
The phosphorylation of RhoA and ROCK1 is necessary for activation of the
RhoA/ROCK1 signalling pathway. RhoA/ROCK1 are involved in multiple important
cellular processes including proliferation, migration and angiogenesis [6, 42],
and its dysregulation is involved in cardiovascular diseases [42]. Moreover,
previous reports have shown that RhoA/ROCK1 is activated in HG-induced HUVECs,
including angiogenic functions [43] and the EndMT [35]. Furthermore, HS can
protects cerebral endothelial cells from oxygen-glucose
deprivation/reoxygenation-induced injury [44], inhibits colonic
smooth muscle contraction [45], inhibits reactive
astrocytes proliferation and promotes neural
functional recovery in cerebral ischaemia/reperfusion injury [46], and improves
erectile dysfunction in bilateral cavernous
nerve injury [47] by inhibiting the RhoA/ROCK pathway. However, researchers have
not yet determined whether DR1 regulates CSE/HS and
attenuates HG-induced endothelial injury by targeting the RhoA/ROCK pathway. In
the present study, SKF38393 and NaHS inhibited the phosphorylation of RhoA and
ROCK1 and reversed the HG-mediated EndMT and alterations in proliferation and
migration. The beneficial effect of SKF38393 was similar to that of Y-27632 (a
ROCK inhibitor). These results suggest that DR1-CSE/HS activation inhibits
the HG-induced EndMT and alterations in proliferation and migration by
attenuating the RhoA/ROCK pathway in HUVECs.
This study has some limitations. First, HUVECs were used to establish an
in vitro model, which may have resulted in some unexpected outcomes, and
other primary endothelial cells and diabetic animal models deserve additional
research. Second, overexpression and knockdown of DR1, CSE, RhoA and ROCK1 were
not performed to further determine the effects of DR1 activation and HS on
HG-induced HUVEC injury. Third, the precise molecular targets
of HS that inhibit RhoA and ROCK1 activity are not clear. Nalli et
al. [45] reported that HS inhibits smooth muscle contraction via
S-sulfhydration of RhoA, resulting in inhibition of RhoA and ROCK activities.
However, additional investigation is required to unveil the precise molecular
mechanism of the interaction between RhoA/ROCK1 and HS in HG-induced
HUVECs.
5. Conclusions
In summary (Fig. 8), this study showed that DR1 and CSE/HS were
downregulated under HG conditions, DR1 activation upregulated the CSE/HS
pathway by increasing [Ca], and DR1 activation attenuated the
HG-induced EndMT and alterations in proliferation and migration by activating
the CSE/HS pathway, which inhibited the RhoA/ROCK1
pathway. These findings help elucidate the role of DR1 as a
significant regulator under HG conditions, and DR1 may be a
beneficial target to improve vascular function in patients with diabetes
mellitus.
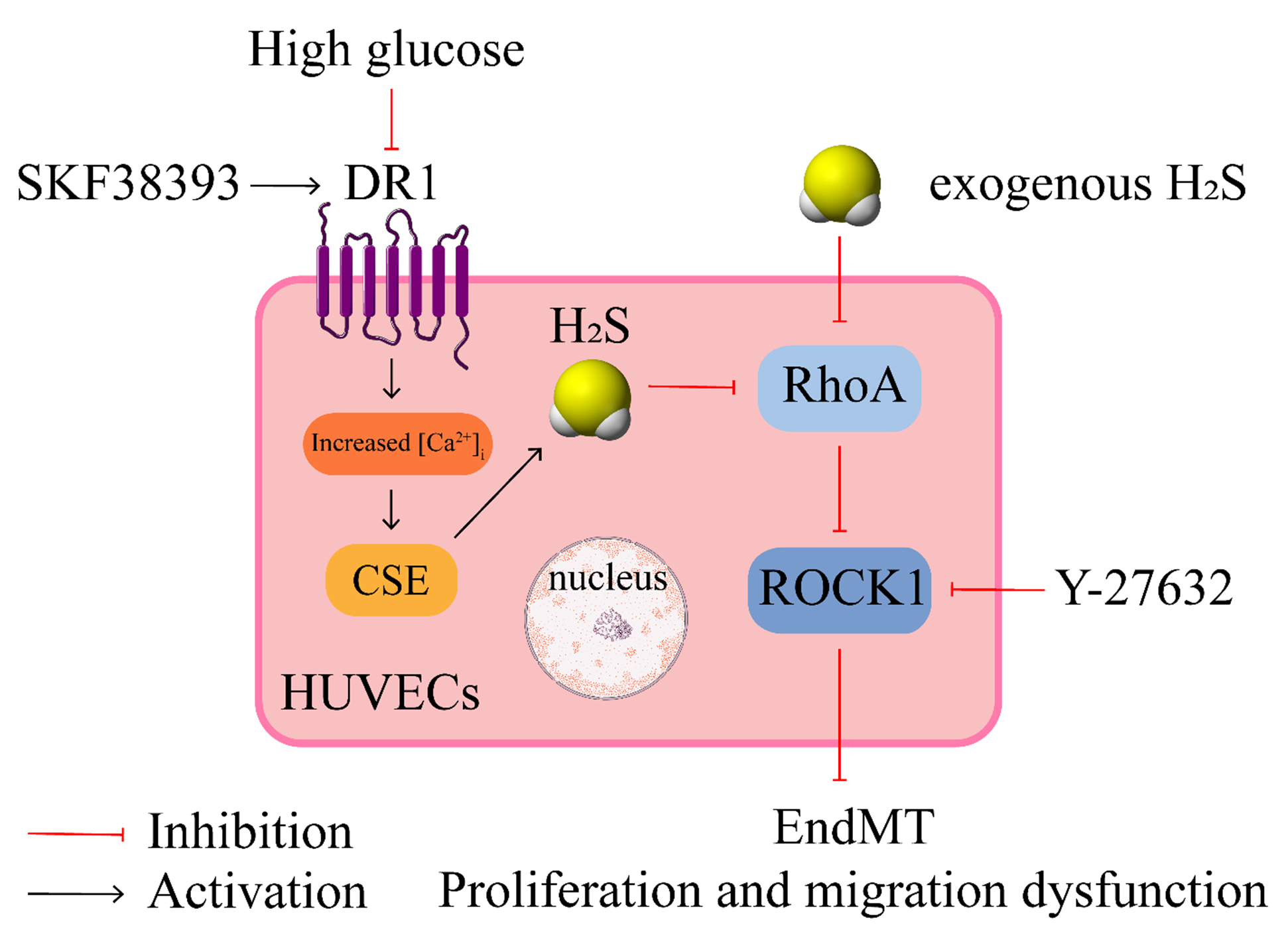 Fig. 8.
Fig. 8.
DR1 up-regulates CSE/HS pathway by increasing
[Ca], which inhibits HG-induced endothelial dysfunction through
down-regulating RhoA/ROCK1pathway. [Ca], intracellular calcium
concentration; EndMT, endothelial to mesenchymal transition; SKF38393, a dopamine
D1-like receptor agonist; Y-27632, a ROCK inhibitor.
Abbreviations
-SMA, alpha-smooth muscle actin; AC, adenylyl cyclases; AzMC,
7-Azido-4-Methylcoumarin; CaMKII, calcium/calmodulin-dependent PK II; CBS,
cystathionine -synthase; Col-1, collagen I; Col-3, collagen III; CSE,
cystathionine -lyase; DAG, diacylglycerol; DM, diabetes mellitus; DR1,
dopamine D1-like receptor; DRs, dopamine receptors; EndMT,
endothelial-mesenchymal transition; HS, hydrogen sulfide; HG, high glucose;
HUVECs, human umbilical vein endothelial cells; IP, inositol trisphosphate;
MMP-9, matrix metalloproteinase 9; MST, 3-mercaptopyruvate sulfurtransferase;
OPN, osteopontin; PCNA, proliferating cell nuclear antigen; PLC, phospholipase C;
PKC, protein kinase C; PPG, DL-propagylglycine; RhoA, Ras homolog gene family
member A; ROCK, Rho-associated coiled-coil containing kinase; TGF,
transforming growth factor receptor ; VECD, vascular endothelial cell
dysfunction.
Author contributions
HZL and SZB conceived the study, designed experiments. GQC, FQS, RW performed
the experiments and analyzed the data. GQC, XW, YXX, JHH and AZ prepared the
figures and performed statistical analysis. GQC wrote the first draft of the
manuscript. CW, AZ and HZL revised the entire manuscript. All authors read and
approved the submitted version.
Ethics approval and consent to participate
Not applicable.
Acknowledgment
Thanks to all the peer reviewers for their opinions and suggestions. Thank you
very much for the help provided by Hong Li and Hong-Xia Li from Department of
pathophysiology in Harbin Medical University.
Funding
This study was supported by the National Natural Science Foundation of China
(No. 81770486, No. 82170268 and No. 81200160).
Conflict of interest
The authors declare no conflict of interest.
Data availability statement
The datasets used and/or analyzed during the present study are available from
the corresponding author on reasonable request.









