
Academic Editor: Paramjit S. Tappia
Background: Thrombospondin (THBS) 3 is an adhesive glycoprotein
involved in cell-cell and cell-matrix interactions. The purpose of this study is
to determine whether THBS3 expression in peripheral blood can be used as a
biomarker to predict the risk of acute myocardial infarction (AMI).
Methods: The peripheral blood of 111 patients with stable coronary
artery disease (SCAD) and 112 patients with AMI was obtained. The experimental
and the control cohorts were the AMI and SCAD groups, respectively. The
expression of THBS3 mRNA and protein in both groups was determined using
reverse-transcription polymerase chain reaction and enzyme-linked immunosorbent
assay. Results: THBS3 expression (range) in the peripheral plasma of
patients in the AMI group was lower than that of patients in the SCAD group
(4.526 (3.748–5.521), 5.511 (4.726–6.334), respectively), which was 0.82 times
lower than the control (p
Coronary artery disease (CAD) is a widely and globally prevalent cardiovascular disease that can result in severe adverse cardiovascular events. If untreated, total occlusion of the coronary arteries can result in myocardial infarction and permanent damage to the underlying myocardium [1]. Atherosclerosis, the main cause of stable CAD (SCAD) and acute myocardial infarction (AMI), is distinguished by arterial wall inflammation and plaque buildup [2]. Acute coronary thrombosis is thought to be caused by the rupture of vulnerable atherosclerotic plaques and is the primary pathological source of AMI. Myocardial ischemia and hypoxia are caused by the rupture of atherosclerotic plaques and intramural thrombosis, which results in myocardial cell necrosis [3, 4, 5]. Thus, early detection and prompt treatment are critical in improving the prognosis of AMI.
Although cardiovascular troponin I, troponin T, myoglobin, and creatine kinase MB are currently used clinically to diagnose AMI [6, 7], these markers are not ideal for identifying the early stages of AMI in patients [8]. Therefore, new, sensitive, and specific biomarkers are required for the early detection of AMI. Protein detection has become indispensable in the diagnosis of CAD in recent years, and high-throughput proteomic studies have been used to identify the markers of early cardiovascular disease [9, 10]. As the pathological basis of SCAD and AMI are similar, and SCAD is an early stage of AMI, it can be difficult to distinguish between the two [11, 12]. Most studies have reported that the abnormal expression of molecular markers in peripheral blood can aid in the diagnosis of cardiovascular disease. For instance, high ADAMTS4 expression in peripheral plasma and mononuclear cells is a key factor aggravating the instability of atherosclerotic plaques [13], and an increase in interleukin-32 expression in peripheral plasma could play a role in the development of AMI from SCAD [14]. Elevated RORA and ABCB1 in leukocytes may be other suitable molecular indicators for the early diagnosis of AMI [15, 16].
Thrombospondin 3 (THBS3) was originally discovered during DNA sequencing upstream of the transcription start site of the mouse Muc1 (episialin) gene [17], a member of the THBS family that mediates cell-cell and cell-matrix interaction. Recent research shows that deleting THBS3 in mice improves the production and stability of integrin membranes, shielding the heart from disease-causing stimuli [18]. Similar to this, fatal cardiac atrophy was caused due to THBS1 (not THBS3) overexpression, which is another member of the THBS family [19]. Although THBS3 is abnormally expressed in cardiomyopathy, there are few studies on its role in CAD. Therefore, this study focuses on patients with AMI and SCAD, which may offer some unique perspectives on the diagnosis and treatment of this condition.
We screened differentially expressed proteins using proteomic studies in the early stage, and the results showed that the expression of THBS3 protein was lower in patients with AMI than in those with SCAD [20]. So, it was reasonable to speculate that AMI and low THBS3 expression in peripheral blood were related. Therefore, the aim of this study was to ascertain whether THBS3 expression was downregulated at the mRNA and protein levels in a larger cohort of patients with AMI, and to further evaluate if the relatively low THBS3 expression was related to the occurrence of AMI. Correlation analysis and logistic regression analysis further demonstrated that THBS3 might be used as a biomarker for identifying AMI.
This is a retrospective study. All patients in this study were enrolled from September 2017 to April 2019 and signed informed consent. The AMI team comprised 112 male patients who were acknowledged to the Department of Cardiology at China-Japan Union Hospital of Jilin University in China. These patients completed a 12-lead ECG as soon as possible at the first medical contact. The diagnosis of AMI was made in accordance with the European Society of Cardiology’s globally accepted definition of myocardial infarction established in 2017 [21], with harsh stenosis and/or occlusion of the foremost coronary arteries (left and right mains) and/or main branches (anterior descending and circumflex arteries).
In addition, 111 males with SCAD were included in the control group. An article posted in the New England Journal of Medicine in 2019 was used to determine the inclusion criteria [22]. The exclusion criteria were as follows: (1) patients with myocardial infarction associated with stent thrombosis and percutaneous coronary intervention/coronary artery bypass grafting infarction; (2) patients with myocardial infarction following cardiac or noncardiac surgery; (3) patients with secondary myocardial infarction; (4) patients with myocardial damage caused by severe stress cardiomyopathy; (5) patients with severe pulmonary embolism, heart failure, and other diseases. Age, history of hypertension, smoking history, diabetes history, blood lipid levels, and other relevant biochemical blood tests were meticulously recorded.
Patients were divided into two groups: AMI and SCAD. Peripheral blood was collected from patients for ELISA and RT-qPCR to evaluate THBS3 expression at the protein and mRNA levels. Lastly, logistic regression and SPSS 25.0 software (IBM Corp, Armonk, NY, USA) were used for statistical analysis (Fig. 1).
 Fig. 1.
Fig. 1.Diagrammatic representation of the experimental scheme.
2.2.2.1 Acquisition of Peripheral Plasma
In the morning, 6 mL of peripheral blood was drawn from subjects who were
fasting, and stored in anticoagulant tubes containing EDTA at 4 °C. Plasma was
extracted within 4 h of specimen collection and centrifuged at
1000
2.2.2.2 ELISA
The instructions listed in the THBS3 ELISA kit (Shanghai Enzyme Link
Biotechnology Co., Ltd., Shanghai, China) were followed. Briefly, samples and
horseradish peroxidase–labeled detection antibodies (3 replicates in each group)
were added to the blank, sample, and standard wells in the microwell plate and
incubated for 60 min before being discarded and washed. Next, 50
2.2.3.1 Acquisition of PBMCs
Peripheral blood was obtained as described in section 2.2.2.1, and mononuclear
cells were extracted using a peripheral blood lymphocyte isolation medium
(Lymphoprep™, STEMCELL Technologies Inc., Vancouver, Washington, Canada). Fresh
anticoagulant was mixed with an equal volume of 0.9% salt solution infusion, and
the mixture was added to an equal volume of lymphocyte separation solution. The
red blood cell layer, clear fluid layer (separated), opalescent mononuclear cell
layer, and plasma layer were separated from the sample after centrifuging at
1000
2.2.3.2 cDNA Synthesis from PBMCs
A total RNA extraction kit (RNAsimple Total RNA Kit, Tiangen Biochemical
Technology Co., Ltd., Beijing, China) was used to extract total RNA. The
manufacturer’s instructions in the kit were meticulously followed to avoid RNA
destruction or pollution during the process. The concentration and absorbance of
items that fit the criteria were determined using a microplate reader. The
A260/A230 value should be
2.2.3.3 Reverse-Transcription Polymerase Chain Reaction (RT-PCR)
RT-PCR was used for the amplification of the 5-fold diluted cDNA samples with
the SYBR fluorescence quantitation kit (Sangon Fluorescence Quantitation Kit, Taq
qPCR Synthesis Premix, Shanghai, China). The dissolution and amplification curves
from 60–95 °C were recorded through multiplication, which comprised
pre-denaturation, denaturation, and annealing steps. The melting curve obtained
using ABI-FAST7500 software (Applied biosystems, Thermo Fisher Scientific, MA, USA) dictated the specificity of the amplification
conditions. THBS3 and GAPDH were the target gene and internal reference gene,
respectively, and the readings for each group were performed in triplicate. Each
sample had a 2
| Genes | Genes Primer sequence (5’-3’) | |
| THBS3 | F |
CTCGGCAGATGGTAGCTGTG |
| R |
TGGATAAGAGGTAGATGTCCCCA | |
| GAPDH | F |
GGAGCGAGATCCCTCCAAAAT |
| R |
GGCTGTTGTCATACTTCTCATGG | |
| F R RT-PCR, Reverse-Transcription Polymerase Chain Reaction. | ||
Data were analyzed using SPSS 25.0 software. The retrieved data were checked for
normality and, if they passed, were expressed as mean standard deviation
(p
Table 2 shows the results of the clinical data analysis. There were significant
changes in age, smoking history, fasting blood glucose, total cholesterol (TC),
low-density lipoprotein cholesterol (LDL-C), and platelet (PLT) levels (p
| Clinical indicators | AMI group | SCAD group | t/z/x |
p |
| N = 112 | N = 111 | |||
| Hypertension (%) | 52 (46.43) | 37 (33.33) | 1.297 | 0.255 |
| Type 2 diabetes (%) | 31 (27.68) | 21 (18.92) | 1.365 | 0.243 |
| TG (mmol/L) | 1.610 (1.110–2.235) | 1.510 (1.078–1.998) | –1.307 | 0.191 |
| HDL-C (mmol/L) | 1.120 (0.890–1.225) | 1.040 (0.918–1.185) | –0.132 | 0.895 |
| Fasting blood glucose (mmol/L) | 7.360 (6.095–9.055) | 5.950 (5.408–6.848) | –4.824 | |
| TC (mmol/L) | 4.730 (4.005–5.440) | 3.650 (3.098–4.055) | –7.063 | |
| LDL-C (mmol/L) | 3.212 |
2.000 (1.728–2.350) | –8.221 | |
| PLT (10 |
237.000 (202.500–284.500) | 223.530 |
–2.288 | 0.022 |
| Age | 59.376 |
50.000 (48.000–53.000) | –6.945 | |
| Smoking history (%) | 65 (58.04) | 52 (46.85) | 7.096 | 0.008 |
| The normal distribution is expressed as the mean | ||||
It can be seen in Fig. 2 that THBS3 protein expression in the AMI group was
4.526 (3.748–5.521), whereas that in the SCAD group was 5.511 (4.726–6.334).
The expression of THBS3 protein in the AMI group was lower than that in the
control group, with a relative expression that was 0.82 times that of the control
group (p
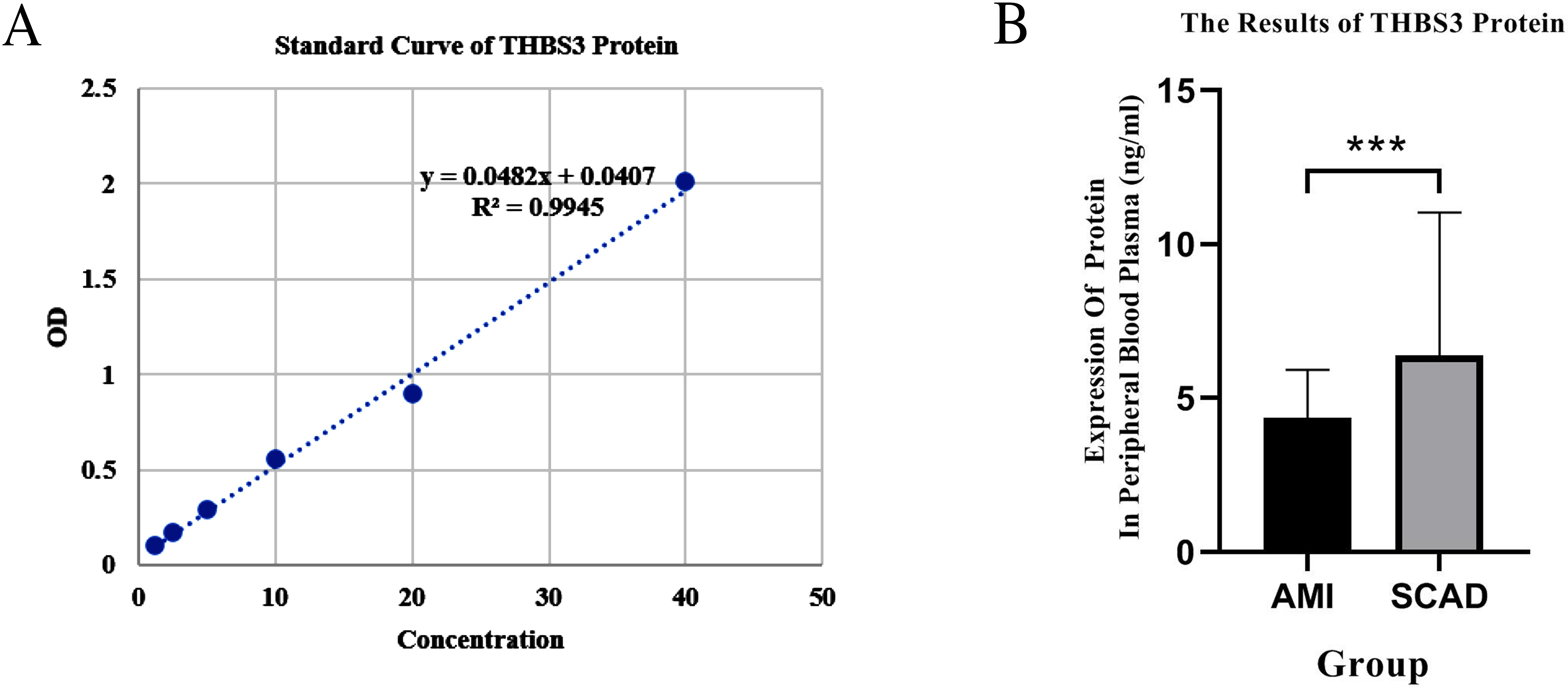 Fig. 2.
Fig. 2.ELISA standard curve and THBS3 protein levels. (A) Standard
curve. (B) THBS3 protein expression. ELISA, Enzyme-linked immunosorbent assay;
*** p
Analysis of peripheral blood RNA and the amplification curve of the
THBS3 gene showed a smooth “S-shape”. The single peak of the
dissociation curve (Fig. 3) is thought to be specific for the amplified product.
The
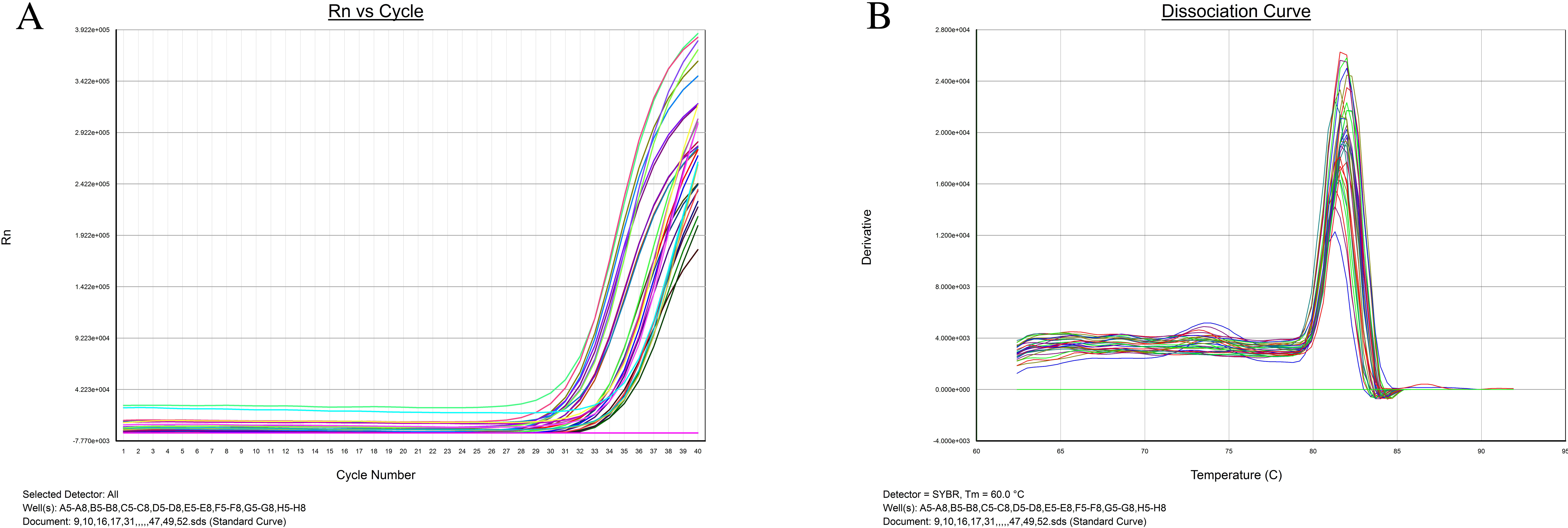 Fig. 3.
Fig. 3.Amplification using RT-PCR and dissociation curve of the THBS3 gene. (A) Amplification curve. (B) Lysis curve.
 Fig. 4.
Fig. 4.Relative expression of the THBS3 gene at the mRNA
level. AMI, Acute myocardial infarction; SCAD, Stable coronary artery disease;
PBMC, Peripheral blood mononuclear cell. * p
THBS3 expression, age, smoking history, fasting blood glucose, TC, LDL-C, and
PLT levels were significantly different between the AMI and SCAD groups (Table 3). Thus, we investigated whether THBS3 protein expression was related to the
aforementioned factors. Patients were divided into two groups: older (
| Groups | THBS3 expression | Z | p value |
| High TC group | 5.316 |
–0.414 | 0.679 |
| Normal TC group | 4.954 (4.259–5.819) | ||
| High LDL-C group | 4.736 |
–1.047 | 0.295 |
| Normal LDL-C group | 4.979 (4.342–5.898) | ||
| High PLT group | 5.006 (4.207–5.802) | –0.274 | 0.784 |
| Normal PLT group | 4.840 (4.093–5.779) | ||
| Smoking group | 4.851 (4.311–5.879) | –1.090 | 0.276 |
| Non-smoking group | 4.703 |
||
| High age | 4.481 (3.896–5.354) | –2.600 | 0.009 |
| Young age | 4.954 (4.259–6.012) | ||
| High fasting blood glucose group | 4.662 (4.039–5.343) | –2.938 | 0.003 |
| Normal fasting blood glucose group | 4.982 (4.200–6.173) | ||
| TC, total cholesterol; LDL-C, low-density lipoprotein cholesterol; PLT, platelet. | |||
Based on the correlation between THBS3 expression and AMI, the maximum value of
sensitivity and specificity was taken as the cut-off value, which was 4.612. We
divided all subjects into two groups: high expression (concentration
| B | Standard variation | Wald | Degree of freedom | p value | OR | 95% CI | ||
| Low expression of THBS3 | 1.405 | 0.456 | 9.502 | 1 | 0.002 | 4.076 | 1.668 | 9.958 |
| High fasting blood glucose | 1.036 | 0.468 | 4.902 | 1 | 0.027 | 2.819 | 1.126 | 7.054 |
| High PLT | 1.874 | 0.722 | 6.730 | 1 | 0.009 | 6.515 | 1.581 | 26.842 |
| High TC | 1.316 | 1.730 | 0.579 | 1 | 0.447 | 3.728 | 0.126 | 110.662 |
| High LDL-C | –1.955 | 1.357 | 2.074 | 0.150 | 0.142 | 0.010 | 2.025 | |
| Smoking History | –0.791 | 0.486 | 2.653 | 1 | 0.103 | 0.453 | 0.175 | 1.174 |
| TC, total cholesterol; PLT, platelet counts; OR, odds ratio; CI, confidence interval. | ||||||||
THBS3 protein expression in peripheral blood was examined between the AMI and SCAD groups. THBS3 protein expression in AMI was significantly lower than that in SCAD, and the relative expression level was 0.82 times that in the SCAD group.
In our previous proteomic research [20], we found that patients with AMI had
lower THBS3 expression in their peripheral plasma than patients with SCAD
(p
Five members make up the glycoprotein family THBS, which codes for the
extracellular matrix (ECM). THBS1 and THBS2 form trimers, whereas THBS3, THBS4,
and THBS5 form pentamers. Their shared domain is intricate [27, 28]. As the five
members are the byproducts of distinct genes on distinct chromosomes, their
regulated DNA and mRNA regions are also distinct. Thus, they can perform several
functional roles in cell adhesion, proliferation, migration, PLT aggregation,
angiogenesis, and wound healing [29, 30, 31, 32]. Single nucleotide polymorphisms in
THBS1, THBS 2, and THBS 4 have been reported to be associated with an increased
risk of early myocardial infarction [33]. As a member of the same family,
Vanhoutte [19] proposed that THBS1 overexpression can atrophy the heart,
ultimately leading to fatal cardiomyopathy. This appears to be a little different
from what we concluded. First off, while both cardiomyopathy and coronary heart
disease were conditions affecting the circulatory system, their pathogenic
mechanisms were different. The former can be caused by genetic variation of genes
and has been the focus of clinical research so far. The latter’s pathophysiology
was based on atherosclerosis, which caused blood vessel stenosis or obstruction
as a result of thrombus formation in blood vessels. Second, the relationship
between myocardial atrophy and myocardial infarction was still unclear. The
control group utilized by Vanhoutte was not used in this investigation. Instead
of a healthy control group, our study included the SCAD group as the control
group. Finally, basic studies of Vanhoutte have confirmed in vivo and in
vitro that overexpression of THBS1 led to myocardial atrophy and that
THBS1
Atherosclerosis is the pathological basis of coronary heart disease. Its
progression and the formation of coronary plaques contribute to the occurrence
and development of various outcomes in CAD. Previous research has shown that the
rupture of vulnerable plaques can result in the formation of an acute thrombus,
leading to acute cardiovascular events [34, 35]. Studies have also shown that the
THBS family can mediate the function of the ECM [36, 37], which is composed of
glycoproteins such as collagen and laminin [38]. Numerous cell receptors,
including integrin and cadherin, interact directly with the components of the ECM
[39]. The common aspects of regulation include cell proliferation, adhesion, and
migration [40, 41, 42]. For instance, THBS1 regulates adhesion and promotes movement
in smooth muscles and endothelial cells by antagonizing the assembly of adhesion
plaques in response to ECM components such as fibronectin [28]. The ability of
laminin to stimulate embryonic retinal cell adhesion and neurite development is
improved by THBS4 [43]. THBS3 also plays a role in cardiomyopathy by
participating in the regulation of integrin expression and function and
increasing disease-induced decompensation to promote myomembrane instability
[18]. Ridley et al. [44] reported that mediating the ECM can regulate
cell migration and advance the onset of atherosclerosis, in which
metalloproteinases (MMPs) play a crucial role [45, 46]. ECM metalloproteinase
inducer (EMMPRIN) can promote plaque instability by inducing ECM degradation and
MMP synthesis, leading to AMI [47]. Moreover, EMMPRIN can stimulate MMP-9 in
monocytes and MMP-2 in smooth muscle cells, which are essential for regulating
MMP activity in cardiovascular diseases [48]. Lindsey et al. [49] also
found that THBS3 expression increased when the collagen I
 Fig. 5.
Fig. 5.THBS3 KEGG pathway diagram.
Baseline analysis revealed a significant difference in fasting blood glucose
levels between patients in the AMI and SCAD groups, where patients in the AMI
group had higher levels than those in the SCAD group. Subsequently, the
correlation between low THBS3 protein expression and high fasting glucose levels
was also found. Elevated fasting blood glucose levels were also a risk factor for
AMI (odds ratio [OR] = 2.819, p
THBS3 is a glycoprotein secreted by PLTs. Although baseline analysis showed that
the PLT counts in the peripheral blood was significantly different between the
two groups (p
Our study has some limitations. As a retrospective study, if a healthy control group is added, it is more meaningful to compare the differential expression of THBS3 among the three groups. At the same time, if the correlation analysis of myocardial markers (troponin I, troponin T, myoglobin, etc.) and THBS3 expression can be carried out, this study will be more comprehensive and valuable. The ECM as well as processes such as cell aggregation, adhesion, and glucose metabolism can contribute to atherosclerosis. Currently, we can only hypothesize how THBS3 might cause AMI and, accordingly, provide a new and reliable biomarker for the diagnosis of AMI. In the future, we intend to further explore disease pathogenesis based on in vitro and in vivo studies and provide new directions and ideas that may offer novel perspectives and suggestions related to THBS3 for the treatment of cardiovascular diseases.
mRNA and protein levels of THBS3 in the peripheral blood of patients with AMI were significantly lower than those of patients in the SCAD group. Low THBS3 expression in peripheral blood was associated with AMI and was an independent risk factor for AMI. Thus, low THBS3 expression in peripheral blood may be a new molecular marker for the early diagnosis of AMI.
YQC, HYM and FBM designed the research study; YQC and FBM performed the research; HYM, XM and ZHY conducted the work and involved in data collection; XM and JRW analyzed the data; YQC and FBM wrote the manuscript; All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Informed consent forms were signed by all patients in this investigation and the guidelines of the Declaration of Helsinki were followed. The protocol was approved by the Ethics Committee of China-Japan Union Hospital of Jilin University (approval number: 2016WJW017).
Not applicable.
This research was funded by grants from the Health Commission of Jilin Province (2018SCZ008); The Special Project For the Development of Medical and Health Industry of Jilin Provincial Department of Science and Technology (20210401054YY); The Double Ten Project of Changchun Science and Technology Bureau (19SS014).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.