






1 Department of Oncology, The First Affiliated Hospital of Zhengzhou University, 450052 Zhengzhou, Henan, China
†These authors contributed equally.
Academic Editor: Alika K. Maunakea
Abstract
DNA methylation plays an important role in the silence of tissue-specific genes to prevent them from being expressed in the wrong tissue. Aberrant DNA methylation (genome-wide hypomethylation and site-specific hypermethylation) are observed in many types of cancer. DNA methylation patterns are established and maintained through the combined actions of methyltransferase and demethylase, such as DNA methyltransferase (DNMT)-1, DNMT-3, and ten-eleven translocation (TET) family enzymes. It is well known that the process of tumor evolution is complicated with different hallmarks. Early findings put forward the model that focal hypermethylation of tumor suppressor genes (TSG) could straightly trigger transcriptional silencing and malignant transformation, whereas varying levels of DNA methylation also occur at other sites and can differently regulate gene expression and biological processes. The interplay of tumor and immune cells in the tumor microenvironment is complex. Understanding the role of DNA methylation in cancer immunity is critical to better navigate epigenetic agents. Furthermore, a greater understanding of the interaction of DNA methylation with tumor metabolic reprogramming would create a bright avenue for pharmacologic managements of malignancies. In this review, we will describe the molecular mechanisms of DNA methylation abnormalities in cancer biology, introduce the roles of DNA methylation patterns on cancer-immunity cycle and metabolic reprogramming, summarize modulators that are used in targeting DNA remodeling, and highlight the importance of combining epigenome-targeting drugs with other cancer therapies.
Keywords
- DNA methylation
- metabolism reprogramming
- cancer-immunity
- novel anti-cancer strategy
Covalent epigenetic modifications made on DNA and histone cooperatively regulate chromatin structure and gene expression [1]. DNA methylation is the most well-characterized epigenetic mechanism. It plays an important role in the silence of tissue-specific genes to prevent them from being expressed in the wrong tissue. The addition and removal of methyl groups to DNA are catalyzed by specific chromatin-modifying proteins (CMP) known as ‘writers’ and ‘erasers’, which is a reversible process and is dynamically regulated [2]. DNA methylation is the covalent addition of a methyl-group (-CH3) to the cytosine (C) base that is subsequently converted to 5-methycytosine (5mC) [3, 4], which is mainly catalyzed by the DNA methyltransferase (DNMT) family, including DNMT1, DNMT3A, and DNMT3B. S-adenosylmethionine (SAM) is the methyl donor provided by the methionine cycle. Conversely, the ten-eleven translocation (TET) family has been proposed to mediate DNA demethylation in an indirect manner through the oxidization of 5-methylcytosine [5].
Genetic alterations of CMPs, such as DNMT3A and TET2, are frequently observed in human cancers [6], leading to aberrant DNA methylation patterns (genome-wide hypomethylation and site-specific hypermethylation). Theoretically, focal hypermethylation of tumor suppressor genes (TSG) could trigger transcriptional silencing and malignant transformation. Varying levels of DNA methylation can also occur at other sites and differently regulate gene expression and biological processes [7, 8, 9, 10]. DNA methylation supports dynamic gene expression, affecting the highly orchestrated trafficking and activation of tumor infiltrating lymphocytes [11, 12]. Therefore, it is capable of modulating anti-tumor immune responses. It is known that DNA methylation is essential for T cell fate and function. A thorough understanding of the role of DNA methylation in cancer immunity is critical to better navigate epigenetic agents. Further, increasing evidence indicates that DNA methylation affects the alteration of metabolic enzymes, which is directly linked to metabolism reprogramming in oncogenesis [13].
Given the extensive alterations and functions in the DNA methylomes, DNA methylation inhibitors form the standard of care for hematological malignancies and the fundamentals of clinical trials in solid tumors [14]. Therapies that combine DNA methylation inhibitors with standard chemotherapy, metabolism modulators, and immunotherapy, will bring novel therapies to clinical decision making.
In this review, we outline the molecular mechanism of DNA methylation abnormalities in cancer biology, highlight the functions of DNA methylation on immune-oncology and metabolic reprogramming, introduce the emerging strategies to target DNA methylation, and explore potential combinations of epigenetic drugs with other therapies.
DNA methylation is the first epigenetic modification observed in human to determine which gene should be turned on or off. Promoter regions contain an increased quality of GC bases that are recognized as CpG islands (CGI). Tumors often present a global DNA hypomethylation with gains of focal DNA hypermethylation at CpG-rich sites [15, 16]. An aberrant DNA methylation landscape has been reported in a variety of tumors, including both hematological malignancies and solid tumors [17, 18, 19, 20, 21, 22, 23]. The mechanisms of carcinogenesis induced by DNA methylation events are displayed in Fig. 1.
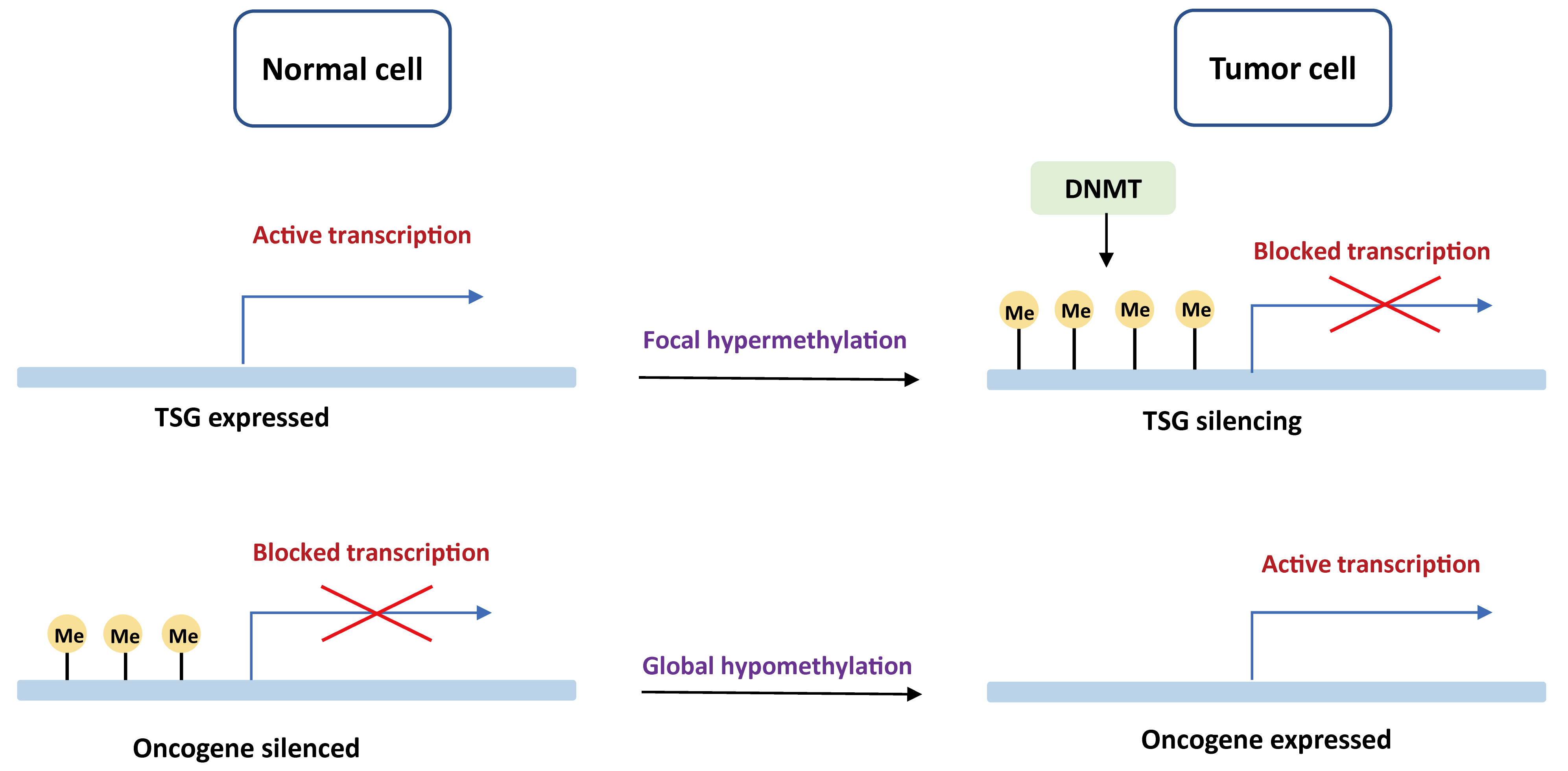 Fig. 1.
Fig. 1.Basic mechanisms of oncogenesis induced by DNA methylation. In normal cells, tumor suppressor genes are demethylated and expressed, while oncogenes are methylated and unexpressed. In the tumor cells, tumor suppressor genes are blocked by DNA de novo methylation with DNMT, whereas oncogenes are actively transcribed due to global hypomethylation. TSG, tumor suppressor gene; DNMT, DNA methyltransferase.
Genome-wide hypomethylation at CpG sites has been reported in many tumor types, such as brain tumor, gastric cancer, liver cancer, and breast cancer [24, 25]. Though global DNA hypomethylation usually occurs in intergenic regions and contributes less to genetic mutations, it can result in genome instability via gene mutations, deletions, inversions, translocations, and amplifications [26]. For example, Long Interspersed Nucleotide Element 1 (LINE-1) is a key component of interspersed DNA repeats. LINE-1 hypomethylation has been reported in several cancer types and portends to a worse prognosis [27, 28, 29, 30]. A recent meta-analysis pointed out that LINE-1 hypomethylation in tissue samples may serve as an epigenetic marker for cancer risk [30]. In addition, several studies highlighted that hypomethylation of cancer-specific CGI may drive the overexpression of oncogenic driver genes [31]. A well-illustrated example is the relationship between BCL-2 and B-cell chronic lymphocytic leukemia [32].
In healthy tissues, the CpG dinucleotides at promoter sites of tumor suppressor genes (TSG) are generally unmethylated and transcriptionally active. However, approximately 5–10% CGI are hypermethylated to prompt TSG silencing in cancer cells [33], such as VHL, RB1, CDKN2A, GATA4, and MHL1 [34, 35]. DNA hypermethylation was first observed in colon cancer. Studies also indicated that CGI methylated phenotype was related to BRAF mutations in colon cancer [36, 37, 38], whereas glioblastoma displaying promoter hypermethylation was significantly associated with IDH1 mutations [39, 40]. Hypermethylation of mismatch repair gene MHL1 has been found in human colorectal cancers, which is related to microsatellite instability [41]. Another example is the hypermethylation of CDKN2A (p16), leading to the inactivation of this gene in esophageal adenocarcinoma [42].
There is a bidirectional interplay between genetic and epigenetic alterations. In addition to genetic mutations driving the dysregulated epigenetic landscape, DNA methylation-induced mutagenesis are also one of the major sources of genetic alterations, resulting in the formation of tumor-associated neoantigens [43]. DNA methylation enables the cytosine base to be more susceptive to deamination, either spontaneous or mutagen motivated, leading to C to T transitions. DNA hypermethylation of transposable elements is critical to epigenetic silencing, as DNA 5-methycytosine is mostly found to locate in the transposable elements [44]. For example, cancer-testis antigens (CTA) could be suppressed by DNA methylation in most somatic cells, but they can be specifically expressed in normal male germ cells [43]. Importantly, reactivation of CTA-coding genes by demethylation can contribute to the presence of neoantigens with immunogenicity and thus enhance immune surveillance [45].
Theoretically, the human immune system
is able to recognize and eliminate tumor cells involving a series of immune
responses, a process called ‘cancer-immunity cycle’ [46].
It has been reported that tumors commonly
hijack various epigenetic remodeling to escape immune surveillance.
DNA methylation not only exerts seminal roles
in the proliferation and effector functions of CD8
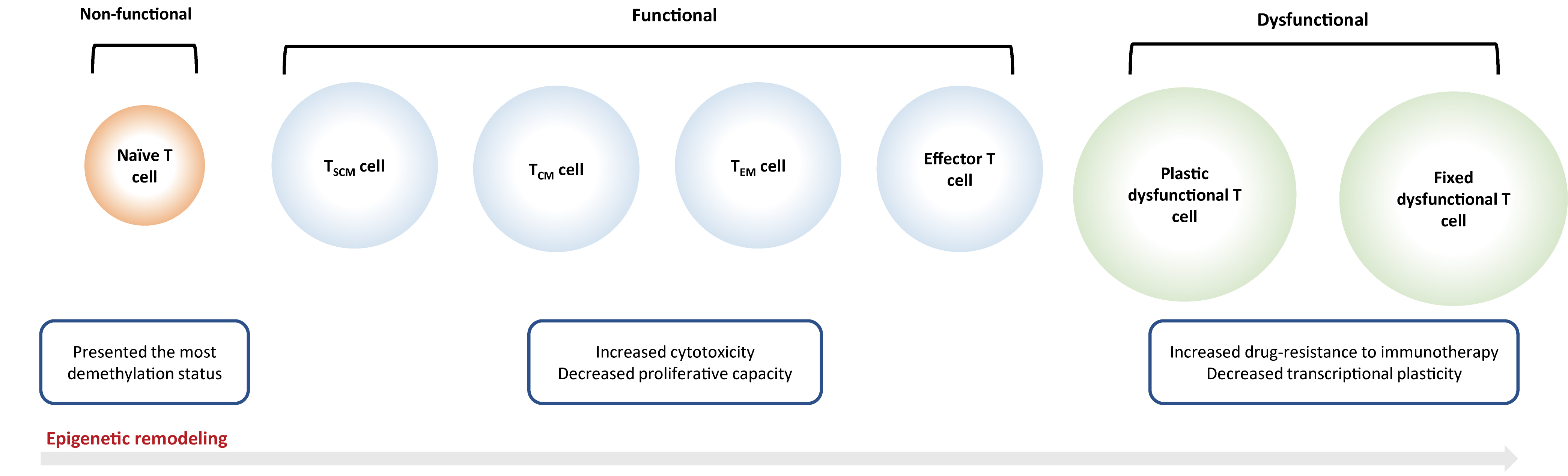 Fig. 2.
Fig. 2.Epigenetic reprogramming on T cells. Naïve T cells will be
activated into an ‘effector’ or ‘functional’ state after antigen exposure, which
is characterized by the rapid secretion of cytokines and effector proteins, as
well as the capacity of infiltration and migration into the tumor
microenvironment. But continuous antigen stimulation would inevitably result in T
cell exhaustion that is subdivided into ‘plastic dysfunctional state’ and ‘fixed
dysfunctional state’. T
DNA methylation changes can regulate the differentiation of
naïve
CD8
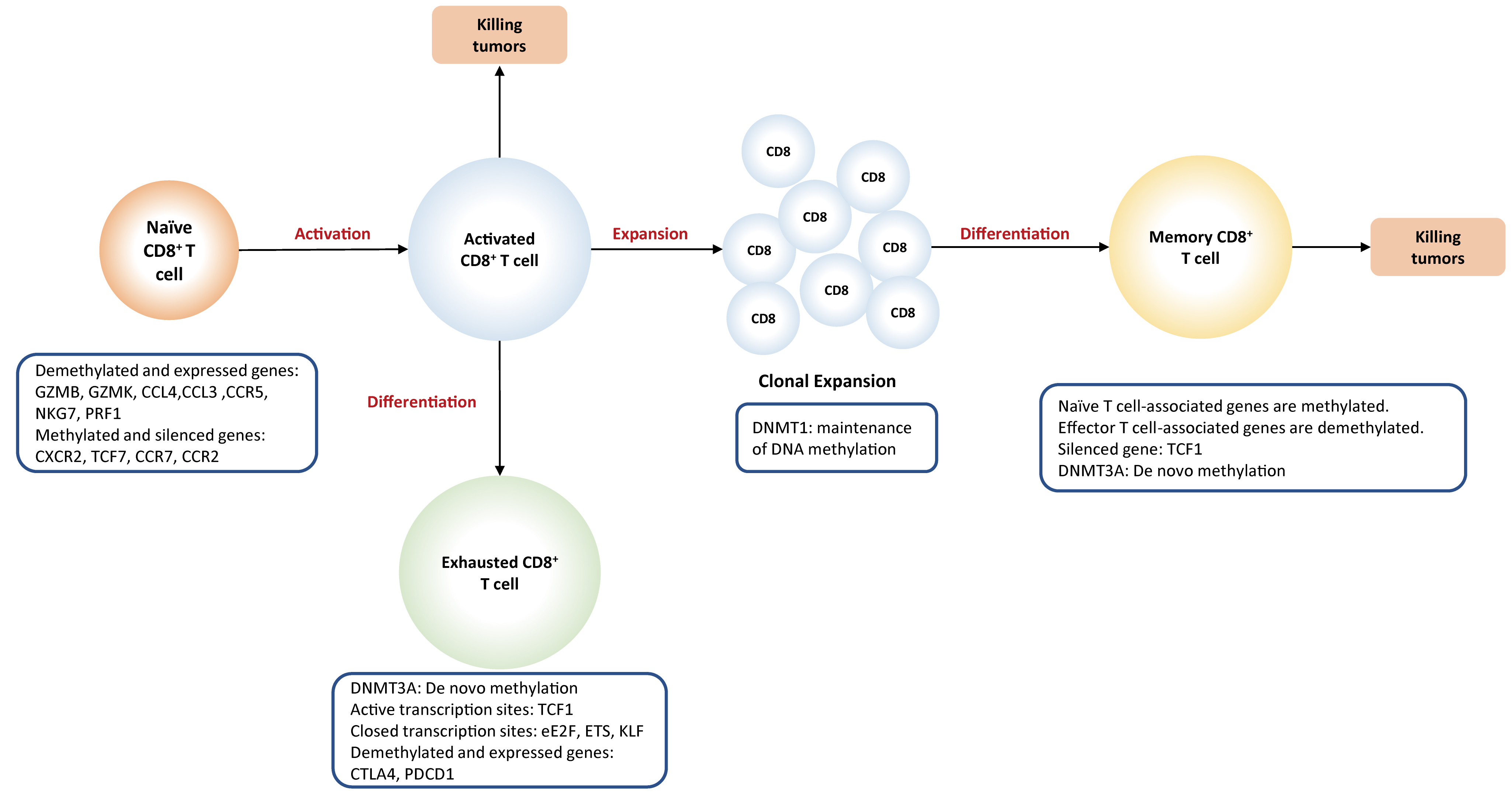 Fig. 3.
Fig. 3.DNA methylation plays a
key role in the differentiation of CD8
Consistently, DNA methylation is responsible for the fate-decision of
naïve
CD4
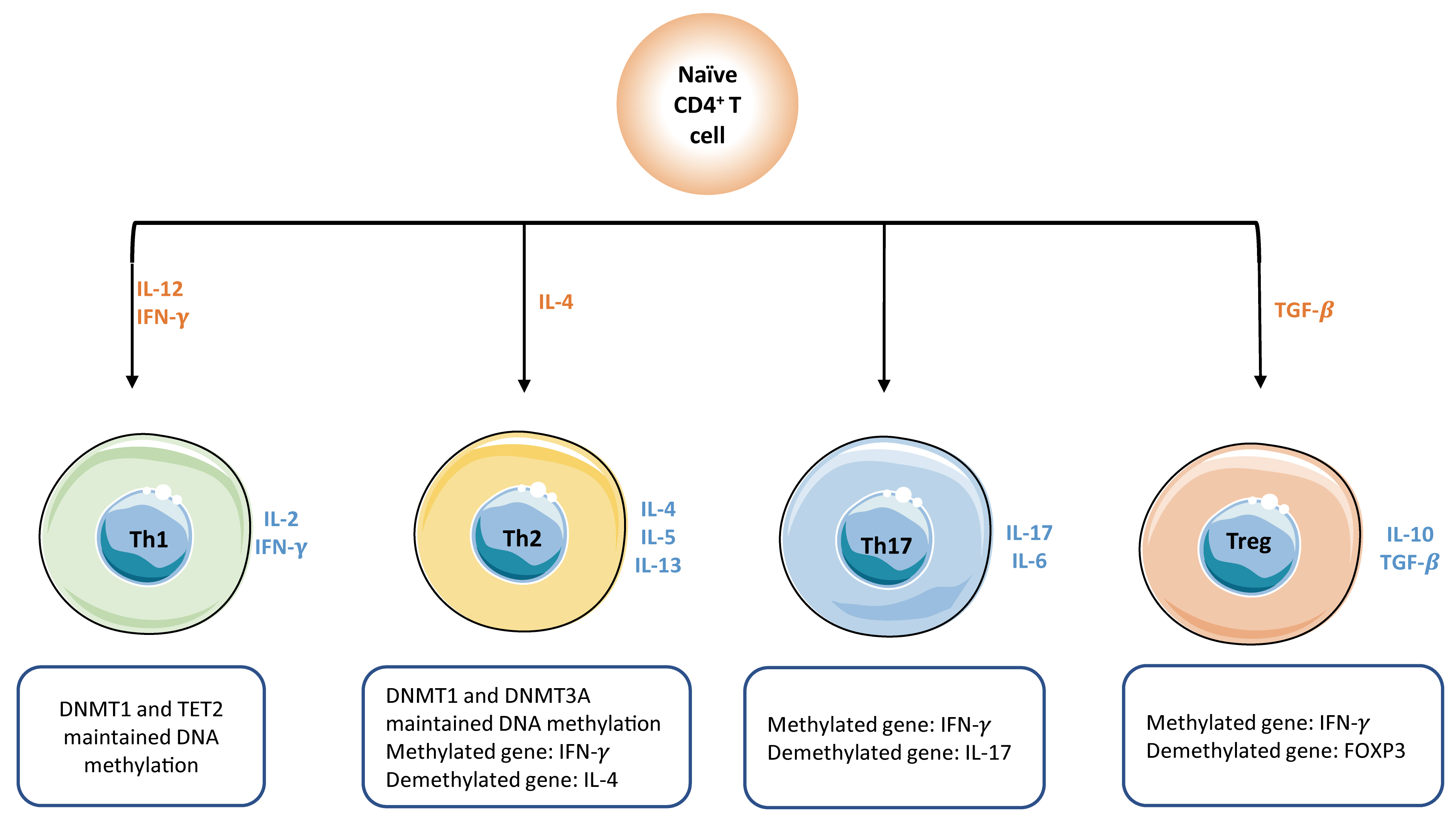 Fig. 4.
Fig. 4.DNA methylation plays a key role in the activation of naïve
CD4
Activation of naïve T cells initially requires the interactions between T cell receptor (TCR) present on T cells and MHC complex (signal 1) expressed on antigen-presenting cells (APC), followed by the activation of co-stimulatory molecules (signal 2) provided by mature dendritic cells. When both signals are present, T cell activation initiates an autonomous intracellular signaling cascade, leading to T cell expansion and differentiation. Global DNA methylation remodeling plays a critical role in priming and activation of cytotoxic effector T cells. For example, demethylation at a promoter-enhancer region of the IL-2 loci, significantly drives the increased levels of IL-2 cytokines that are required for T cell activation and proliferation [62]. Promoter demethylation is observed in several effector genes, such as GZMK and GZMB [53].
T cells become dysfunctional or
exhausted when they are exposed to continuous antigen stimulation, resulting in
the failure to produce effective immune response. Generally, exhausted T cells
display high levels of inhibitory receptors, such as PD-L1, leading to reduced
effector function and suppressed T cell proliferation [63, 64, 65]. It is recognized
that DNA methylation contributes to regulating PD-L1 expression.
Compelling evidence implicates that
PD-L1 promoter is demethylated within exhausted
CD8
Dysregulated metabolism represents a hallmark of malignancy [72, 73]. Recently, accumulating evidence suggests that DNA methylation affects malignant cells’ metabolism and vice versa [13]. The metabolic pathways that DNA methylation participates in mainly involve glycolysis, methionine cycle, and tricarboxylic acid (TCA) cycle [74, 75, 76] (Fig. 5).
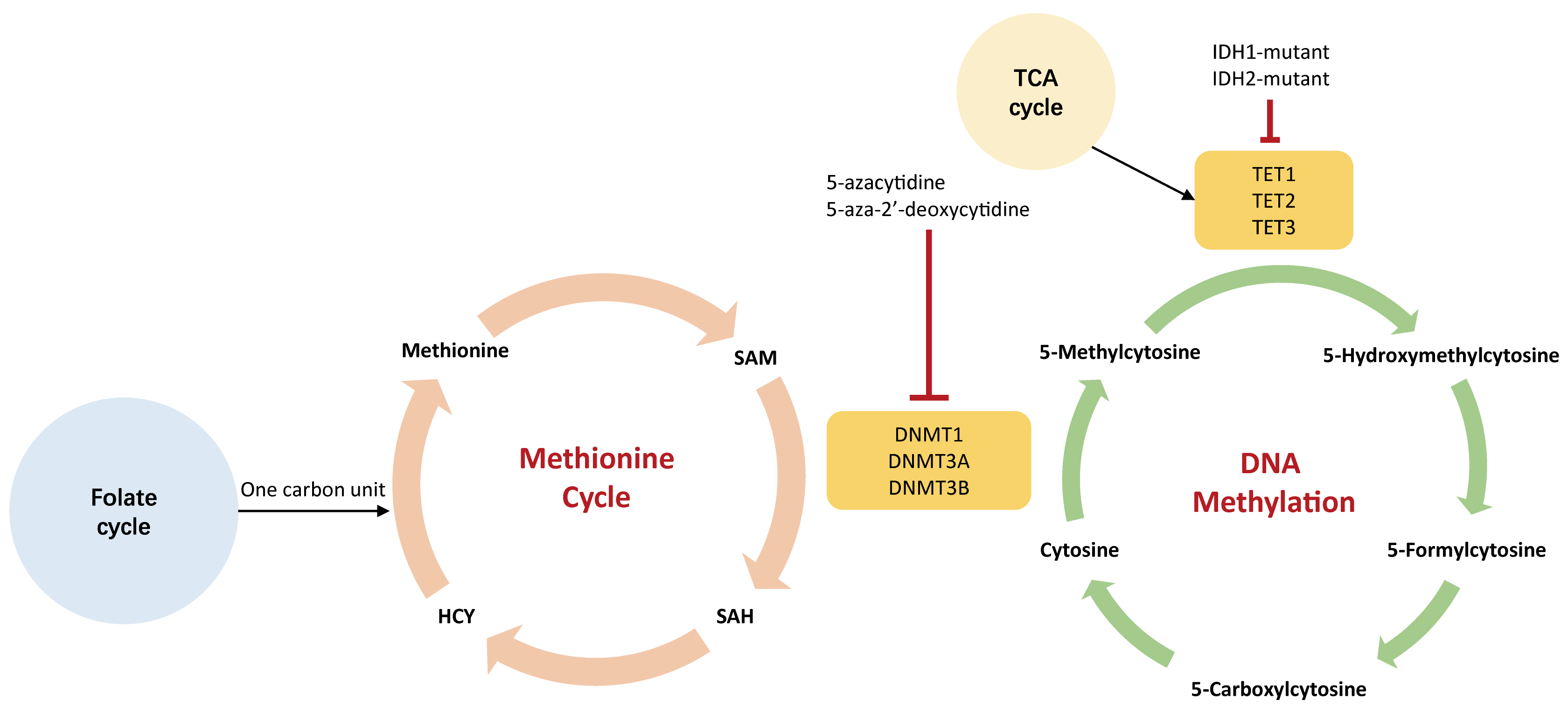 Fig. 5.
Fig. 5.The association between DNA methylation and metabolism. The green cycle represents DNA methylation that occurs on genome; the orange cycle indicates methionine cycle that generates SAM in the cytoplasm. The 5-carbonxylcytosine could be methylated to form 5-methylcytosine by DNA methyltransferases (DNMTs), including DNMT1, DNMT3A, and DNMT3B. Small molecules agents, such as 5-azacytidine and 5-aza-2’-deoxycytidine, could inhibit the activity of DNMTs leading to hypomethylation. These hypomethylating agents are used to treat a variety of human tumors. IDH1-2, isocitrate dehydrogenase 1-2; HCY, homocysteine; SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; DNMT, DNA methyltransferase; TET, ten-eleven translocation demethylase.
DNA methylation indirectly controls the preferential use of aerobic glycolysis (the Warburg effect) even with an abundant supply of oxygen, contributing to a higher glycolytic influx. Several TSGs that regulate aerobic glycolysis are silenced by DNA hypermethylation, such as VHL which is crucial to TSG in the HIF pathway [77]. Therefore, VHL deficiency reinforces glycolytic activity [78]. Also, studies have demonstrated that hypermethylation of the VHL promoter induces gene silencing and consequently reduces HIF1-mediated proteolysis in multiple myeloma [77]. Additionally, promoter demethylation is associated with overexpression of HK2 and PKM2, both of which favor enhanced glycolysis [79, 80, 81]. Oppositely, promoter hypermethylation leads to the gene silencing of FBP-1 and FBP-2 that encode rate-limiting enzymes in glucogenesis. The silence of FBP-1 and FBP-2 could limit gluconeogenesis but support glycolysis, which is beneficial for the proliferation of tumor cells [82, 83].
The methionine cycle gives rise to SAM which is a methyl donor required by DNMT to methylate DNA. It is thought that navigating the methionine cycle may counter leukemogenesis, especially for DNMT3A-mutated AML [84]. Authors have confirmed the findings that alteration of SAM metabolism and DOTL1 inhibition exert a synergistical effect on inhibiting leukemogenesis [84, 85].
Epigenetic enzymes require TCA
metabolites as either substrates or co-factors for post-translational
modifications of DNA.
For
example, the TET family (TET1, TET2, and TET3) is a group of DNA demethylases
that are flavin adenine dinucleotide (FAD)- and
Metabolism not only orchestrates epigenetic marks but also functions in cancer immunity [94, 95]. Several studies supported that metabolism reprogramming impacts intercellular signaling cascade and epigenetic landscape to regulate the longevity and functionality of T cells [96]. Both epigenetics and metabolism are able to regulate T cell exhaustion [97]. Specifically, exhausted T cells would go through nutrient deficiency, along with the altered epigenome.
Abnormalities in DNA methylation are related to tumor initiation and progression [98]. Therefore, a large number of epigenetic modulators are currently being developed to target DNA hyper- and hypo-methylation (Table 1). Several trials are ongoing to investigate potential combinations of therapies [15, 99]. Epigenetic modulators can be broadly divided into ‘reprogramming agents’ and ‘targeted agents’. The former includes DNMT inhibitors (DNMTi) that may reverse abnormal DNA methylation patterns, whereas the latter specifically targets genetic alterations in the epigenetic pathways, such as targeting IDH1/2 mutations in AML.
| Target | Genetic alteration | Inhibitors | Status | Indication | |
| DNA methylation | DNMT | LOF-mutations; Dominant-negative mutations at Arg882 | Decitabine | Approved | MDS, AML |
| Azaciditine | Approved | MDS, AML | |||
| Guadecitabine (SGI-110) | Phase-III | MDS, AML, CMML | |||
| TET2 | LOF-mutations | NA | NA | NA | |
| Abbreviations: CMP, chromatin-modifying protein; DNMT, DNA methyltransferase; TET, ten-eleven translocation dioxygenase; LOF, loss-of-function; MDS, myelodysplastic syndrome; AML, acute myeloid leukemia; CMML, chronic myelomonocytic leukemia; NA, not available. | |||||
TSG silencing caused by promoter hypermethylation is a common mechanism of tumorigenesis, which greatly facilitated the discovery of DNA methyltransferase inhibitors (DNMTi) in the past few decades [100, 101, 102, 103]. DNMTi, also termed as hypomethylating agents, are the most widely utilized epigenetic drugs, especially for treating hematologic malignancies. Analogues of the nucleoside cytidine involve 5-azacytidine (5-AZA), 5-aza-2’deoxycytidine (decitabine), and SGI-110 (guadecitabine), which make DNMT inaccessible to DNA and consequently cause DNA hypomethylation [104, 105, 106]. Both 5-AZA and decitabine have been approved by the Food and Drug Administration (FDA) [107]. These two drugs are currently used as first-line therapy for myelodysplastic syndrome (MDS), bringing improved response rates and prolonged survival [108, 109]. Impressive success has also been achieved in the treatment of acute myeloid leukemia (AML) and chronic myelomonocytic leukemia (CMML) (Table 1). In addition, DNMTi has shown improved outcomes in solid tumors, such as ovarian cancer and non-small cell lung cancer [110, 111, 112].
There is a frequently overlooked phenomenon that the genome-wide DNA
hypomethylation occurs in several solid tumors [113], which is related to tumor
initiation and progression. TET2 mutations are detected in approximately
20–25% of MDS, 7–23% AML, and over 53% CMML [114, 115, 116]. Further, previous
studies indicated that TET1 acts as a suppressor of hematological
malignancies [117, 118]. However, there
are no approved TET inhibitors to reverse DNA hypomethylation, though several
agents are being tested at preclinical stages. For example, the novel compound
Bobcat339 has been shown to directly block the enzymatic activity of TET1/2
[119]. Since the JAK/STAT pathways are involved in TET1 transcription,
STAT inhibitor UC-51423 may avoid aberrant TET1 function [120]. An alternative
approach is to target SAM that donates the methyl group and manipulates the level
of methylation. More importantly, emerging studies suggest that SAM could
modulate cancer immunity by affecting the functionality of CD8
Since DNA methylation and histone modification usually work in parallel,
considerable efforts are being made to explore various combinations that may
significantly increase the efficacy of a single agent and decrease drug
resistance. For instance, in a murine ovarian cancer model, the combination of
DNMTi and histone deacetylase (HDACi) is capable of increasing the cytotoxic
activity of CD8
Numerous clinical trials are evaluating the combination of epigenetic modulators with immune checkpoint inhibitors (ICIs) [125]. The rationale for this proposal is that epigenetic drugs may assist immunotherapy to increase the ability of cytotoxic T cells to attack malignant cells [126, 127, 128, 129]. ICI, an exciting cancer therapy which has emerged in the past several years, has been approved by FDA for non-small cell lung cancer (NSCLC) , melanoma, and renal cancer [130, 131, 132]. There was an unexpected finding that a group of advanced NSCLC patients who had relapsed after a combination of azacytidine (DNMTi) and entinostat (HDACi), received a robust and durable response when they were subsequently enrolled in a clinical trial of ICI [129]. However, it is unknown whether the favorable outcomes reflect the effectiveness of combined therapy or are merely responses to the exposure to ICI. Numerous efforts have been made to figure out the underlying mechanisms as to how epigenetic therapy mediates the augmentation of ICI. It is thought that DNMT inhibitors could reverse immune evasion, as they are able to upregulate the expression of tumor-associated antigens (TAAs) and MHC molecules on the tumor cell surface [133, 134]. Recent studies highlighted that ‘viral mimicry’ produced by DNMTi was tightly associated with increased interferon that may trigger immune attraction [126, 127]. Interests have been switched into the activation of endogenous retroviruses (ERV) which are generally methylated and suppressed in most somatic cells. Mounting evidence suggests that ERV expressed in clear cell renal cell carcinoma could encode peptides that induce T cell and B cell immunoreactivity [135]. As cytosine methylation is critical to the regulation of ERV, reactivation of ERV could be achieved by exposure to DNMTi, and might be effective in reversing immune tolerance for ICI therapy [126, 127, 136, 137].
Aberrant DNA methylation is an epigenetic memory signature that is central to the pathobiological of IDH-mutant tumors, such as gliomas and AML [138]. DNMTi could be used in combination with isocitrate dehydrogenase (IDH) inhibitors, reducing the production of oncometabolites. More specifically, inhibition of mutant forms of IDH, an enzyme in the TCA cycle, could impact the demethylation status of DNA and histone. Findings also demonstrated that DNMTi might be more effective than IDH inhibitors [139], but the efficacy of combination therapy is still unknown. Another example is the combination use of DNMTi with serine metabolism that has been shown to more aggressively kill liver kinase B1-deficiency tumors carrying KRAS [140]. Overall, despite great attentions in targeting both metabolism reprogramming and epigenetic remodeling, it is unknown whether these two hallmarks function in a synergistical manner.
Preclinical studies suggest that DNMTi may improve outcomes when combined with standard cytotoxic drugs. Epigenetic regulation might be a potential mechanism leading to drug resistance to cytotoxic drugs, and thus DNMTi could reverse these pathways and improve the durability of clinical responses [141]. One ongoing clinical trial is being tested to combine DNMTi with cytotoxic agents, making ovarian cancers re-sensitive to these standard drugs [142, 143, 144, 145, 146]. Furthermore, a series of clinical trials are underway to explore combinations of DNMTi with chemotherapy, in an attempt to restore chemosensitivity among patients whose disease have relapsed.
A triple combination of DNMTi, HDACi, and ICI, has also been shown to trigger a stronger anti-tumor effect. The application of ICI following combined inhibition of DNMT1 and EZH2 would make ovarian cancer cells to express CXCL9 and CXCL10, which could activate T helper 1 cells and attract tumor-infiltrating lymphocytes (TILs) [147].
There is a remarkable association between epigenetics and environmental exposure. Lifestyle factors, such as diet, body weight, smoking, and physical activity, constitute the majority of cancer causes, playing an essential role in cancer prevention [148]. One research put forward that lifestyle may modify DNA methylation status leading to genome reprogramming [149]. For example, a study illustrated that the intake of ‘healthy food’ is positively associated with LINE-1 methylation level, which was negatively related to the genome instability [150]. Besides, it has been proven that increased BMI was associated with a lower LINE-1 methylation, particularly for obese females [151]. Increasing evidence suggested that nutritional status may have a lasting effect on metabolism via DNA methylation. A prospective study found that higher dietary folate intake was associated with less LINE-1 hypomethylation in colon cancer, whereas higher alcohol consumption is related to a higher risk of LINE-1 hypomethylation [152]. It is notable that the degree of LINE-1 hypomethylation had a dose-response relationship with poor prognosis and higher mortality in colon tumors [153]. Therefore, modulating DNA methylation may hold the potential to reverse the harmful effects of dietary and lifestyle on tumorigenesis.
In summary, both hypomethylation and hypermethylation of DNA coexist in the process of oncogenesis. DNA hypomethylation is capable of activating proto-oncogenes that leads to genomic instability, while DNA hypermethylation could silence the promoters of TSGs that are frequently associated with tumor suppression. Epigenetic alterations may reactivate several genes that are normally limited to immune-privileged organs, such as CTA. These neoantigens are immunogenic and thus enhance the visibility to immune surveillance. In addition, DNA methylation plays critical roles in the regulation of metabolic reprogramming and the cancer-immunity cycle, and thus intensive investigations have focused on expanding the application of DNMTi to rational combinations with other cancer treatments. The efficacy of single DNMTi is likely to be enhanced by combinatorial therapies. Combining DNA methylation inhibitors with ICIs might sensitize less-immunogenic tumors and overcome immune escape. Combing DNMTi and HDACi can synergistically increase the expression levels of TSG. A combination of DNMTi with chemotherapy may restore sensitivity and reverse drug resistance in relapsed patients. Finally, the reciprocal connection between metabolism and epigenetics will continue to motivate this novel combination strategy. Identifying the most effective epigenetic targeting strategies and exploring rationale-based combinational strategies to boost durable anti-tumor response will play an important role in future clinical practice.
YQ designed the study and reviewed the manuscript. CC and ZW participated in study design and wrote the original draft of the manuscript. CC was mainly responsible for the design of tables and figures. YD, LW, SW, and HW contributed to the conception of the paper. All authors agreed to the submission of the final manuscript.
Not applicable.
Not applicable.
This study was supported by the National Natural Science Foundation of China (grant no. 81872264).
The authors declare no conflict of interest.