
Academic Editor: Graham Pawelec
Background: Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) first occurred in Wuhan (China) in December of 2019. Since the outbreak, it has accumulated mutations on its coding sequences to optimize its adaptation to the human host. The identification of its genetic variants has become crucial in tracking and evaluating their spread across the globe. Methods: In this study, we compared 320,338 SARS-CoV-2 genomes isolated from all over the world to the first sequenced genome in Wuhan, China. To this end, we analysed over time the codon usage patterns of SARS-CoV-2 genes encoding for the membrane protein (M), envelope (E), spike surface glycoprotein (S), nucleoprotein (N), RNA-dependent RNA polymerase (RdRp) and ORF1ab. Results: We found that genes coding for the proteins N and S diverged more rapidly since the outbreak by accumulating mutations. Interestingly, all genes show a deoptimization of their codon usage with respect to the human host. Our findings suggest a general evolutionary trend of SARS-CoV-2, which evolves towards a sub-optimal codon usage bias to favour the host survival and its spread. Furthermore, we found that S protein and RdRp are more subject to an increasing purifying pressure over time, which implies that these proteins will reach a lower tendency to accept mutations. In contrast, proteins N and M tend to evolve more under the action of mutational bias, thus exploring a large region of their sequence space. Conclusions: Overall, our study shed more light on the evolution of SARS-CoV-2 genes and their adaptation to humans, helping to foresee their mutation patterns and the emergence of new variants.
The emergence of the human pathogen Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) in China and its rapid spread poses a global health emergency. On March 11, 2020, WHO publicly declared the SARS-CoV-2 outbreak as a pandemic.
SARS-CoV-2 belongs to the Coronaviridae family, with a genome of approximately 30 kb in length and a structure characteristic of known coronaviruses. The virus genome encodes for structural, non-structural, and accessory proteins [1]. The four structural proteins are the envelope protein (E), the membrane protein (M), the nucleocapsid protein (N), and the spike glycoprotein (S). The overlapping open reading frames ORF1a and ORF1ab encode for the two isoforms of the polyprotein complex pp1a and pp1ab, respectively, where the latter is further proteolytically cleaved into 16 different non-structural proteins (NSP) [2, 3, 4].
The nucleocapsid protein plays an important role in maintaining the RNA
conformation stable for replication, transcription, and translation of the viral
genome, as well as protecting it. It is highly immunogenic and capable of
modulating the metabolism of an infected cell [5]. The envelope protein
acts as a viroporin [6] and plays multiple roles in viral replication
and signalling pathways that affect inflammatory and type 1 INF-
The spike protein S is responsible for receptor recognition and membrane fusion, that leads to viral entry into the host cells [8]. Finally, the membrane protein is associated with the spike protein and is responsible for the virus budding process [9].
Characterization of viral mutations can provide valuable information for
assessing the mechanisms linked to pathogenesis, immune evasion and viral drug
resistance. In addition, viral mutation studies can be crucial for the design of
new vaccines, antiviral drugs and diagnostic tests. The mutation rate of RNA
viruses (10
In this study, we investigate the evolution of SARS-CoV-2 genomes and codon usage patterns over time, by analysing the virus adaptation to the human host. The main hypothesis is that the codon usage should be more optimized with respect to the host’s one [13], to maximize the efficiency of translation. This is linked to the exploitation of the host’s resources, such as tRNA, which is tightly related to codon usage bias [14, 15].
For this purpose, we focused on the four SARS-CoV-2 genes encoding for the structural proteins membrane (M), envelope (E), spike surface glycoprotein (S) and nucleoprotein (N), as well as RNA-dependent RNA polymerase (RdRp). We also decided to consider the polyprotein complex ORF1ab taking the two segments ORF1a and ORF1b separately as they code for many different proteins. Of note, RdRp is encoded by a segment of the ORF1ab, but it has been isolated due to its key role in viral replication. In Table 1 (Ref. [16]) can be found the lengths of the considered genes, along with their position in the viral genome.
| Gene | Slope | x-Intercept | R-squared |
| M | 0.2951 |
–0.0027 |
0.086 |
| N | 1.9015 |
–0.0657 |
0.904 |
| S | 2.1984 |
–0.0071 |
0.784 |
| RdRp | 0.5477 |
–0.1692 |
0.161 |
| ORF1a | 1.648 |
0.012 |
0.525 |
| ORF1b | 1.0354 |
–0.0265 |
0.369 |
All available SARS-CoV-2 genomes reported across the world were obtained from GISAID (available at https://www.gisaid.org/epiflu-applications/nexthcov-19-app/), on February 7, 2021. Then, the sequences were classified according to their isolation dates. Only complete genomes (29–30 Kb) were included in the present analysis, with a list of 320,338 SARS-CoV-2 genomes. We used the SARS-CoV-2 coding DNA sequences (CDSs) deposited in January 2020 by Zhu and co-workers [17], formerly called “Wuhan seafood market pneumonia virus”, also referred to as “Wuhan-Hu-1” (WSM, NC 045512.2), as reference sequence. We retrieved these sequences from NCBI public database at https://www.ncbi.nlm.nih.gov/. The CDSs of the reference SARS CoV-2 genome (NC 045512.2) were used to retrieve the homologous protein-coding sequences from the genomes under study, by using the Exonerate tool (v 2.2.0, https://www.ebi.ac.uk/about/vertebrate-genomics/software/exonerate) with default parameters [18]. A threshold in length of 95% the total reference gene length has been used to filter out defective sequences.
Relative Synonymous Codon Usage (RSCU) is the fraction of the observed codon frequency to expected codon frequency, given that all the codons corresponding to a precise amino acid are used equally. RSCU value for each gene is calculated using Eqn. 1 for each codon i, as described by Sharp and Li [19]:
where X
The two indices Codon Adaptation Index (CAI) and Similarity Index (SiD) are both used to quantify the extent of codon usage adaptation of SARS-CoV-2 to the human coding sequences.
The principle behind CAI [20] is that the codon usage in highly expressed genes
can reveal the optimal (i.e., most efficient for translation) codons for the
translation of each amino acid. Hence, CAI is calculated based on a reference set
of highly expressed genes to assess, for each codon i, the relative synonymous
codon usages (RSCU
w
Finally, the CAI value for a given gene g is calculated as the geometric mean of the usage frequencies of codons in that gene, normalized to the maximum CAI value possible for a gene with the same amino acid composition, as shown in Eqn. 3.
where the product runs over the l
On the other hand, SiD is the cosine of the solid angle between the two RSCU vectors of the virus and the host, without taking into account the expression rate of the genes.
Formally, SiD is defined in Eqn. 4 [21]:
where a
Both indices values range from 0 to 1; the higher the value, the more adapted the codon usage of SARS-CoV-2 to the host. They are fundamental estimators for this research, as they are useful to quantify the codon usage bias viral adaptation to the host’s one.
The reference set of highly used genes used for CAI has been retrieved from DAMBE 5.0 [22], while the codon frequencies used to estimate human RSCU, from CUBAP [23].
Principal Component Analysis (PCA) [24] is a multivariate statistical method to transform a set of observations of possibly correlated variables into a set of linearly uncorrelated variables (called principal components), spanning a space of lower dimensionality. The transformation is defined so that the first principal component accounts for the largest possible variance of the data, and each subsequent component in turn has the highest variance possible under the constraint that it is orthogonal to (i.e., uncorrelated with) the preceding components.
We use this technique on the space of RSCU vectors, so that each gene of SARS-CoV-2 is represented as a 59-dimensional vector with codons as coordinates. Such coordinates are separately normalized to zero mean and unit variance over the whole genome. We then obtain the associated covariance matrix between the dimensions of codon bias and diagonalize it. The eigenvectors of the covariance matrix, ordered according to the magnitude of the corresponding eigenvalues, are the principal components of the original data. PCA has been performed by using Python scikit-learn (v 0.20.3)(https://pypi.org/project/scikit-learn/0.20.3/).
ENC is used to evaluate the degree of bias of a gene or genome, in respect to codon usage. It quantifies the difference from a random usage of all codons. Its value ranges from 23, when there is a strong bias and only one codon per amino acid family is used, to 61, when all codons are equally used. 6 codon-families have been considered as distinct subfamilies of 2 and 4 codons, as explained in Sun et al. (2013) [25].
An ENC plot analysis was performed to estimate the relative contributions of mutational bias and natural selection in shaping the CUB of the SARS-CoV-2 genes. In this plot, the ENC values are plotted against GC3 values. If codon usage is dominated by mutational bias, then a clear relationship is expected between ENC and GC3, given by the formula in Eqn. 5,
where s represents the value of GC3 [26]. If the mutational bias is the main force affecting the CUB of the genes, the corresponding points are expected to fall near Wright’s theoretical curve. Conversely, if CUB is mainly affected by natural selection, the corresponding points should fall considerably below Wright’s theoretical curve [27]. To quantify the relative extent of the natural selection, for each gene, we calculated the Euclidean distance d of the corresponding point, from the curve. We then showed the average values of the distance over time with a heatmap.
To study the mutational rates of genes under study, we performed an analysis by using Forsdyke plots [28]. Each gene of the SARS-CoV-2 sequences under investigation was compared to its orthologous gene in the reference Wuhan-Hu-1 genome. Each pair of orthologous genes is represented by a point in the Forsdyke plot, wherein protein divergence is correlated with RNA divergence. The protein sequences were aligned using Biopython. The RNA sequences were then aligned using the protein alignments as templates. Then, both RNA and protein divergences were assessed, as explained in Methods in [28], by counting the number of mismatches in each pair of aligned sequences. Thus, each point in the Forsdyke plot measures the divergence between pairs of orthologous genes in the two viruses, as projected along with the phenotypic (protein) and nucleotidic (RNA) axis. The first step in each comparison is to compute the regression line between protein versus RNA sequence divergence in the Forsdyke plot for getting values of intercept and slope for each variant of genes.
The records of virus genomes according to geographical location and month of isolation are shown in Fig. 1. A great percentage of the annotated SARS-CoV-2 genomes (about 62% and 22% respectively) are distributed in Europe and North America. The number of complete viral genomes is non-homogeneous, ranging from 200–300 in the first months, reaching a peak of around 50,000 in December 2020. In the other months, the values fluctuate between 7000 and 40,000.
 Fig. 1.
Fig. 1.Distribution of the 320,338 genomes used in this study. Distribution of available genome sequences, subdivided per month and geographical region.
We performed PCA over the space of RSCU vectors measured for each SARS-CoV-2 gene in all sequences. The two first principal components (PC1 and PC2) appear to represent as much as 78% of the total variance of RSCUs (respectively, 52% for PC1 and 26% for PC2). The projections of the first two principal components on the individual codons show that none of the codons predominantly contributes to the data variability (see Fig. 2). Thus, the placement of a gene on the PC1-PC2 plane depends on a weighted contribution of all codons.
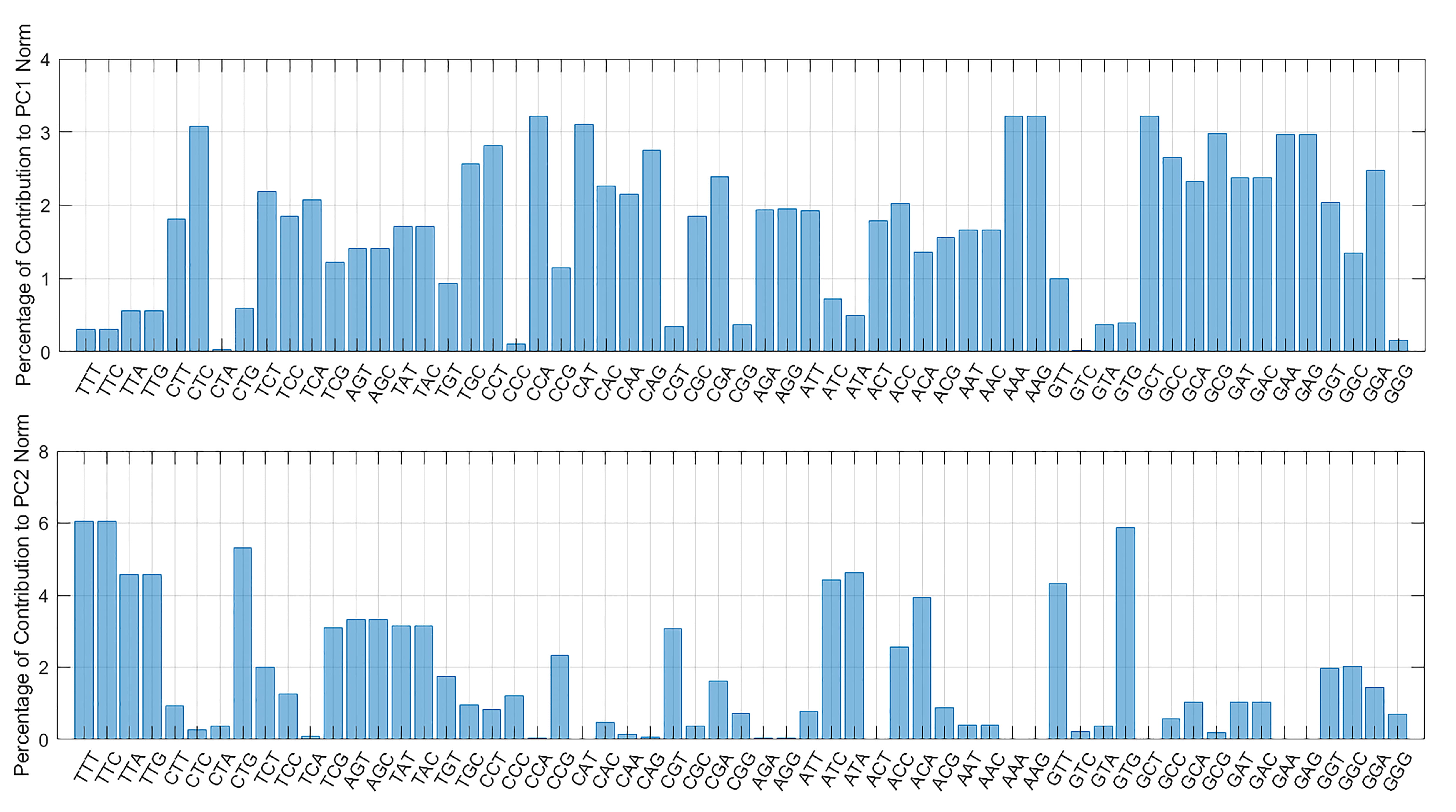 Fig. 2.
Fig. 2.Projection of the first two PCA components on the RSCU vectors. The distribution of codons in PC1 is more uniform, resulting in a weighted and coherent contribution of all codons, whereas in PC2 the contribution is more focused on specific codons encoding phenylalanine, leucine, isoleucine, lysine and valine.
Most codons highly affect PC1, whereas PC2 is mostly affected by triplets that code for phenylalanine (TTT and TTC), leucine (TTA, TTG and CTA), isoleucine (ATT, ATC), lysine (AAG) and valine (GTA).
The PCA plot of all considered genes is shown in Fig. 3. Interestingly, the genes coding for the polyproteins and S protein are very close, in the PCA plane at the bottom of the graph, far apart from the others. Regarding the N protein, the coordinates in PC1 is similar to those of the S-ORF1ab cluster, while is quite distant in PC2. The M protein is closer to N in PC2, denoting a similar usage of codons translating the five amino acids displayed above. Finally, the E protein is the mostly separated and dispersed cluster. The difference in RSCU of the protein E from the others and the dispersion in PCA is due to its shortness (see Table 1) for it does not allow a consistent repetition of amino acids and thus the use of different codons. Taken together, this analysis shows that the longer genes carry a signature RSCU of the virus, suggesting that SARS-CoV-2 genes tend to use the same set of codons. For the two proteins M and N, this set differs in the codons encoding the five aforementioned amino acids.
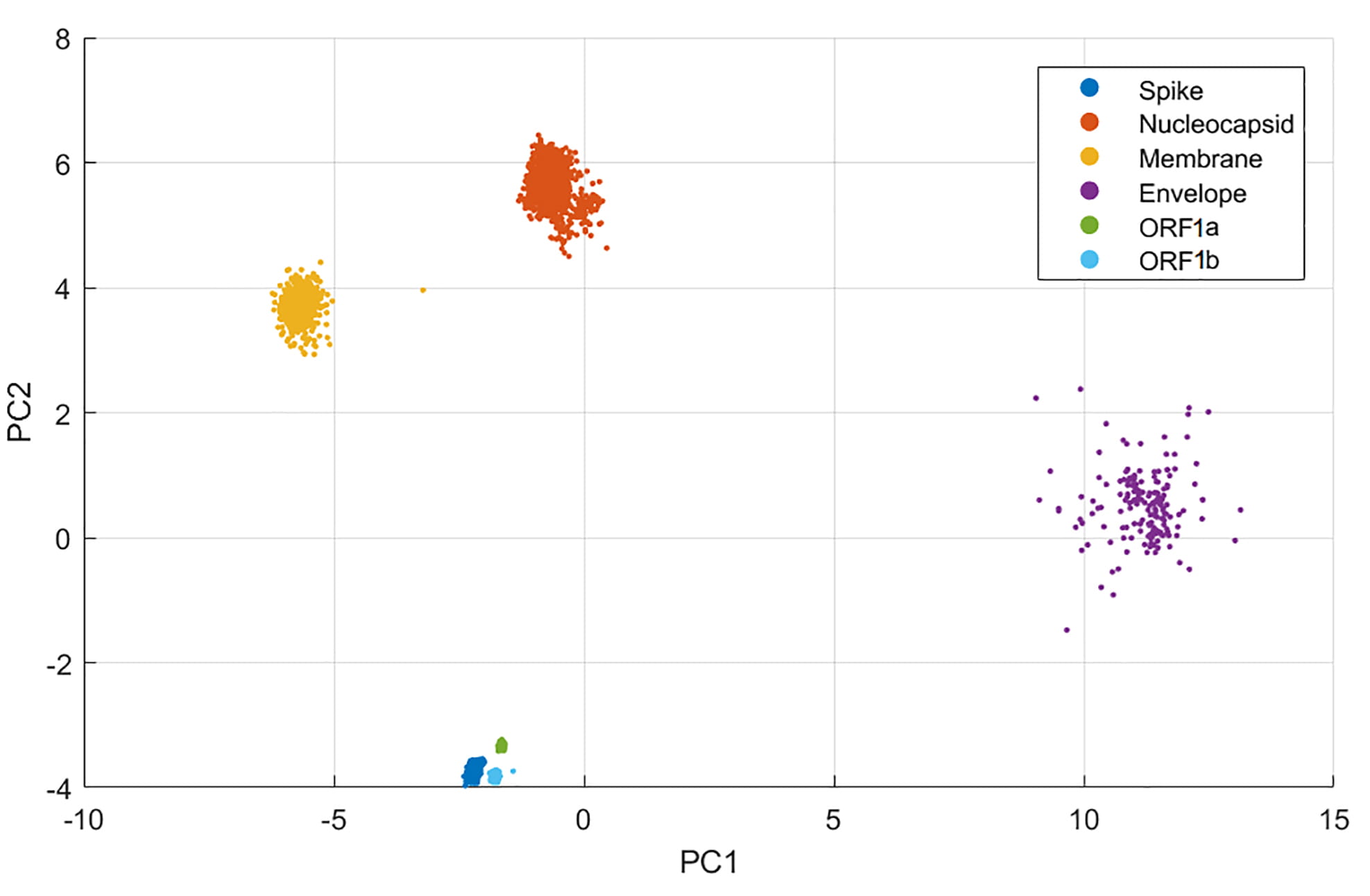 Fig. 3.
Fig. 3.PCA of SARS-CoV-2 Genes. All available sequences for each gene have been used in this study, by calculating the RSCUs and performing PCA for each of them. Approximately 320,000 sequences were analysed per gene.
To measure SARS-CoV-2 codon usage bias adaptation to the human host, we first evaluated CAI for all genes under investigation. For each gene, we performed a daily mean of the CAI values (see Fig. 4). In line with the PCA analysis, the CAI starting values are very similar for genes S, ORF1a, ORF1b and RdRp. The N protein has the highest CAI value, implying a larger adaptation of its codon usage to that of highly expressed genes in humans. Conversely, the M protein exhibits the lowest CAI value, indicating a less adapted codon usage with respect to the host.
 Fig. 4.
Fig. 4.CAI values daily averages in SARS-CoV-2 genes. Daily averaged
CAI values estimated from all the available sequences and the standard errors on
their assessments (
The fastest evolving gene appears to be the one coding for the N protein with a globally descending trend. Similarly, the S protein displays the same trend but with a down-peak in the month of July 2020. In the timeline of sequence evolution shown in Supplementary Fig. 1, where the sequences are divided by region, that this down-peak is mostly due to Oceania sequences. Other geographical regions display a smoother decrease.
The M protein evolves in a constant manner until August, where a steep ascent occurs. On the other hand, RdRp presents a sudden decrease in the first months and a subsequent stabilization on a lower CAI value. This observation suggests an initial adaptation of RdRp to the new host with the emergence of a new variant that rapidly superseded the older, less optimized one. Finally, the two CDSs of gene ORF1ab manifest a lower variability over time, in line with the fundamental role of these proteins in replication.
Overall, the SARS-CoV-2 genes display a descending trend over time, which is in contrast with the hypothesis that a virus evolves to optimize its codon usage to the one of the human host. Our observations suggest a general evolutionary trend of SARS-CoV-2 according to which it has been evolving towards a sub-optimal codon usage bias, which may favour the host survival and, therefore, its spread. Indeed, if the virus replicates efficiently, then it is more likely to be successful at invading a given host, eventually killing it. This finding is in accordance with the virulence trade-off hypothesis [29].
In line with our previous study [30], we calculated the SiD of SARS-CoV-2 genes with respect to the human host. To understand the rationale behind these results, the higher the value of SiD, the more adapted the codon usage of SARS-CoV-2 to the host under study [31]. Specifically, we calculated here the SiD values for each gene under study, and performed a daily average of all the available sequences (see Fig. 5).
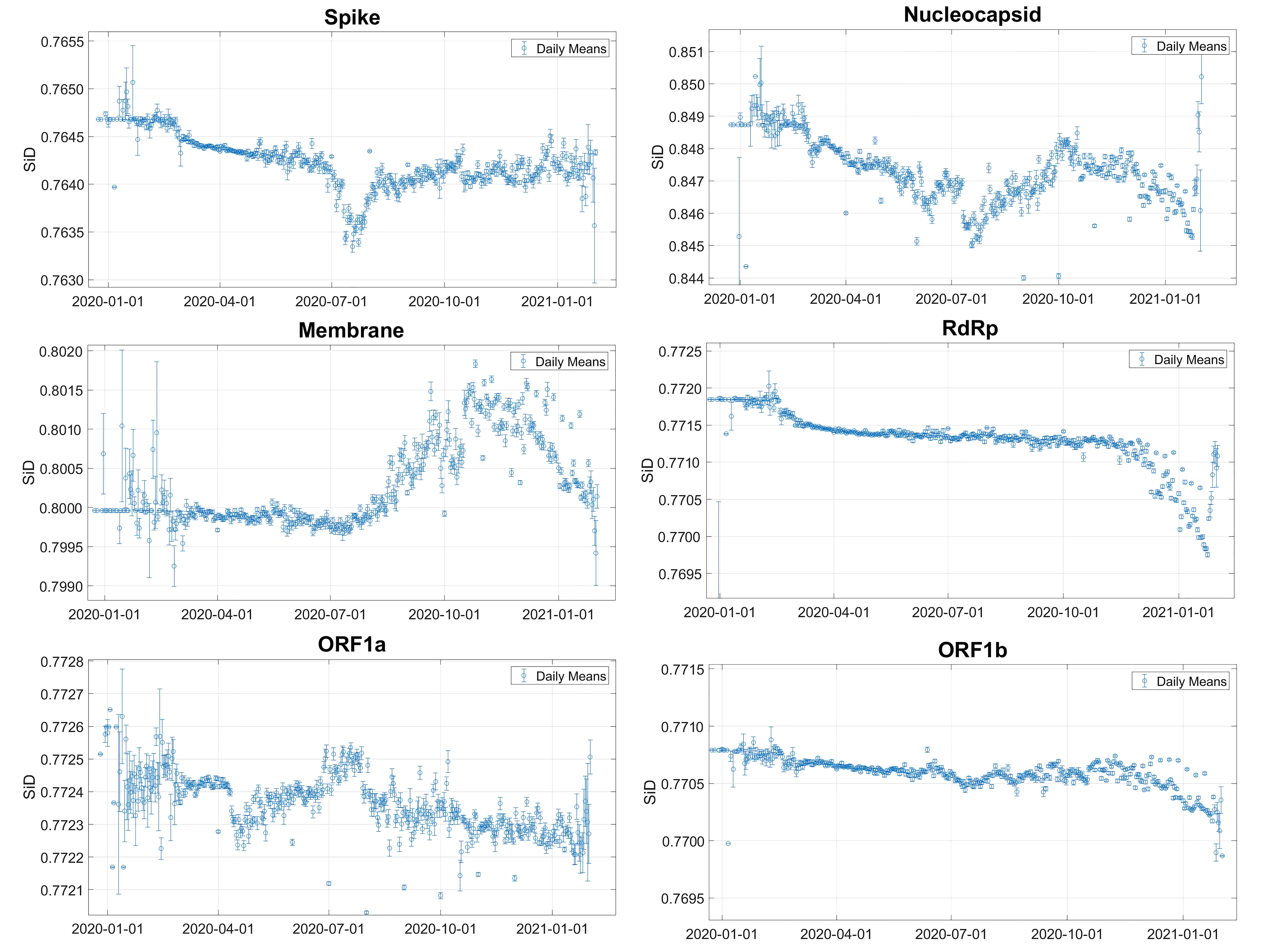 Fig. 5.
Fig. 5.SiD values daily averages in SARS-CoV-2 genes. Daily averaged
SiD values estimated from all the available sequences and the standard errors on
their assessments (
In accordance with the CAI temporal evolution (see Section 3.3), the N protein corresponds to the most adapted gene; in contrast, the gene encoding the S protein is the least adapted.
Also in this case, the RdRp, ORF1ab, and protein S, show similar SiD values. Moreover, the temporal trends are mostly descending, remarking the previous statements on CAI about a reduced optimization of SARS-CoV-2 codon usage to the human host over time. As observed in Section 3.3, the averaged SiD value for the S protein shows a down peak in July, which is mainly due to the Oceania sequences (Supplementary Fig. 1).
To further investigate the evolutionary forces that affect the SARS-CoV-2 codon usage, an ENC-plot analysis was conducted separately for each gene here considered. In Fig. 6, we show the ENC-plot obtained for the Wuhan-Hu-1 genes, together with Wright’s theoretical curve (see [27] and Section 2.4 for details). This plot was performed to have a reference for the next temporal analysis.
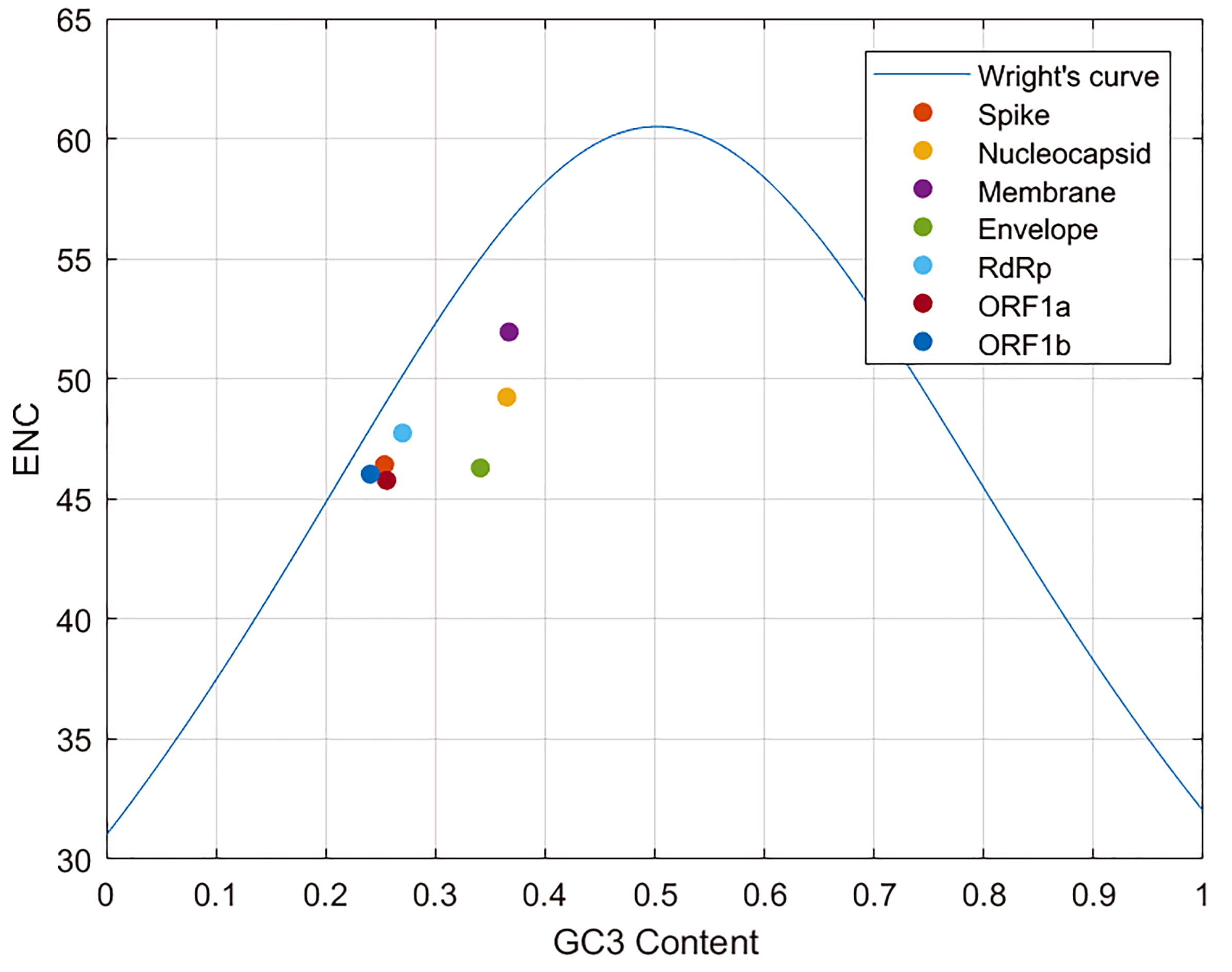 Fig. 6.
Fig. 6.ENC plot of Wuhan-Hu-1 genes. Representation of Wright’s Theoretical curve together with Wuhan-Hu-1 genes ENC Plots (GC3 vs ENC). ENC values span from 23 to 61. GC3 is the fraction of GC content at the third position of the codon, and ranges from 0 if no codon ends in C or G, to 1 if all codons do.
All genes are found below the curve, in accordance with Ayan (2020) [32], an indication that not only mutational bias but also natural selection plays a non-negligible role in their codon choice. The M, E and N proteins have the greatest distance from Wright’s curve, pointing out that the codon usage of corresponding genes is the most subject to natural selection.
By using Wuhan-Hu-1 genes as reference, we analysed variations over time in the relative extent to which natural selection and mutational bias affects SARS-CoV-2 genes codon usage by generating ENC plots for each gene under study at different time points. To illustrate more clearly the temporal diversities, 10–day averages of the distances from Wright’s theoretical curve have been estimated. In Fig. 7 is shown the heatmap of the differences between these values calculated over time and the reference values of Wuhan-Hu-1. In Fig. 7, a positive difference means that the distance is lower with respect to that of the reference gene (lighter colours), while a negative difference implies that the points are getting farther from the curve (darker colours).
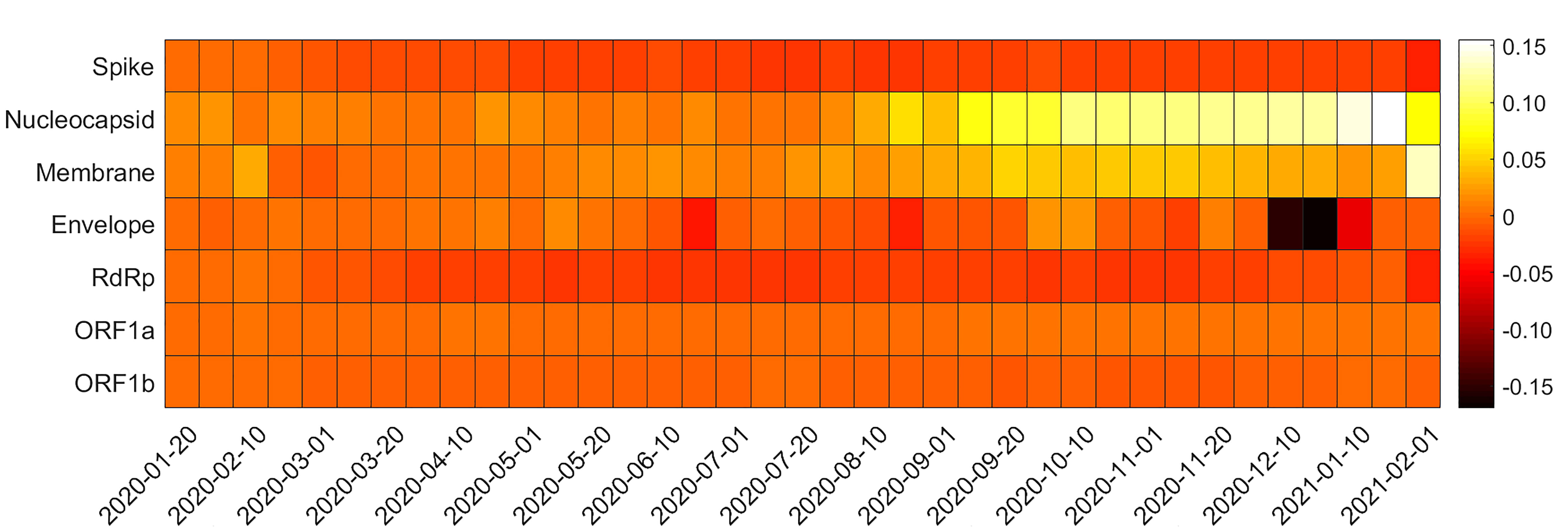 Fig. 7.
Fig. 7.Heatmap of ENC plots. Each square of the heatmap represents the difference between the distance of Wuhan-Hu-1 gene from Wright’s curve and the distance of the examined gene from the same curve; 10-day averages have been calculated. Darker colours represent an increase of the distance from Wright’s theoretical curve with respect to the reference genes distance, while lighter colours indicate a closer distance.
These differences reflect the action of natural selection and mutational bias in shaping codon usage of SARS-CoV-2 genes over time. Interestingly, the average distances from Wright’s theoretical curve increase over time for genes S and RdRp. This indicates that the codon usage of these genes is more affected by natural selection as time goes on. This observation is consistent with the fact that the Spike protein and the RdRp are the key proteins for infecting host’s cells, controlling the binding and fusion with the host cell membrane, and the subsequent viral replication.
In contrast, the N and M proteins get closer to Wright’s theoretical curve, suggesting that their codon usage is more affected by mutational bias than Wuhan-Hu-1 genes.
These observations, in a nutshell, indicate that S and RdRp have a tendency to explore increasingly more focussed regions of their viable sequence space, whereas M and N are prone to a wider, less contained exploration.
Next, we estimated the evolutionary divergences of the seven genes considered herein. For this purpose, the RNA and protein sequence divergences of these genes were analysed by comparing the nucleotide sequences of the reference Wuhan-Hu-1 genes and their corresponding protein sequences with the other SARS-CoV-2 sequences extrapolated over time (Fig. 8). In this Fig. 8, each panel refers to a single gene, and each gene sequence is represented by a single point. Because of the redundancy of mutations, many gene sequences characterized by the same pattern of mutations at the nucleotide and amino acid level are often superimposed on this plane. To take into account this redundancy, we report a bubble plot, in which the radius of each circle is proportional to the number of genes with the same coordinates (i.e., same nucleotide and amino acid divergence). Each bubble in these plots measures the divergence between a gene sequenced at different time points and the reference Wuhan-Hu-1 gene, as projected along with the phenotypic (protein) and nucleotide (DNA) axis. Thus, the slope is an estimation of the fraction of RNA mutations that results in amino acid substitutions [27].
 Fig. 8.
Fig. 8.Forsdyke plots of the considered genes. Forsdyke Plots of structural proteins (panels on the top) and replicase polyprotein complex (panels on the bottom). The radius of each circle is proportional to the number of data points in the same spot. Values span from 0, when the sequence under study is identical to the reference one, to 1, when they are different in every nucleotide/amino acid, for both axes. The inferred data from linear regressions are shown in Table 2.
Overall, protein and RNA sequence divergences are linearly correlated, and these correlations correspond to slopes and intercepts of the linear regressions in Table 2.
| Gene | Length (aa) | Location |
| ORF1a | 4405 | 266–13 483 |
| ORF1b | 2696 | 13 483–21 555 |
| RdRp | 932 | 13 442 –16 236 |
| S | 1273 | 21 563–25 384 |
| E | 75 | 26 245–26 472 |
| M | 222 | 26 523–27 191 |
| N | 419 | 28 274–29 533 |
Specifically, gene M displays an accumulation of points around the x-axis, suggesting that the corresponding protein tends to evolve slowly by accumulating mutations on its sequence. Conversely, the steeper slopes for genes N and S suggest that these genes are fast-evolving compared to the other genes. This is in line with our previous study [30], according to which the N and S proteins are the main drivers of the speciation process among Coronaviruses. Considering the results from the ENC plots (section 3.5), the evolution of S protein is characterized by a purifying selection, while the N protein mutates under the action of mutational bias.
Moreover, ORF1a and ORF1b have a lower slope, pointing out that the phenotypic divergence of those is not as fast as that of the structural proteins. This is in line with the role of ORF1ab in viral replication and, therefore, with its higher sequence conservation. Interestingly, RdRp has a lower slope than ORF1b, pointing out the great importance of this protein in the replication process, that is, it tends to accumulate more synonymous mutations than missense, conserving the protein structure and functionalities.
It is worth noting that all the x-intercepts are close to 0. This observation suggests that genic differences (i.e., amino acid changes) have played a significant role in the gene divergence.
In this study, we performed a comprehensive analysis of the evolutionary divergence and codon usage of SARS-CoV-2 genes over time, considering all genomes available in GISAID up to February 7, 2021. After filtering out incomplete genomes, we retained 320,338 complete genomes, with the purpose of investigating their divergences from the first sequenced SARS-CoV-2 genome in Wuhan (NC 045512.2). We focused on seven SARS-CoV-2 genes and their encoded proteins that are crucial for virus structure, synthesis, transmissibility and virulence. Specifically, we considered Spike, Nucleocapsid, Membrane, Envelope, RdRp and the two segments of ORF1ab separately, ORF1a and ORF1b. The SARS-CoV-2 genomes have a tendency to diverge constantly from the reference genome, accumulating mutations over time [12]. We assumed herein that the accumulation of these mutations could affect the codon usage bias of the SARS-CoV-2 genes. Thus, we investigated the codon usage preference of SARS-CoV-2 in relation to the human host, using classical measures of codon usage bias such as RSCU, CAI, SiD and ENC.
First, we performed PCA in the space of RSCUs to investigate the major sources of variations in the codon usage of SARS-CoV-2 genes (Fig. 3). The two first principal components (PC1 and PC2) represent as much as 78% of the total variance of codon bias. Focusing on the PC1-PC2 plane, all the genes considered herein form distinct clusters, which indicate a different usage of synonymous codons in their sequence. Specifically, ORF1a, ORF1b and Spike exhibit very similar RSCUs and quite compact clusters. In contrast, Envelope protein corresponds to the most dispersed cluster. As this protein is the shortest one, we suppose that every mutation has a greater impact on the RSCU, resulting in the dispersed cluster that we observed in the PC1-PC2 plane. Finally, the Nucleocapsid protein shows a diffused cluster which is mainly due to the high rate of mutations occurring in its sequence. Notably, except for E, all the genes have similar values of PC1, which is affected by all codons uniformly (see Fig. 2). Conversely, the clusters separated along PC2 reflect a different usage only of codons encoding for phenylalanine (TTT and TTC), leucine (TTA, TTG and CTA), isoleucine (ATT, ATC), lysine (AAG) and valine (GTA).
Next, we analysed the temporal evolution of the codon usage bias by investigating CAI (Fig. 4) and SiD (Fig. 5) values as a function of the isolation dates of genomes across the world up to February 7, 2021. We observed that most SARS-CoV-2 genes show a tendency to use a broader set of synonymous codons over time. This is in contrast with the initial hypothesis of adaptation of the viral codon usage to the human host, which would lead to a greater tendency to use the synonymous set of codons preferred in highly expressed genes. We suppose here that a well-adapted codon usage to the host might be counterproductive, increasing the efficiency of translation, speeding up viral replication, and potentially leading to the death of the host. For this reason, a less-adapted codon usage over time could favour the adaptation of the virus to the human host while ensuring its survival through its spreading across the human population. Interestingly, RdRp shows a sudden decrease in both CAI and SiD values during the first months, to stabilise afterwards. We suggest that the first adaptation can be linked to the use of antiviral drugs, that mainly target this protein.
To understand the evolutionary forces modelling the differential usage of codons over time, we performed an ENC-plot analysis. We compared the position of genes isolated at different time points across the world with the first sequenced genomes in Wuhan, taken here as reference (see Fig. 6 for the reference ENC plot). This analysis revealed that the codon usage of the viral genes are subject to different balances between mutational bias and natural selection as a function of the isolation date. We found that the codon preference of the genes S and RdRp is mainly determined by natural selection. This may be linked to the crucial role of their corresponding proteins in the infection (S) and replication (RdRp) processes. RdRp is considered less vulnerable to mutations [12] due to its vital role in maintaining viral genome fidelity; the mutations that occur in RdRp likely promote the virus adaptive flexibility and enhance its resistance to antiviral drugs [33].
Conversely, the codon usage of the genes M and N is strongly affected by mutational bias, leading to a broader exploration of the mutation spectrum.
Performing a Forsdyke plot analysis (Fig. 8), we obtained results confirming those of our previous study [30], showing that the speciation process of Coronaviruses is predominately driven by the genes N and S. According to Rehman et al. (2020) [34], the spike protein, which mediates the virus interaction with the human host cells, is more prone to mutations, although the amino acids implicated in the spike and the angiotensin-converting enzyme 2 (ACE2) interface tend to be more conserved [35], due to the importance of their function [36, 34]. In particular, we have found about one missense mutation for each of the 2700 analysed sequences in the Receptor Binding Domain. These divergences though, considering the results from ENC plots, are profoundly different, that is, while S is mutating under the action of a purifying pressure, N evolution is mainly due to mutational bias.
Therefore, the N and S genes accumulate beneficial mutations that would increase the evolvability and transmissibility of SARS-CoV-2, allowing it to continuously adapt to different populations, like spilling over from bats, or other candidate natural reservoirs, to humans. The greatest rate of mutation of these two proteins may be linked to the high number of epitopes found on them [37, 38, 39].
In the present study, we have studied the adaptive features of SARS-CoV-2 genomes, taking into account the location and time those genomes were isolated and sequenced. The main question addressed in this study is whether the human codon usage bias, during the pandemic, has affected and optimized the codon bias of SARS-CoV-2.
We focused on the RNA genes coding for M, E, S, N, RdRp and ORF1ab. All these viral genes appear to evolve toward a codon usage pattern that is less efficient than it could be with respect to the human codon usage, at least in the time sequel we have considered. This is in contrast with the hypothesis of a greedy viral adaptation in codon usage bias to the host. On the other hand, it is in agreement with the virulence trade-off hypothesis, according to which the virus would not adapt to the host at the optimal level, to keep it alive and consequently spread the infection, a hypothesis that has been extensively discussed in a recent paper [29].
Moreover, from Forsdyke and ENC plots, we observed that the S and N proteins evolve faster and drive the speciation process. The S protein is subject to positive purifying selection, while N is under mutational bias. ORF1ab, and in particular RdRp, present a lower number of non-synonymous mutations (i.e., they have a low value for the slope in the Forsdyke plots). As a matter of fact, according to the ENC plot in Fig. 7, RdRp mutations are mostly driven by natural selection, consistently with its key role in the replication process.
It is very important to extend the type of investigation presented herein, so as to have a clearer perspective on the adaptation of viral codon bias to the host, a theme of great relevance for the control of epidemics.
EP, MD, SF, AP, AGG and AG designed the research study and performed the research. EP, MD, SF wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
The authors warmly thank the anonymous referees whose constructive comments were of great help in revising and focusing our paper.
This research received no external funding.
The authors declare no conflict of interest.