


 , Sylvia Mansilla 1, Benjamin Piña 1
, Sylvia Mansilla 1, Benjamin Piña 11 Instituto de Diagnóstico Ambiental y Estudios del Agua (IDAEA-CSIC), E-08034 Barcelona, Spain
Academic Editor: Guohui Sun
Abstract
Environmental toxicogenomics aims to collect, analyze and interpret data on changes in gene expression and protein activity resulting from exposure to toxic substances using high-performance omics technologies. Molecular profiling methods such as genomics, transcriptomics, proteomics, metabolomics, and bioinformatics techniques, permit the simultaneous analysis of a multitude of gene variants in an organism exposed to toxic agents to search for genes prone to damage, detect patterns and mechanisms of toxicity, and identify specific gene expression profiles that can provide biomarkers of exposure and risk. Compared to previous approaches to measuring molecular changes caused by toxicants, toxicogenomic technologies can improve environmental risk assessment while reducing animal studies. We discuss the prospects and limitations of converting omic datasets into valuable information, focusing on assessing the risks of mixed toxic substances to the environment and human health.
Keywords
- adverse outcome pathways
- risk assessment
- molecular toxicology
- transcriptomics
- proteomics
Toxicogenomics aims to obtain and understand gene expression data and the associated protein activity, within an organism in response to toxic substances [1, 2]. It is known as Environmental Toxicogenomics when including compounds involved in environmental pollution.
Toxicogenomics can produce large-scale datasets with information on genes, proteins, and metabolites by globally measuring the changes occurring at the molecular, cell, tissue, and whole-organism levels [3, 4]. Compared with earlier approaches based on toxicant-induced molecular alterations, toxicogenomic technologies represent an improvement in environmental risk evaluation, while reducing animal testing [5, 6, 7]. The rising of DNA sequencing technologies has boosted our knowledge of the genomes of humans and hundreds or thousands of other organisms, including several animal models traditionally used to evaluate chemical toxicity. However, genome sequences provide information on genes and their locations within the genome in a chromosomal context, but they do not reflect individual variations that would account for the different responses to environmental toxicants [2, 8].
The wide variety of resources available for analyzing the genome, together with the required bioinformatic tools have provided unprecedented insights into the structure and function of the genome, transcriptome, proteome, and metabolome [9, 10]. Diverse omic analytical methods have been adapted to environmental research, but some limitations and challenges remain that need to be considered to fully reveal the potential and acceptance of toxicogenomic data by industry and regulatory agencies [5, 11].
At present, data on human risk assessment rely heavily on the study of comparable effects in model species for which there is extensive knowledge of their physiology, biochemistry, and genetics [10]. Those species are easy to use in well-designed experiments to clarify the effects and consequences of exposure to potentially dangerous external compounds. The limited genomic resources available for non-model species make it difficult to interpret the data and constrain the possibility of integration with different omic technologies to obtain environmentally relevant information. Rodents (mainly mice), the zebrafish Danio rerio, the fruit fly Drosophila melanogaster, the water flea Daphnia magna, the cress Arabidopsis thaliana, and the budding yeast Saccharomyces cerevisiae are worthy examples of such organisms. Toxicogenomics aims to identify common targets between species and determine common molecular processes associated with altering these targets. Using human cells in culture can further refine common responses across species. Cell lines are widely used to gain mechanistic, toxic, and therapeutic insights [12]. They are a highly useful tool for the generation and testing of hypotheses in environmental toxicology [13, 14, 15], but it is often difficult to extrapolate the effects of the dose applied to cells to the effects due to the number of substances reaching epithelial cells and organs in animals and humans.
Comparisons among species are eased by omic technologies and by the availability of massive databases [5, 10]. Omic procedures might answer key questions in risk assessment, including the identification of species-specific effects and the demonstration of human health relevance [11, 16]. However, it is necessary to extend the reproducibility of omics data and data analysis [7].
Toxicogenomics refers to genome-wide studies (omics) applied to toxicology [1]. There are special omics terms in areas of toxicological research based on a single omic technology, such as toxicogenomics, toxicoproteomics, and toxicometabolomics [5, 7, 17], and bioinformatics tools used to examine the databases they generate (Fig. 1).
 Fig. 1.
Fig. 1.Schematic representations of various omic technologies. The omics and bioinformatics tools embraced by the term toxicogenomics can be used to assess the risks of toxic substances to the environment and living beings.
Genomics involves sequencing and analyzing genomes using high throughput DNA sequencing and bioinformatics to assemble and analyze the function and structure of entire genomes. While genomics is based on the sequence information of genes and proteins, transcriptomics, proteomics, and metabolomics provide information about the biological function of genetic information. The widespread use of omics can increase the efficiency and timeliness of environmental assessment, gaining new insights into the mechanisms that contribute to environmental degradation [3, 4, 5, 10], yet omics have to identify common targets across species and determine common molecular events.
The purpose of toxicogenomics is to derive toxicological mechanisms and identify adverse outcome pathways (AOPs), which are defined as a direct association between specific molecular networks and harmful effects on biological systems [18]. AOPs are designed to provide a clear mechanistic representation of critical toxicological effects that spread through different levels of biological organization from the initial interaction of a chemical with a molecular target, to the adverse result at the individual or population level. Specialized bioinformatic tools use omic datasets to create or refine existing adverse outcome pathways (AOPs) [3, 18, 19]. AOPs can be compared between species to identify specific and shared mechanisms of toxicity, and this information can evaluate and manage environmental risks [3, 10, 18].
The analysis of the effect of toxicants on the relative abundance of mRNA of specific genes has become a major technique for assessing their physiological impact on a variety of environmental and toxicology-relevant animal and plant species [2, 20, 21]. The simplest and most widely-used technique for this is the real-time quantitative polymerase chain reaction (qPCR). The process of selecting and validating “target” genes and reference (housekeeping) genes is the first step in the study of gene expression by qPCR. Relative quantification is easy to undertake as the amount of the gene studied is compared to the amount of a control reference gene. Although there are commercial settings that make it possible to analyze up to a few hundreds of genes by qPCR, they can quantify a limited number of genes, with great accuracy, repetitiveness, and robustness. Furthermore, qPCR may offer the possibility of identifying new genes activated by toxic agents [22], providing sufficient information on the physiology of the species tested and the toxic effect. The two major limitations of qPCR are, on the one hand, the limited number of genes that can be analyzed, and on the other hand, the requirement for prior knowledge of target genes. The solution to these two limitations is the application of genome-wide transcriptomic methods that can target thousands of distinct mRNAs in a single run. Theoretically, these methodologies permit the quantitation of every single transcript expressed by a given organism, tissue, or cell.
The first comprehensive method for transcriptomic analyses was DNA microarrays, consisting of a solid matrix surface bearing thousands of different DNAs, which are hybridized against a pool of RNA to measure gene expression. This generates a large amount of data, but it is limited to evaluating genes included in the microarray. Microarrays are commercially available for humans, murine, zebrafish, and rainbow trout, among other standard model organisms. The most significant contribution of microarrays in complex studies was to provide paramount information for risk assessment, where traditional limited-endpoint studies fail [2, 10, 23]. However, the requirement for high-quality manufactured arrays, the complex methodologies used to label the nucleic acid samples, and the cumbersome process of hybridization, washing, and array scanning make the whole technique very time-consuming. Except for very specific purposes, RNA-Seq has replaced it.
The possibility of simultaneously sequencing thousands of nucleic acid molecules has fueled transcriptomics in a way completely unimaginable a couple of decades ago. RNA-Seq uses high-throughput sequencing methodologies to detect at any time the presence and quantity of RNA in a biological sample, analyzing the continuously changing cellular transcriptome. The method involves obtaining total RNAs from a biological sample and copying it to obtain double-stranded cDNA. cDNAs are then sequenced as short reads, aligned, and mapped against a known genomic reference sequence. RNA-Seq provides much more data than microarrays, thereby a complete list of pathways can be elaborated. The new generation sequence techniques offer an unparalleled view of the transcriptome of a specific tissue or cell type. RNA-seq lets us investigate the transcriptome, as well as the total cellular content of RNAs including mRNA, rRNA, tRNA, lncRNA, and miRNA. When comparing RNA-Seq and Microarray platforms for the toxicogenomic evaluation of hepatic toxicity in rats, it was observed that RNA-Seq identified more differentially expressed protein-coding genes and provided a wider quantitative range of changes in expression levels than microarrays [24].
Nonetheless, RNA-Seq data are more cumbersome to analyze and interpret, and it requires high levels of genomic annotation. A huge effort to adapt statistical procedures to omics data has resulted in several software packages specifically designed to handle this data (see, for example, [25, 26]). The usual statistical analyses include the identification of differentially expressed genes (DEG), as indicators for the biological mechanisms implicated in the toxic response or effect. Many tools and algorithms for identifying DEGs have been developed, although the three most popular ones are the R-packages ‘DESeq2’ [25], ‘edgeR’ [26], and ‘limma’ [27]. The normalized RNA-seq count data is necessary for edgeR and limma but is not necessary for DESeq2. There is a consensus that data analysis tools have to be further simplified to alleviate the need for overly sophisticated bioinformatics support.
A relatively recent development of transcriptomic techniques has been their use on a single-cell scale [28]. Single-cell transcriptomics involves first the unequivocal isolation of a cell, or at most, a few hundred cells of the same type, extraction of total nucleic acids, reverse transcription of mRNA, and sequencing of the results; all of this done using a few pg of RNA [28, 29]. Unlike “conventional” transcriptomics, the result of the process consists of hundreds to thousands of low-depth (20–50,000 reads) transcriptomic profiles, each of them associated with a single cell. In this way, a ‘transcriptomic atlas’ can be made to be used to describe with unprecedented accuracy the organic or tissular response to a variety of stimuli, from the development of cancer to toxic effects [28, 30].
Proteomics aims to identify the structure and function of proteins and their
modifications [2, 31]. The proteome encompasses all the products of gene
translation. It is essentially the final product of gene expression and, it can
provide a genuine overview of the actual condition of cells upon toxicological
damage [2, 6, 32]. Recent advances in sample processing, separations, and MS
instrumentation make it possible to quantify
Toxicoproteomics applies global protein expression analysis technologies to toxicological and clinical research. Some proteins respond to exposure to toxicants and display subtle variations in different cells and tissues. Technological development in mass spectrometry, bioinformatics, and protein databases rises new opportunities for mechanistic and effects-oriented research [2, 6]. Proteomic experiments generally collect, in a sample, data on three properties of proteins: location, abundance/turnover, and post-translational modifications. The first truly proteomic technique was 2D-Polyacrylamide gel electrophoresis, which allowed monitoring, and partial identification, of a few hundred proteins, at most. Currently, matrix-assisted laser desorption/ionization (MALDI) techniques coupled to a Time-of-Flight mass spectrometer (MS-TOF) have increased enormously the yield and quality of the proteomic analysis, but some caveats still exist. For example, no technology can amplify proteins and, therefore, they have to be analyzed at their native abundances. The identification of peptide and protein sequences from MS uses a variety of bioinformatics tools, which search protein and nucleotide sequence databases [34] (see section 7). Therefore, our knowledge of the proteome of a given species is limited by our parallel knowledge of its genome. A targeted complementary technique, microarrays of immobilized antibodies can be used to specifically recognize proteins in complex mixtures. In principle, this should allow for a certain level of comparison between species, as long as two species are evolutionarily close.
Modified forms of proteins may be more critical to their function than the absolute levels of the protein, as their function can be modulated by post-translational modifications (PTM). For some cellular functions, like those depending on MAPK pathways, this is the main form of regulation [35, 36]. Proteomics embraces methods for monitoring PTMs and studying proteins in the context of the environment [37]. Because of the large number of modifications, which include phosphorylation, ubiquitination, and acetylation of lysines, dynamic studies of PTM can provide information about the role of PTM in the biological process and identify substrates of PTM-regulating enzymes. Future challenges in PTM proteomics include achieving increased sensitivity and a wider dynamic range of protein abundance, detecting low-abundance modifications, improving the accuracy in the identification and localization of modifications, and developing robust quantitative methods.
Metabolomics embraces identifying and quantifying metabolites, generally considered the downstream elements of any toxicological mechanism. There are two main technical approaches to metabolomics. On the one hand, chemical fractionation (either by liquid or gas chromatography, LC or GC) followed by MS or MS/MS, allows both identification and targeted quantification (i.e., the search for metabolites of known mass and MS/MS spectra). On the other hand, NMR allows for unambiguous identification as well as relative quantification of metabolites, but it requires larger sample quantities. These techniques provide structural and quantitative data and allow us to identify hundreds of potentially biologically active metabolites at their physiological levels, sometimes only amounting to a few parts per billion.
The metabolome includes a chemically diverse collection of compounds, which range from small peptide, lipid, and nucleic acid precursors and degradation products to chemical intermediates in biosynthesis and catabolism, as well as compounds derived from diet and environmental sources [38]. The chemical diversity of metabolome components makes it difficult for a comprehensive analysis with any single analytical tool.
Lipidomics can be defined as the comprehensive identification and quantification of all lipid molecular species in a biological system. It is an extension of the classical lipid profiling performed in an “omic manner”, and it can be considered a subdiscipline of metabolomics. Lipids are typically hydrophobic and the distinct solubility properties of many lipids dictate their separate analysis in metabolomics/lipidomics experiments. Lipidomics is still developing in environmental toxicology studies, although it is known that many environmental pollutants are lipid-soluble and interact with lipid metabolic pathways. Therefore, lipidomics could be very useful as an environmental toxicological tool to study chemically triggered health disorders, a field of research that needs to be developed more widely. For example, lipidomics has been used in high-profile biomedical research, owing to the link between metabolic dysfunction and some lipidic diseases [2, 3]. The rising number of publications on metabolomics and/or lipidomics show that technical improvements together with the experimental results obtained in recent years are convincing researchers of the value of these omic technologies [2, 39].
Although some complex regions of the genome are still difficult to sequence, almost all of them can be sequenced and annotated. This is not the case with metabolome or proteome, which are difficult to fully identify from mass spectrometry data. Unlike single-cell genomic analyses, determining protein expression using samples from a single mammalian cell is much more difficult because sample losses have to be carefully minimized during sample preparation and analysis. In addition, among the technical difficulties of such trace analyses, MS instruments had to become much more efficient [33]. Even worse, no currently available method can identify and quantify the great diversity of molecules that configure the metabolome of a single cell or determine protein expression from a single mammalian cell. Metabolomics faces the fact that the metabolism changes rapidly, so even collecting a sample for analysis can change the native state of metabolism [10].
Epigenomics examines the epigenome of a cell or organism. The epigenome consists of the set of chemical modifications to the DNA and DNA-associated proteins in the cell, which alters gene expression, and is heritable through meiosis and/or mitosis. The modifications occur as a natural development process and tissue differentiation and can be altered in response to environmental exposures [40]. Epigenomics embraces chromatin modifications that directly influence chromatin conformation, such as DNA methylation and histone posttranslational modifications, as well as the activity of noncoding RNAs (ncRNAs) [41]. While these regulatory pathways converge to regulate gene expression, the mechanisms may be diverse. It is quite clear that there is a widespread cross-talk between DNA methylation, histone modifications, and ncRNAs [41, 42].
Epigenomics is being applied to the study of the effects of environmental pollution [43]. These studies show the significance of exploring environmental samples with a potentially high impact on gene expression and its regulation. Following the frequent inclusion of epigenetics as a parameter in environmental epidemiology studies, numerous reports have linked changes in DNA methylation to various environmental factors, including biological agents, dietary habits, and air pollution [41].
New advances in molecular epigenetics, including single-cell epigenomics and transcriptomics, and in the editing of epigenetic marks, ease the investigation of environmental and endogenous factor-dependent epigenetic mark dynamics in an integrative manner [40]. Thanks to experimental tools used for the gene-scale analysis of DNA methylation, such as genome-wide bisulfite sequencing, it is now possible to examine the effect of exposure to environmental toxic substances on stable DNA signals. Similarly, experimental methods catalog changes in miRNA and histone signals. Nevertheless, the relevance of specific epigenetic changes in environmental toxicology is not yet sufficiently established. It is known epigenetic processes can be significantly modulated by exposure to complex environmental samples [2, 43, 44], although further studies are required on the mechanisms by which epigenetic changes are involved in toxicity and to identify epigenetic biomarkers associated with negative outcomes. Epigenetic marks typically remain for days, months, or years after the end of exposure, and in some cases may be inherited through successive generations; thus, they might be useful in situations where the environmental toxicants tested have short in vivo half-lives or to evaluate long term effects of environmental toxicants [41, 42].
A single-omics technique could not even detect the total of biomolecules involved in a toxicological response, but only a smaller subset of compounds with similar physicochemical properties. Even the detection of short and long RNAs or short peptides and proteins requires diverse transcriptomic or proteomic approaches. A particular omic technology can directly detect a significant fraction of any pathway involved in response to pollutants, but multi-omics strategies are required for toxicological assessment [2, 7, 8, 10]. Fig. 2 (Ref. [10]) summarizes the mutual interactions between the different outputs of various omic techniques. The metabolome is considerably more complex than the genome (made up of four bases) or proteome (made up of twenty amino acids) because it shows variability at the atomic level. To deconvolute this complexity the metabolome can be subdivided into, for example, the lipidome, glycome, and peptidome, this then leads to lipidomics, glycomics, and peptidomics [45]. The expansion of multi-omics research has encouraged the development of new online tools and applications that ease their integration for deeper interpretations that go beyond the correlation between biological molecular features and biological response, while they might consider data analysis for regulatory application [46].
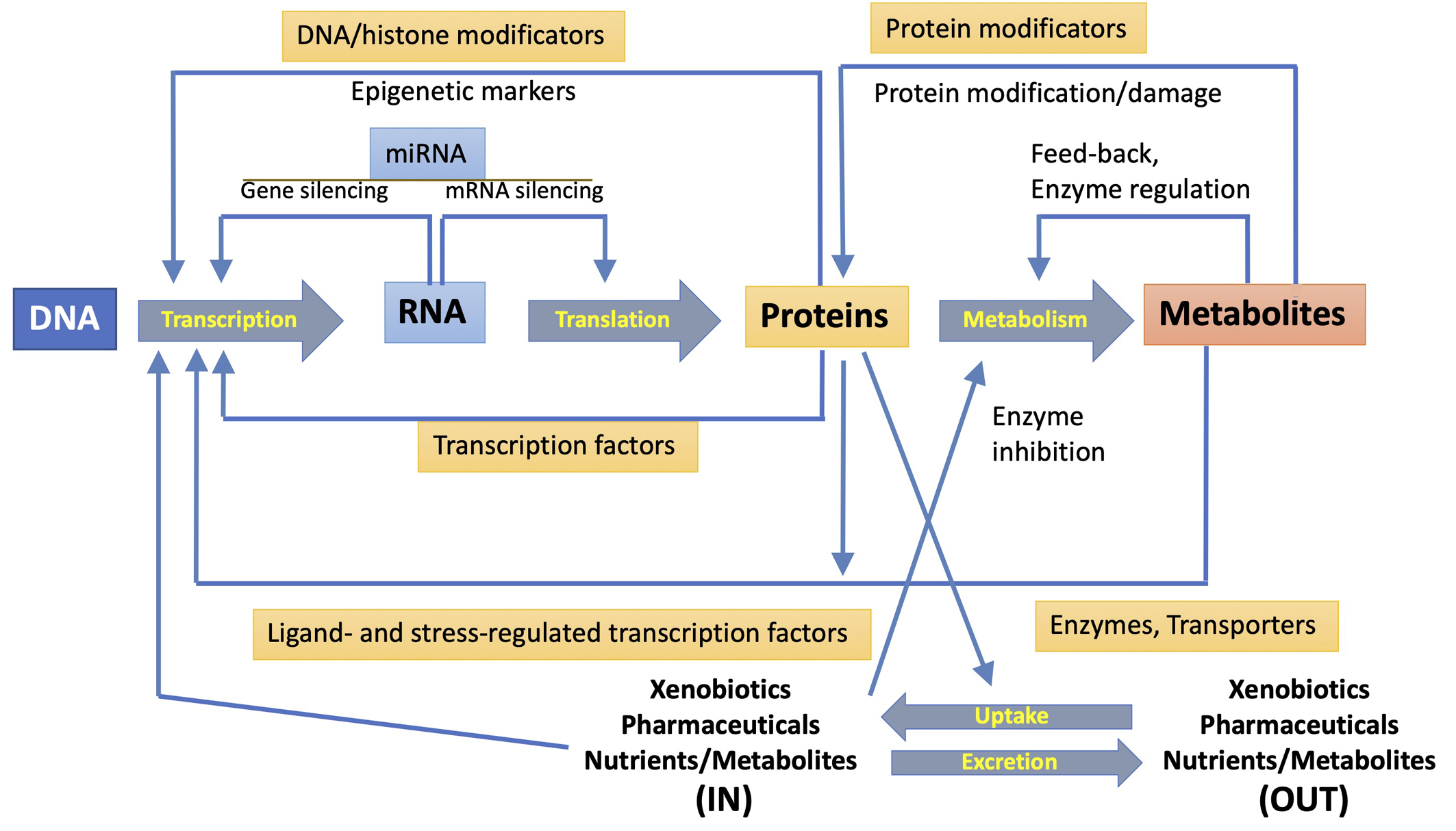 Fig. 2.
Fig. 2.Mutual interactions between transcription, translation, and metabolism functions. The diverse interactions and regulatory mechanisms (represented by large central arrows) are shown. They involve transcription factors, protein and DNA modifications, miRNA expression, regulation of enzyme activity and uptake of exogenous compounds, and secretion of xenobiotics and metabolites. Reproduced with permission from [10].
The relationship between toxicology and genomics has created novel informatic challenges [10, 47]. These challenges are addressed by collecting data in well-designed toxicogenomic databases. There are several publicly accessible toxicogenomics databases [10, 48, 49, 50]. Most of the relevant functional information about genes, proteins, and metabolites is deposited in those public databases, commonly providing taxonomic information. Evaluating and integrating different omics data is essential to determine adverse outcome pathways (AOPs) [10].
The Gene Ontology Database (http://www.geneontology.org/) comprises (as of August 2022) more than 7.5 million publicly-available annotations corresponding to more than 5000 species. Around 1.5 million annotations correspond to H. sapiens, M. musculus, and R. norvegicus, but many eukaryotic and prokaryotic organisms are also present. It comprises more than 20,000 annotations per species. These annotations correspond exclusively to genes that are organized in hierarchical GO (gene ontology) categories, from the more general (such as “metabolic process” or “cytoplasm”) to more specific ones (such as “histone lysine demethylation” or “telomeric loop formation”), including information about subcellular localization, metabolic activity, roles in cell division and development, among others. The different GO categories are identified by a key number, which simplifies the functional annotation of omics results. GO databases are publicly available, and computer analysis can be done online by using pages like the DAVID database (https://david.ncifcrf.gov) or the AmiGO database (http://amigo1.geneontology.org/cgi-bin/amigo/term_enrichment). Furthermore, algorithms embedded in those web pages can undertake the so-called “term enrichment”, which associates every gene of an input list to one or more GO terms, uncovering what biological processes would change in a given experimental context [10].
The Kyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.genome.jp/kegg/) is a database resource that allocates biological pathway data for a large number of species [50]. It is a resource for understanding high-level functions and utilities of the biological system, such as the cell, organism, and ecosystem, from molecular-level information.
The Genomics of Drug Sensitivity in Cancer database (GDSC) (https://www.cancerrxgene.org) combines genomics data from the Cancer Genome Project and drug activity data. It displays information about over 100 cell lines and contains information on 450 compounds targeting 24 pathways. Average-linkage dendrograms have been obtained, based on the activity of 118 drugs over 60 human cancer cell lines. Gene expression and drug sensitivity profiles have provided comprehensive databases [49, 51], which delimit genes whose expression is significantly correlated with patterns of drug sensitivity [52]. Lists of genes related to a specific mechanism of action can be obtained for almost any compound.
The innovative, and publicly available, Comparative Toxicogenomics Database (CTD; http://ctdbase.org/) relates toxicological information for chemicals, genes, phenotypes, diseases, and exposures to advance understanding of the effects of toxicants on human health [48]. Manually-curated interactions, which are based on bibliographic information, are integrated into a database that matches cross-species heterogeneous data for chemical exposures with their biological effects. To date, CTD provides about 45 million toxicogenomic relationships for over 16,300 chemicals, 51,300 genes, 5500 phenotypes, 7200 diseases, and 163,000 exposure events, from 600 comparative species.
Expression Atlas is an open resource (http://www.ebi.ac.uk/gxa) that provides users with a powerful way to find information about gene and protein expression in different species and contexts, such as tissue, developmental stage, diseases, or cell type [53]. Data from large-scale RNA sequencing studies can be visualized next to each other. It is possible to undertake gene set overlap queries that result in a list of comparisons from differential experiments enriched for the group of genes of interest.
A protein-specific database is SWISS-Prot (https://www.uniprot.org/), which can facilitate domain/homology search, and is incorporated within DAVID. The annotation of any specific metabolite to a metabolic pathway is eased by publicly accessible databases like KEGG (http://www.kegg.jp/), mentioned above, which is included in DAVID analyses, or MetaboAnalyst (http://www.metaboanalyst.ca/MetaboAnalyst/), which includes data from metabolically-relevant genes and proteins, as well as term-enrichment algorithms. For the analysis of the lipidome, there are some resources such as LipidMaps (http://www.lipidmaps.org), with encompasses a mass spectra database that could be used for interpreting lipidome data [54]. Other resources are Lipid Library (http://www.lipidlibrary.co.uk/), and LipidBlast (http://fiehnlab.ucdavis.edu/projects/LipidBlast).
Hopefully, toxicogenomic datasets should be useful for determining certain mechanisms of action and for assessing sensitivity differences between species. This might ensure the validation needed to overcome the regulatory hesitations for accepting genomic-based evidence of certain environmental risks [8]. Requirements for the analysis of transcripts, peptides, metabolites, lipids, and carbohydrates are very different and, in many cases, incompatible one to each other. It is almost impossible to gather information from different omics by using a single sample, and, in many cases, it is difficult to do so with different samples in a unique experiment. Therefore, the combined assessment of the complex omic databases is essential for understanding many biological processes [10, 55]. In summary, multi-omic studies involve a deeper understanding of biological processes and pathways by simultaneously examining the genome, transcriptome, proteome, and metabolome. Collecting multiple toxicological data can explore the pathways of response to environmental toxicants [10, 55]. It is also interesting to reanalyze the published data, as the integration of multi-omic data can significantly improve confidence in detecting response pathways. Different methods of sample preparation can cause difficulties in omic analysis, and each subsequent step introduces potential sources of variation. In the multi-omics approach, new sample preparation techniques progress together with instrument development and annotated databases, facilitating the collection of new information on AOPs.
In the following paragraphs, we outline some cases that corroborate the feasibility of omic techniques in environmental toxicology. No attempt will be made to survey the vast bibliography on toxicogenomics. Our choice of research examples is quite restrictive and somewhat arbitrary. It aims to illustrate the deep relationship between toxicogenomics and environmental risk assessment.
Zebrafish (Danio rerio) embryos and larva are utilized as standard models for many toxicity tests because they share 71% genetic homology with humans. Toxicogenomic was used to evaluate cadmium toxicity in zebrafish [17]. Cadmium is a toxic heavy metal posing several environmental concerns. An omics approach to the toxic effects of the metal concluded that cadmium inhibits the activity and synthesis of antioxidant enzymes and proteins. Moreover, the levels of reactive oxygen species rose in various organs, causing the liver to accumulate fat [17].
Zebrafish were exposed to mixtures of
Various pollutants are known to alter the hormonal balance of organisms [20]. Gene expression patterns induced by different mixtures of endocrine-disrupting chemicals were analyzed using a commercial microarray designed to study the transcription of the rat brain. A combination was found of altered pathways associated with cell death and inflammation [57], consistent with the known ability of many pollutants to alter the hormonal balance of organisms [20].
There is a worldwide demand for freshwater for human consumption, agriculture, and industry [58]. Sources of fresh water are limited and often contaminated, thus reclaimed wastewater is an additional resource used to irrigate crops in agriculture [59]. Crops may become contaminated with toxicants, pathogenic bacteria, or bacterial genetic elements, thus developing a risk for consumers [59, 60]. Indeed, employing reclaimed water in agriculture can favor the dissemination of antibiotics, antibiotic-resistant bacteria, and antibiotic-resistance genes in the environment [61]. Quantitative PCR has been used to analyze the distribution of antibiotic resistance genes in soils and crops, allowing their quantification [61, 62].
Pollution of the aquatic environment is a worrying problem because of industrial waste, farming effluents, sewage, and wastewater spillage. Organic pollutants found in those waters trigger plant response to oxidative stress and, therefore, promote transcriptomic, metabolomic, and morphological changes in plants [16, 59, 63]. Metabolomic analyses indicate that exposure to contaminants of emerging concern at environmentally relevant concentrations can cause substantial metabolic alterations in plants that are linked to changes in chlorophyll content and some morphological parameters [16, 63]. The presence of antibiotics in fertilizers used in irrigation of lettuce (Lactuca sativa) and radish (Raphanus sativus) under agronomic plot-scale conditions would indicate, in keeping with transcriptomics (qPCR) and metabolomics data, that osmotic/hydric stress can be the primary factor of phenotypic variability [16, 63].
Narcosis, or basal toxicity, has been linked to the lipophilicity of chemicals, but the mechanism behind narcosis remains unclear [64]. Omic approaches, which include a multistep modeling strategy, suggest that integrating information about chemical structure with a mechanistic view of genomic studies can identify a mechanism for narcosis toxicity in Daphnia magna linked to calcium signaling. Altered calcium homeostasis may be a key early event in narcosis induced by lipophilic compounds [64].
Oxidative stress has been suggested in humans and fish as a relevant mechanism for the toxicity of organophosphorus compounds [65]. Danio rerio (zebrafish) and PCR arrays for genes associated with oxidative stress have been used to evaluate the mechanisms of toxicity of organophosphorus compounds. After treatment with those substances, most genes associated with oxidative stress and mitochondrial dysfunction pathways were altered, suggesting that these pathways are involved in developing the damaged phenotype [66, 67]. Nevertheless, although oxidative stress is produced during the development of the phenotype, this process might not be essential to the pathologies triggered by organophosphorus compounds [10].
The liver and gonads of male Carassius auratus (goldfish), which had been exposed for 10 days to environmental-relevant concentrations of mixtures of some contaminants of emerging concern [68], were analyzed by NMR-based metabolomics. In this work, the authors retrieved various metabolites from each organ matched them to KEGG identities, and unveiled a series of modulated intracellular signals [68], demonstrating the sensitivity of metabolomics in the risk-assessment of environmental toxicant mixtures.
Neonates of the crustacean Daphnia magna (water flea) were exposed to water from the Akaki river (Ethiopia) and tested for mortality and the expression of genes involved in different biological pathways. Despite the poor quality of the Akaki river water, no mortality was seen. Sublethal toxicogenomic responses indicated that exposure to Akaki water changed the expression of genes involved in immune response, oxidative stress, respiration, reproduction, and development. Toxicogenomic data provided insights into the mechanisms that could cause potential adverse effects on aquatic organisms, suggesting the accumulation of stress response [69].
The toxicity of the main components of electronic cigarettes has been examined in Sprague-Dawley rats exposed via inhalation [70]. Lipidomics, which was used together with transcriptomics and proteomics, detected and quantified the major classes of lipids in blood serum and lungs including changes in molecular and metabolic levels [70]. Despite the relatively low throughput from the lipidomics analyses, it was concluded that aerosols showed only very limited biological effects without signs of toxicity.
Proteomics has been used to assess the response of Scrobicularia plana clams to contamination in three sites of Guadalquivir Estuary at the southern end of the National Park of Doñana (Spain). Silver-stained gels detected nearly 2000 well-resolved spots in a soluble fraction of S, plana gills. The more intense protein spots were analyzed by MALDI-TOF MS. Different protein expression signatures were found at each site, with the highest number of more intense spots observed in animals having the highest metal content [71].
Traffic-related air pollution is a potential risk factor for many respiratory disorders [72]. In an epigenomic study, differentiated levels of global DNA methylation between lungs and blood cells were observed in rats that were exposed to traffic-related air for up to seven days. Gene-specific increases and decreases in methylation occurred [44].
The use of silver nanoparticles (AgNPs) in foods and cosmetics has been associated with toxicity. Global gene expression profiles have been evaluated in human HepG2 liver cells exposed to AgNPs [73]. HepG2 cells responded to the toxic insult by transiently upregulating stress response genes such as metallothioneins and heat shock proteins. Functional analysis of the altered genes showed several biological processes were affected. Human HepG2 cells represent an alternative model to whole animals for assessing the cell’s response to toxicants.
Toxicogenomics seeks to determine toxicological mechanisms and identify adverse outcome pathways (AOPs) in biological systems [18]. AOPs are important for developing the use of toxicological data for risk assessment (RA) and regulatory applications, including environmental management. By comparing AOPs between species, we can identify general and specific mechanisms of toxicity that we can use to understand and manage environmental risks [19].
Modes of Action (MOAs) and Adverse Outcome Pathways (AOPs) describe, at different levels, our mechanistic knowledge of a biological organism. While the experience gained in MOA analysis is focused primarily on the later stages of cellular, biochemical, and tissue events, the AOP approach concentrates on the eco-toxicological community and describes mechanistic knowledge at varying levels of biological organization to facilitate its assimilation, integration, and evaluation for research and regulatory applications [2, 3]. AOPs are important for risk estimates and regulatory applications, including environmental management. Using omics technologies in environmental toxicology has produced large amounts of data from a variety of technical approaches that need to be analyzed and integrated. AOPs are useful for distinguishing initial (central) events from toxic effects and indubitable adverse effects that affect the health of human beings, and ecosystems [10, 18, 19]. Mainly, the possibility of linking molecular effects and functions between species and levels of biological complexity has increased the usefulness of model systems such as animals, cells, and embryos in medical sciences and toxicology [10]. Several of the illustrative examples presented above, in section 8, are in keeping with this statement.
Implementing animal-free toxicity studies has resulted in using the lowest possible number of animals and in the development of a diversity of cellular models that might preserve most of the characteristics of the actual target cells within an animal [12]. The most commonly used cell culture approach is growing cells in two-dimensional (2D) monolayers (an illustrative case is presented above in section 8.13), but those cultured cells do not mimic the natural cell environment, as cultured cells are mostly deprived of cell-cell interactions. For this, three-dimensional (3D) cultures are considered better in vitromodels. Advances in 3D cell culture and cell analysis have enabled the development of more physiologically relevant engineered models of human organs with accurate control of the cellular microenvironment [74], forming organoids, structures that simulate the physiological environment of the native organ ant that may provide new insights into risk assessment. As the liver is a potential target organ in toxicology, a novel in vitro 3D model generates liver organoids from human pluripotent stem cells as an alternative model of the human liver. These organoids were used to explore the harmful biological effects of exposure to contaminant polystyrene microbeads [74]. Organoids provide new insights into risk assessment. Furthermore, many cell lines can self-assemble and form spheroids. Prostate epithelial cell lines can form spheroids with evidence of glandular differentiation in three-dimensional cultures [75]. Prostate cancer spheroids are a cellular model capable of mimicking the mechanical tensions of tumor tissues, providing a more representative pathophysiological model than conventional cell cultures. They have been used, for example, to study the effects of arsenic on human prostate [75].
The purpose of AOPs is to provide a clear mechanistic representation of critical toxicological effects that extend across different levels of biological organization, from the initial interaction between chemicals and their molecular targets to adverse outcomes at the individual or population level. Omics datasets can be used to create or refine existing adverse outcome pathways [3, 18, 19]. They can be compared across species to identify specific and shared mechanisms of toxicity. Therefore, this information can evaluate and manage environmental risks [3, 5, 10, 18]. Omic tools provide valuable clues about potential key events involved in the development of complex phenotypes induced by toxic substances. However, the potential key events identified using omic methodologies should be further confirmed through biochemical approaches, thus establishing whether the identified pathways are involved in developing the phenotype [10, 18]. At present, it is relatively easy to generate many millions of sequences that exceed the capacity of computing systems; thus, the challenge may be to use them efficiently. This situation of “excess of experimental data” is suitable for basic research, where the emphasis is on obtaining as much information as possible, without limiting the time spent analyzing the data to achieve meaningful results. On the contrary, a large amount of data can be a great obstacle when considering clinical practice.
Toxicogenomics has provided profuse mechanistic data applicable to hazard identification, dose-response analysis, and quantitative risk assessment of benzo[a]pyrene, a known human carcinogen [76], which can be considered a proof-of-principle for a well-established mode of action (MOA) that affects multiple tissues. Based on that work, some useful lessons for both basic research and RA rose that underline how toxicogenomics can help risk assessment. These lessons are: (a) we need to obtain biologically relevant data that are adequate to establish an MOA for toxicants, (b) we should examine the importance of MOAs in humans from animal tests, or better by using alternative living models, and (c) we have to propose appropriate quantitative values for RA. Toxicogenomics can be a tool in RA, especially if combined with other short-term toxicity tests (apical endpoints), increasing confidence in the proposed MOA. It emphasizes the need for further studies on other MOA to define best practices for applying toxicogenomics in risk assessment [10, 18]. In addition, the study of the metabolic responses of organisms can help regulatory policy and decision-making processes during chemical risk assessment. There are several challenges in using that data for regulatory purposes, such as interpreting the information and presenting adverse outcomes, but there is little doubt about the potential of metabolomics to unravel the molecular processes involved in chemical toxicity. Some studies in this direction are already available for human cell lines, fish, and mussels [10].
We should address the evaluation of risks associated with combined exposure to multiple chemicals for both humans and the environment. Most times, the problems that arise when testing single toxicants or mixtures are the same, but sometimes, it may be necessary to take certain peculiarities into account, thereby different approaches may be applied to the risk assessment of mixtures on humans or the environment [2]. The Organization for Economic Co-operation and Development (OECD) has launched a project to develop guidance for reporting omics data for regulatory use [77]. Using a toxicogenomic approach instead of endpoint assays has many advantages for regulatory agencies. Toxicogenomics enables a systems biology proposal to understand the interactions between genes, proteins, and metabolites within a resulting phenotype. It simultaneously measures effects on a wide range of biological pathways and can be used to study the mode of action of chemicals, predict toxicological effects, and characterize and understand species relevance. Omic approaches permit the simultaneous study of several biological pathways and measuring changes in those pathways before changes in traditional toxicological endpoints occur, and they may reveal biomarkers that predict adverse outcomes. In any case, transparent documentation on assumptions and uncertainties related to the potential impact of the assessment is recommended to enable risk managers to make informed decisions. Considering the many combinations of chemicals in mixtures, prioritization is necessary to first address the mixtures of greatest concern and the chemicals that create the risk of the mixture. Chemicals with different applications are usually regulated separately but can cause similar toxicological effects, thus it is important to take into account the specificities of chemical mixtures in any legislative initiative.
It is not always clear how exactly toxicogenomic data have to be used to support human health risk assessment, yet there are some paradigmatic case studies evaluating the utility of toxicogenomics in RA, such as the study, mentioned above, on benzo[a]pyrene (BaP) [76], and some of the Illustrative Cases described in section 8. If we focus on regulatory organizations, risk managers should make informed decisions and develop transparent documentation of toxicogenomic case studies, as well as of the associated uncertainties. Due to a large number of potential chemical combinations, it is necessary to start considering the most worrisome chemicals because they could increase their risk in mixtures. Given that chemicals with different uses and controlled separately can cause similar toxicological effects, chemical mixtures must be taken into account experimentally and through all legislative sectors [78]. Risk assessment of combined exposures to multiple chemicals should be addressed for humans, and the environment, taking advantage of surrogate models, including cultured cells. There may be specific features to consider, and different approaches, including the wider use of toxicogenomic tools, which can be applied to assess the risk of environmental mixtures to humans [2, 7, 20, 43].
Omic methods, which include the measurement of genes, proteins, and/or metabolites (Figs. 1,2), permit the evaluation of the response to environmental insults of tens of thousands of genes and their products in a single sample [5]. Toxicogenomics measures changes occurring at the molecular, cellular, and organism levels qualitatively and quantitatively, allowing us to fully assess the consequences of releasing toxic substances into the environment [1, 10].
DNA/RNA sequencing methods and metabolomic- and proteomic-based approaches provide measurable functional outputs in environmental toxicology. When used with advanced data analysis, omics datasets allow us to dissect the structure of biological pathways involved in responding to environmental toxic substances. The size and scope of omics datasets have increased the prospects of what these data can achieve in toxicology and risk assessment, particularly by facilitating the identification of changes at the molecular level that trigger responses to contaminants. Omic approaches offer a proper opportunity to inform risk assessments if they are used as part of an integrated systems biology approach [2, 4, 10]. Currently, omics datasets cannot provide sufficient evidence to fully characterize environmental risk, but they improve knowledge of toxicological effects.
Interestingly, the integrative toxicogenomics approach is a key point in the current knowledge of the environmental hazards, including the elucidation of taxon-specific potential effects, reached through comparative genomics, the characterization of multiple cell targets for a single substance, or the development of new and more accurate methods to monitor toxic effects in humans and other organisms [1, 79]. Active research in toxicogenomics is being conducted to include predictive studies based on high-performance omic technology, providing valuable information for the assessment of human and environmental risks [3, 11]. Comparing AOPs across different species makes it possible to identify specific and common toxicity mechanisms [10, 18, 19]. The ultimate goal is to develop robust strategies to evaluate the safety of complex mixtures of chemicals together with human and environmental risks.
JP designed the contents of the review and wrote the first draft of the manuscript. SM and BP provided help and advice. All authors wrote the final version of the manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This work has been funded by the Spanish Ministry of Science, Innovation, and University (MCIN/AEI/10.13039/501100011033, grant RTI2018-096175-B-I00), and the Generalitat de Catalunya (2017SGR902). IDAEA-CSIC is a Centre of Excellence Severo Ochoa (Spanish Ministry of Science and Innovation, Project CEX2018-000794-S, ERDF A way of making Europe).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.