
†These authors contributed equally.
Academic Editor: Igor Lavrov
Background: Postoperative cognitive dysfunction (POCD) is a common complication after surgery and anesthesia. In this study, we aimed to determine the neuroprotective mechanism of Sirtuin 3 (SIRT3) and propofol in POCD. Methods: The cognitive dysfunction models in C57BL/6J mice were induced and treated, then cognitive function of mice were tested using morris water maze and novel object recognition tests. Primary neurons were stimulated by lipopolysaccharide (LPS) to mimic neuroinflammation during POCD. Meanwhile, cells were treated with propofol. 3-methyladenine (3-MA) was administrated to inhibit autophagy in neurons. SIRT3 overexpression vector was constructed to upregulate SIRT3. Biomarker changes in inflammation, oxidative stress and autophagy were determined in vivo and in vitro. Results: Propofol enhanced the spatial cognitive ability and novel objective recognition of POCD mice. Inflammation and oxidative stress were observed in the hippocampus, which were inhibited by propofol treatment. During POCD, SIRT3 expression and autophagy in the hippocampus was decreased; propofol activated autophagy and upregulated SIRT3. In LPS-stimulated neurons, SIRT3 upregulation enhanced the anti-inflammation and anti-oxidative stress roles of propofol; SIRT3 elevated propofol-activated autophagy in neurons undergoing LPS administration. Moreover, 3-MA reversed propofol-induced biomarker changes in inflammation, oxidative stress and autophagy in LPS-stimulated neurons. In POCD mice, SIRT3 upregulation enhanced the cognitive function during propofol treatment; SIRT3 overexpression elevated the inhibitory role of propofol in inflammation, oxidative stress and autophagy. AMPK/mTOR pathway was activated in response to propofol treatment and SIRT3 enhanced the signaling activation. Conclusions: SIRT3 enhances the protective effect of propofol on POCD by triggering autophagy that eliminates oxidative stress and inhibits the production of pro-inflammatory cytokines.
Postoperative cognitive dysfunction (POCD) is a common complication characterized by cognitive dysfunction lasting weeks to years after surgery and anesthesia, which results in the increased risk of mortality and poor outcome [1, 2]. POCD is found in 10–25% of patients who undergo anesthesia and surgical operation at 3–6 months [3]. There are potential risk factors of POCD such as age, lower educational attainment and cerebrovascular accident history [4]. POCD pathogenesis is complex and heterogeneous, and inflammation in hippocampus contributes to the occurrence and development of POCD. Pro-inflammatory cytokines are enriched in the brain to trigger neurotoxicity and systemic inflammation, thereby developing to pathological alterations such as neuroinflammation, oxidative stress and autophagy disorder [5]. Autophagy is associated with the removal of impaired cells/organelles and excessive pro-inflammatory cytokines, which functions as the neuroprotective role in POCD [6]. The core connection between autophagy-mediated inflammation and POCD suggests a potential therapeutic strategy of drugs.
Anesthetic drugs have the dual effect during POCD progression. On the one hand, anesthesia evokes changes in cholinergic system and cell apoptosis, thereby contributing to cognitive impairment [7]. On the other hand, appropriate anesthesia strategy protects patients from cognitive impairment during perioperative period [8]. It is an urgent requirement of POCD to explore the optimal anesthetic administration based on drug kinds and dosages. Propofol, an anesthetic drug, has the potential of POCD improvement. Recently, a randomized controlled preliminary trial showed that propofol resulted in a lower risk of POCD compared with dexmedetomidine and midazolam [9]. Also, propofol can reduce the incidence of delayed neurocognitive recovery after major cancer surgery [10]. At the molecular level, propofol can protect blood-brain barrier from the neuroinflammation condition via regulating the POCD-associated miRNAs and the inflammation-related pathways [11, 12]. Understanding the functional mechanism of propofol will promote its application in POCD.
Sirtuin 3 (SIRT3), a member of sirtuin family, is a histone deacetylase expressed in mitochondria that contributes to the protection of mitochondrial integrity and energy metabolism via its biological activity involved in the catalysis of deacetylation and ADP ribosylation [13]. SIRT3 is related to the production of reactive oxygen species (ROS) and neuron death in hippocampus [14]. Mounting studies suggest the neuroprotective role of SIRT3 in POCD. In aged mice undergoing cognitive impairment, SIRT3 upregulation mitigates neuroinflammation and oxidative stress in hippocampus, which is the key event of POCD improvement [15]. SIRT3 activation is the key event to resist POCD via the anti-inflammation and the anti-oxidative mechanisms [16]. Thus, SIRT3 may be the vital therapeutic target of POCD. Moreover, SIRT3 plays the crucial role of autophagy during POCD. SIRT3 can modulate autophagy activation via numerous signaling pathways. For example, SIRT3 deficiency results in the inactivation of adenosine-monophosphate activated-protein kinase (AMPK) /mammalian target of rapamycin (mTOR) pathway, which fails to trigger autophagy in cells [17]. SIRT3 can catalyze the deacetylation of autophagy-related proteins such as forkhead box protein O1 (FOXO1, contributing to autophagosome formation) and FOXO3A (associated with mitochondrial fission) [18]. The intriguing connection between SIRT3 and autophagy shows the potential mechanism in controlling autophagy-mediated neuronal damage during POCD.
Thus, we provide a potential hypothesis that propofol upregulates SIRT3 to activate AMPK/mTOR pathway that can evoke neuronal autophagy to remove the neuroinflammation during POCD. SIRT3 contributes to the protective role of propofol in POCD via activating AMPK/mTOR pathway to evoke autophagy in neurons. This study aims to provide a novel insight into the therapeutic mechanism of propofol in POCD based on neuronal autophagy. Also, our findings will be instrumental in understanding the role of SIRT3-mediated autophagy in inflammatory neurons during POCD.
Animal experiments were approved by the Ethics Committee of Beijing
Stomatological Hospital, Capital Medical University (Approval No. MDKN-2021-055).
18 months old male C57BL/6J mice, weighing 26–34 g, were purchased from the
Model Animal Research Center of Nanjing University (Nanjing, China) and housed at
24
The spatial memory of mice was determined by Morris water maze. Morris water
maze testing was carried out in a round white pool (diameter: 100 cm; depth: 38
cm), in which a platform was placed. The pool contained 30-cm-depth water at 20
After a 7-day habitation in cages, each mouse was placed in a box (25
After the last behavior test, mice were performed with the euthanize via
decapitation. Hippocampus tissues were isolated and embedded into paraffins after
dehydration using ethanol. Tissues were cut into 3-
The euthanize in C57BL/6J was processed by decapitation after the disinfection
of 75% Alcohol. Brain tissues were isolated and then placed in D-hank’s solution
(Solarbio, Beijing, China) without irons of calcium and magnesium. Hippocampus
was separated from brain tissue, followed by the incubation with 0.25% Trypsin
(Solarbio) for 15 min at 37 °C in a 15 mL conical tube. During incubation, the
tube was inverted every 5 min. Then, supernatant in the tube was carefully
removed. Cell precipitate was washed by D hank’s solution for 3 times and then
resuspended in D hank’s solution. After the 2-min standing, cell suspension was
centrifugated at 4 °C and 1000 rpm for 10 min. Cells resuspended in DMEM-F12
(Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS; Gibco)
were filtered using a 100-
6 groups were formed: control, LPS, LPS + propofol, LPS + propofol + pcDNA3.1,
LPS + propofol + SIRT3 and LPS + propofol + 3-MA. To induce inflammation in
neurons, cells were stimulated by 1
The fluorescent LC3 antibody was used to determine autophagy in neurons. First, neurons were incubated in 100% ice-cold methanol (Sigma) for 15 min at –20 °C after cell medium was remove. Cells were washed using PBS solution for 3 times. Then, cells were incubated with PBS solution (#9808, Cell Signaling Technology) containing 5% normal goat serum (#5425, Cell Signaling Technology) and 0.3% Triton X-100 to be blocked for 60 min at room temperature. LC3 antibody (#4108, 1:200, Cell Signaling Technology) diluted in PBS containing 1% bovine serum albumin (BSA; #9998, Cell Signaling Technology) and 0.3% Triton X-100 was added to incubate cells at 4 °C overnight. After that, cells were incubated with diluted secondary antibody (#4412, 1:500, Cell Signaling Technology) for 1 hour at room temperature, protected from light. Finally, washed cells were incubated with ProLong® Gold Antifade Reagent (#9071, Cell Signaling Technology) to avoid light quenching. Fluorescent LC3 was observed under a fluorescence microscope (Olympus, Tokyo, Japan).
We measured pro-inflammatory cytokines in hippocampus and neuron based on ELISA.
According to the guideline of kits, pro-inflammatory cytokines including
TNF-
Before the measurement, cells or hippocampal tissues were incubated in RIPA buffer for lysate. Then, MDA and SOD level in lysates were measured by MDA kit (E-BC-K025-M, Elabscience, Wuhan, China) and SOD kit (E-BC-K020-M, Elabscience), respectively. Procedures of the measurement was followed by the product manual. The absorbance of each sample was read by the microplate reader at 532 nm (for MDA detection) and 450 nm (for SOD detection).
After the medium was removed, neurons were incubated with Trizol regnant
(Solarbio) to extract total RNA. Based on One Step RT-qPCR Kit (Sangon, Shanghai,
China), RNA was reversely transcribed into cDNA and quantified under a real-time
PCR system (Applied Biosystem, Foster City, CA, USA). The procedure of
reverse-transcription and quantification was as follows: reverse-transcription
for 5 min at 50 °C; pre-denaturation for 3 min at 95 °C; denaturation for 10 s at
95 °C and Annealing/extension/acquisition of fluorescent signals for 30 s, 40
cycles. The relative quantitative expression was determined using the
2
Cell and tissue samples were incubated with the lysis buffer (Beyotime,
Shanghai, China) consisting of 20 mM Tris, 150 mM NaCl and 1% Triton X-100 to
extract proteins. The concentration of protein samples was measured by a BCA
Protein Assay Kit (E-BC-K318-M, Elabscience). Protein samples were then separated
using SDS-PAGE under an electrophoresis apparatus (Bio-Rad, Hercules, CA, USA),
followed by the transport of blots from gels to nitrocellulose membranes (Cell
Signaling Technology). The membranes were blocked by 1
Experimental data were shown as mean
The spatial memory of mice was described by escape latency, time spending on the
quadrant and the times crossing the platform based on water maze assay.
Anesthesia/surgery increased escape latency (Fig. 1A) but declined spending time
(Fig. 1B) and crossing times (Fig. 1C), indicating the poor capability of spatial
memory. After propofol treatment, the escape latency, spending time and crossing
times of mice with POCD were close to that of mice in sham group (Fig. 1A–C).
Learning memory was tested by new object recognition index. Significantly,
propofol enhanced the anesthesia/surgery-reduced recognition index (Fig. 1D),
showing the improvement of learning memory in mice with POCD after propofol
treatment. In the brain of mice, anesthesia/surgery elevated levels of
TNF-
 Fig. 1.
Fig. 1.Propofol enhanced SIRT3 and autophagy in mice with POCD. (A) Escape latency in Morris water maze assay. (B) Time
spending on the target quadrant based on Morris water maze. (C) Times
crossing the platform in Morris water maze assay. (D) Recognition index
was tested by novel object recognition test. (E) Levels of
TNF-
Primary neurons were isolated (Fig. 2A) and incubated with 1
 Fig. 2.
Fig. 2.SIRT3 enhanced the inhibitory effects of propofol on
inflammation and oxidative stress in LPS-stimulated hippocampus neurons.
(A) Images of isolated neurons. (B) SIRT3 expression in
primary neurons was detected using qPCR. (C) SRIT3 expression in
primary neurons was detected using western blot. (D) The expressions of
pro-inflammation factors including TNF-
The phosphorylation of AMPK/mTOR pathway was enhanced in LPS-stimulated neurons during propofol treatment (Fig. 3A), suggesting that propofol could induce the activation of AMPK/mTOR pathway in inflammatory neurons. SIRT3 further elevated the propofol-enhanced phosphorylation level of AMPK and decreased mTOR phosphorylation level in inflammatory neurons (Fig. 3A). Moreover, propofol could reverse LPS-induced altered expressions of autophagy-related proteins including LC3, Beclin-1 and p62. Propofol caused the increases of LC3 and Beclin-1 and the decrease of p62 (Fig. 3B). SIRT3 upregulation enhanced the role of propofol in alterations of autophagy-related proteins (Fig. 3B). Collectively, SIRT3 upregulation enhanced propofol induced autophagy mediated by AMPK/mTOR pathway in LPS-treated neurons.
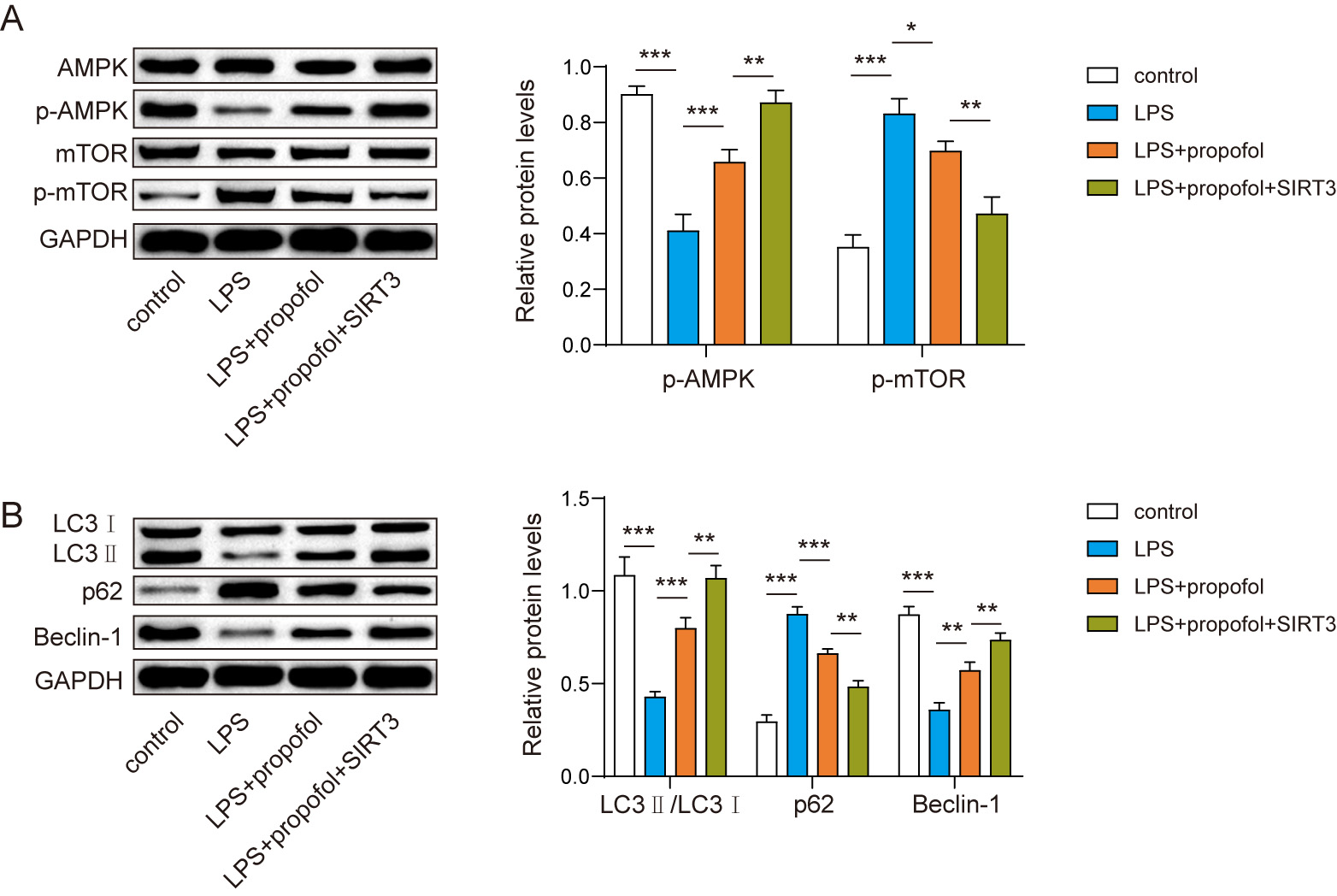 Fig. 3.
Fig. 3.SIRT3 increased the role of propofol in AMPK/mTOR pathway
mediated neuronal autophagy. (A) Expressions of AMPK/mTOR
pathway-related proteins were measured using western blot. (B)
Autophagy-related protein expressions were determined in neurons, including LC3,
p62 and Beclin-1 via western blot. n = 3. * p
To demonstrated whether autophagy mediated the anti-inflammatory role of
propofol in neurons, 3-MA, an inhibitor of autophagy, was administrated in
LPS-stimulated neurons during propofol treatment. For pro-inflammatory cytokines,
3-MA reversed propofol-decreased levels of TNF-
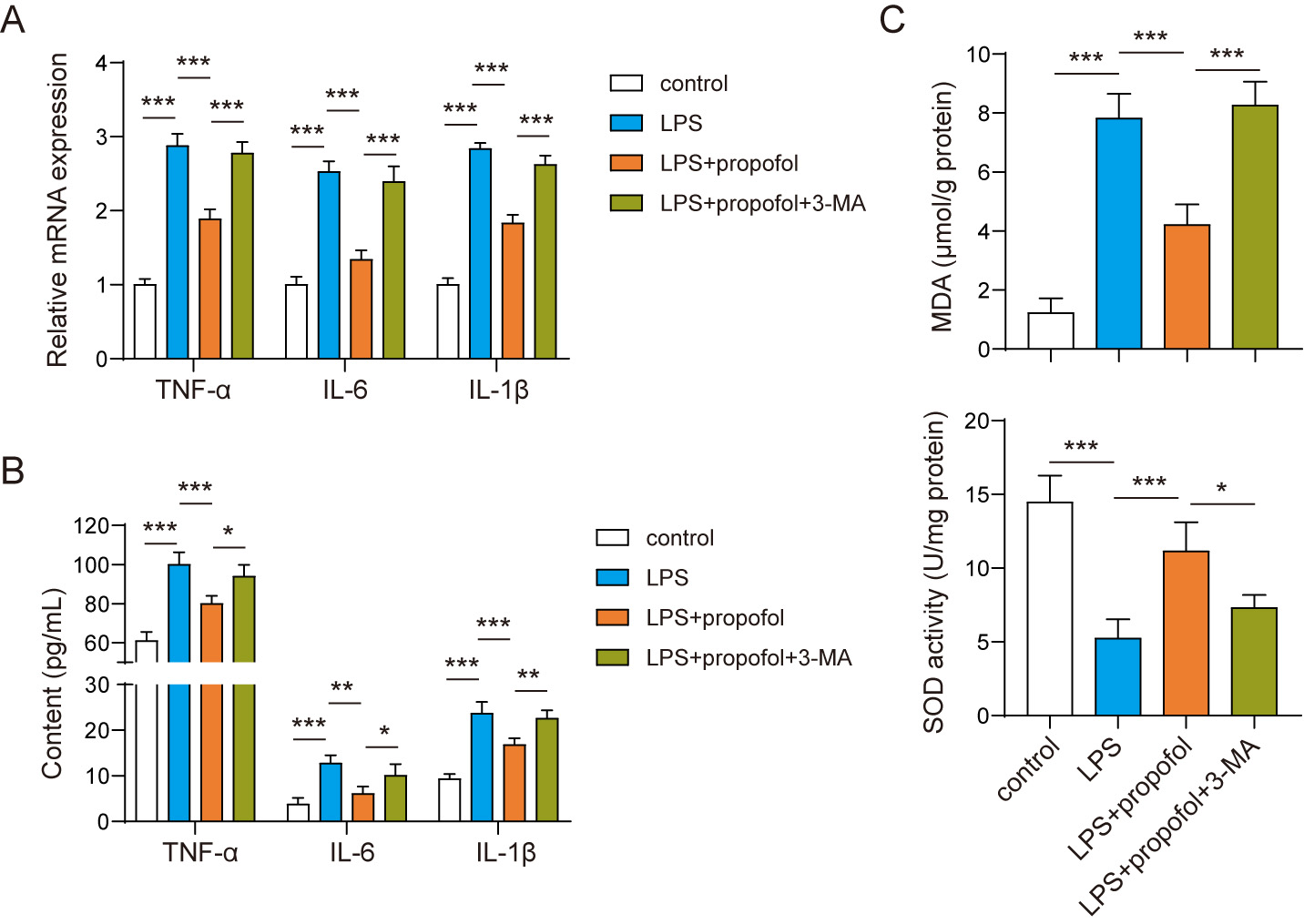 Fig. 4.
Fig. 4.Autophagy contributed to the protective role of propofol in
LPS-stimulated neurons. (A) Expressions of TNF-
Compared with propofol group, escape latency (Fig. 5A), spending time (Fig. 5B), crossing times (Fig. 5C) and recognition index (Fig. 5D) in propofol + SIRT3 group were closer to these in sham group. Also, further decreases of inflammation (Fig. 5E) and oxidative stress (Fig. 5F) were observed in propofol-treated mice after SIRT3 upregulation. In addition, SIRT3 upregulation also enhanced the regulatory role of propofol in autophagy-related proteins including LC3, Beclin-1 and p62 in the hippocampus of mice with POCD (Fig. 5G,H). Therefore, SIRT3 promoted the protective role of propofol in mice with POCD via activating autophagy.
 Fig. 5.
Fig. 5.SIRT3 promoted the protective role of propofol in mice with
POCD. (A) Escape latency in Morris water maze assay. (B)
Time spending on the target quadrant in Morris water maze assay. (C)
Times crossing the platform in Morris water maze assay. (D) Recognition
index in novel object recognition test. (E) Levels of TNF-
POCD is a global healthy problem, and propofol administration declines the occurrence of POCD although its therapeutic mechanism remains unclear. For the first time, we provide the interesting neuroprotective role of propofol based on SIRT3-mediated autophagy during POCD. In neurons, propofol can target SIRT3 expression to trigger autophagy that eliminates oxidative stress and inhibits the production of pro-inflammatory cytokines.
In mice with POCD, propofol can inhibit pro-inflammatory cytokines and oxidative stress to improve the cognitive function of mice. Based on water maze assay and novel object recognition, we found propofol contributed to the enhancement of cognitive function in mice with POCD. There is a relationship between propofol and POCD in clinical. Propofol is a short-acting intravenous anesthetic associated with the decreased risk of POCD and protecting patients from cognitive impairment injury [19, 20]. Pro-inflammation cytokines and oxidative stress are the major cause of inflammation in cellular. Neuroinflammation is possibly the pathological marker of POCD [21]. Thus, the anti-inflammatory role of propofol can rescue neurons undergoing inflammation condition, which avoids the further inflammatory injuries such as blood-brain barrier disruption and subsequently immunocyte recruitment and microglia activation that mediate synaptic plasticity related to learning and memory [22]. Impressingly, we found propofol resulted in SIRT3 upregulation and autophagy activation in the hippocampus of mice undergoing cognitive impairment. These findings suggest SIRT3 and autophagy may be the especial and significant links of the therapeutic path of propofol. Thus, we stimulated neurons with LPS to explain the roles of SIRT3 and autophagy during propofol treatment.
SIRT3 contributes to enhancing the neuroprotective role of propofol in
vivo and in vitro during POCD. We constructed the overexpression vector
of SIRT3 to upregulate SIRT3 expression. In LPS-stimulated neurons, SIRT3
upregulation further decreased cytokines, SOD and MDA during propofol treatment.
This finding indicates SIRT3 is instrumental in the anti-inflammation of propofol
in impaired neurons. We verified the indication based on SIRT3-overexpression
POCD mice treated with propofol. SIRT3 deficiency results in the poor remote
memory with neuron decrease in a mouse model [23]. In other words, SIRT3 is need
for neurodevelopment and cognitive function. Besides, SIRT3 is a nicotinamide
adenine dinucleotide (NAD
There is a novel mechanism of propofol in autophagy based on SIRT3. We found propofol activated autophagy in neurons under inflammation condition. Autophagy responds to neuroinflammation and then inhibits the production and section of pro-inflammatory cytokines via modulating microglia activation [25], which mitigates neuroinflammation-associated injury and/or disease. Thus, autophagy may mediate the anti-inflammation of propofol during POCD. SIRT3 plays the significant role when propofol activates autophagy in neurons. We observed that SIRT3 enhanced propofol-activated autophagy via AMPK/mTOR pathway. AMPK/mTOR pathway is the common positive modulator of autophagy [26]. SIRT3 is able to elevate the activation of AMPK/mTOR pathway and then trigger autophagy in cells [26, 27]. Thus, SIRT3 functions as the core role of autophagy during propofol treatment. We have determined that SIRT3 upregulation promote the neuroprotective role of propofol during POCD. Autophagy may contribute to explain why SIRT3 is instrumental in propofol treatment. Based on LPS-stimulated neurons, we found SIRT3-mediated autophagy elevated the anti-inflammation of propofol. Surprisingly, our results are inconsistent with the results of a previous report, Yang et al. [28] showed the anesthesia using propofol impaired autophagy to cause POCD in aged mice. The autophagy-mediated by propofol may depend on the administration strategy. They used propofol to anesthetize animal at 50 mg/kg, which resulted in cognitive dysfunction. However, we treated mice with POCD with propofol at 2.5 mg/kg for 8 weeks. Obviously, propofol administration has the risk of POCD as anesthetic drug but it also contributes to POCD treatment.
Our findings provide the potential therapeutic role of propofol during POCD. However, the administration strategy may require more researches to ensure its protective role in POCD, which is not investigated in our study. Also, this study fails to discuss how propofol regulate SIRT3 expression in mice with POCD. And limitations in this study need to be verified by subsequent experiments.
We determine propofol can activate SIRT3-mediated autophagy to repress inflammation and oxidative stress in neurons during cognitive impairment after anesthesia/surgery (Fig. 6). Our findings indicate the significant therapeutic value of propofol in POCD. This novel role of propofol suggests the potential insight into the management and treatment of POCD based on SIRT3-mediated autophagy. Also, SIRT3 expression may be utilized to describe the alterations in neuronal damage during POCD at molecular level.
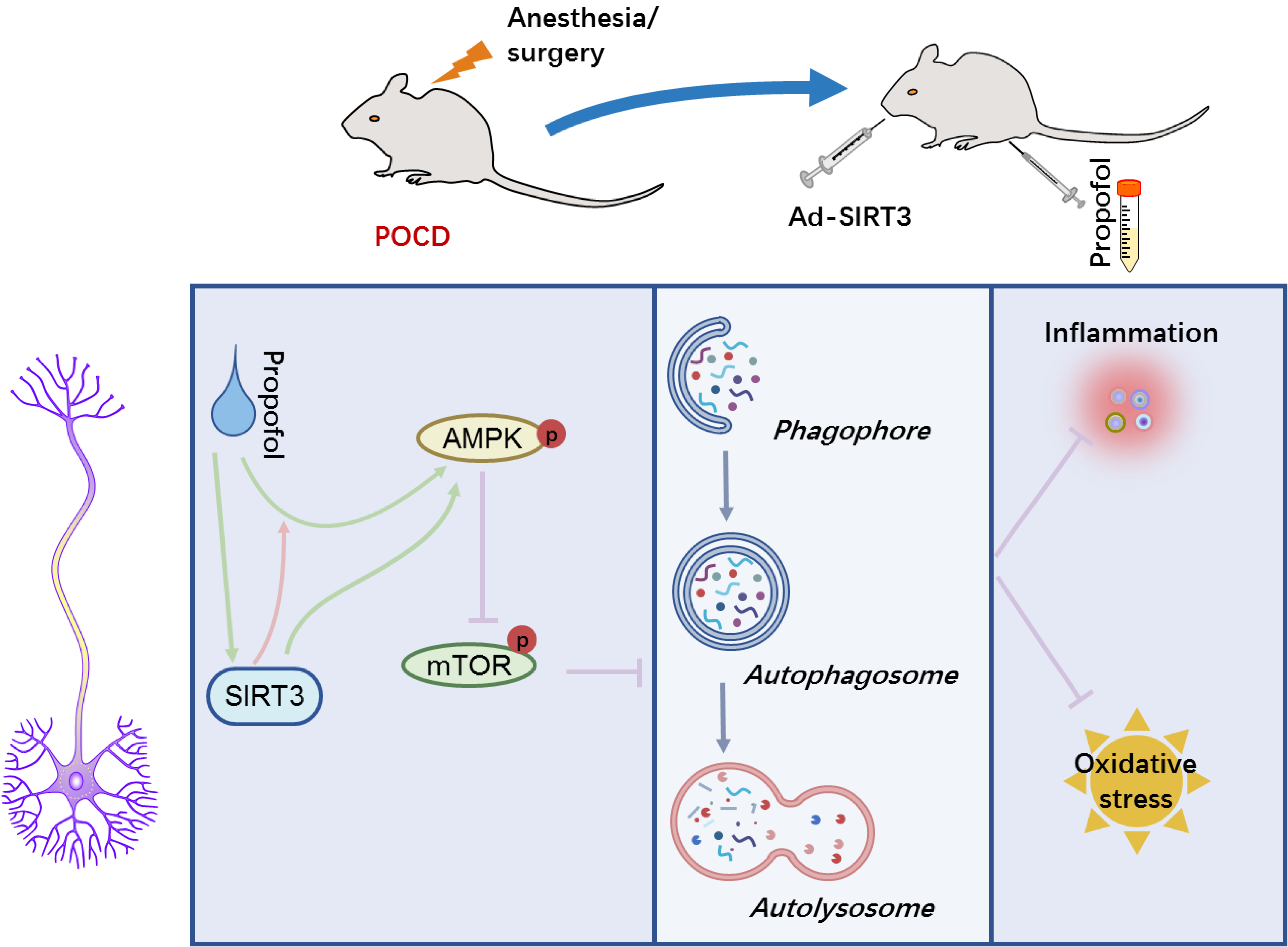 Fig. 6.
Fig. 6.A schematic model for propofol functional role and its underlying mechanism in POCD. SIRT3 mediates autophagy activation via AMPK/mTOR pathway during propofol treatment, and enhances the inhibitory effect of propofol on neuroinflammation and oxidative stress via autophagy in POCD.
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
DZ—guarantor of integrity of the entire study; DZ, JY and SL—study concepts; DZ and JY—study design; YZ, SL, CB and JX—experimental studies; HH and XW—data acquisition; YZ and CB—data analysis; HH and XW—statistical analysis; SL and YZ—manuscript preparation; DZ and JY—manuscript editing; DZ and JY—manuscript review.
Animal experiments were approved by the Ethics Committee of Beijing Stomatological Hospital, Capital Medical University (Approval No. MDKN-2021-055). This article does not contain any studies with human participants performed by any of the authors.
We would like to thank the anonymous reviewers who have helped to improve the paper.
This work was supported by the National Natural Science Foundation of China (No. 82071552).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.