


 , Xiaobin Liu 4,*, Yijie Ding 2,*
, Xiaobin Liu 4,*, Yijie Ding 2,*1 Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, 610054 Chengdu, Sichuan, China
2 Yangtze Delta Region Institute (Quzhou), University of Electronic Science and Technology of China, 324000 Quzhou, Zhejiang, China
3 Department of Oncology Radiology, Beidahuang Industry Group General Hospital, 150000 Harbin, Heilongjiang, China
4 Department of Nephrology, The Affiliated Wuxi People's Hospital of Nanjing Medical University, 214023 Wuxi, Jiangsu, China
Academic Editor: Graham Pawelec
Abstract
Introduction: The electron transport chain is closely related to cellular respiration and has been implicated in various human diseases. However, the traditional “wet” experimental method is time consuming. Therefore, it is key to identify electron transport proteins by computational methods. Many approaches have been proposed, but performance of them still has room for further improvement. Methodological issues: In our study, we propose a model stacking framework, which combines multiple base models. The protein features are extracted via PsePSSM from protein sequences. Features are fed into the base model including support vector machines (SVM), random forest (RF), XGBoost, etc. The results of base model are entered into logistic regression model for final process. Results: On the independent dataset, the accuracy and Matthew’s correlation coefficient (MCC) of proposed method are 95.70% and 0.8756, respectively. Furthermore, we show that the model stacking framework outperforms single machine learning classifiers statistically. Conclusion: Our models are better than most known strategies for identifying electron transport proteins. Our model can be used to more precisely identify electron transport proteins.
Keywords
- Electron transport chain
- Ensemble learning
- Model stacking
- Logistic regression
- Transport protein
Protein is a vital component of all human cells and tissues, and it is intimately linked to life and many forms of biological activity, such as cellular respiration. Cellular respiration is the process by which organic matter passes through a series of oxidative breakdowns inside cells to form inorganic or small molecules of organic matter, releasing energy and producing Adenosine triphosphate (ATP), which is the most direct source of energy for most cellular reactions [1]. In this process, the electron transport chain is critical for storing and transferring electrons. Five protein complexes make up the electron transport chain and are named complex I, II, III, IV, and V. Electron transport proteins are made up of many electron carriers and serve a variety of molecular functions [2, 3, 4]. In studies, electron transporter abnormalities have been found to be associated with diseases such as idiopathic diabetes [5, 6, 7, 8], Parkinson’s disease [4], and Alzheimer’s disease [9, 10, 11, 12]. Therefore, the identification of electron transporter proteins is helpful in exploring the causes of human diseases and may help prevent and treat human diseases. Due to the high time and cost of identifying protein functions by traditional experimental techniques, computational approaches must be developed. The construction of meaningful feature sets and selection of appropriate classification algorithms are considered to be the two most important steps in protein classification. When constructing feature sets, some studies had taken advantage of the biochemical properties of proteins. Le et al. [13] extracted protein characteristics through biochemical characteristics, which improving the accuracy of identification of electron transporters. Khatun et al. [14] developed a model for predicting anti-inflammatory peptides using computational methods by using binary as the characteristic representation of proteins. Others employed position specific scoring matrix (PSSM) to extract evolutionary information of protein. In SulCysSite [15], binary, PSSM profiles, and pCKSAAP are combined as feature information to predict protein S-sulfenylation sites. Hasan et al. [16] used PSSM profiles to preserve the evolution information in proteins and developed pbPUP, a model for identifying protein pupylation sites, which had a good performance. Le et al. [17] proposed ET-CNN, which fed PSSM profiles into the convolutional neural networks (CNN) for electron transport protein classification. PSSM profiles and amino acid composition (AAC) were utilized by Chen et al. [18] to extract protein features, which were fed into radial basis function networks. In Mishra’s study [19], features including amino acid composition, biochemical properties, and PSSM profiles were fed into the support vector machines (SVM) to predict transporters including electron transporters.
ET-GRU is a model proposed by Le et al. [20] to identify electron transport proteins. CNN is used to extract features from PSSM matrix before using deep-gated recurrent unit (GRU) for classification. In addition, traditional machine learning algorithms have been widely used. Gromiha et al. [21] and Ru et al. [22] used machine learning methods, including support vector machine, logistic regression, decision tree, random forest, and naive Bayes, to perform functional recognition of electron transport proteins. Single machine learning classification has both disadvantages and advantages. For example, the random forest has good performance on the unbalanced dataset but has high feature requirements. When XGBoost is used, excellent model performance can be achieved by adjusting a large number of complex parameters, which is totally difficult. Ensemble learning can flexibly combine various classifiers, train different models as base classifiers, and then combine them with ensemble strategies for final prediction. Common integration strategies include stacking, majority voting, bagging, boosting.
In our study, a model of stacking framework (MSF), which combines multiple base models is proposed. We represent protein features using the PSSM matrix produced by PSI-BLAST. Then, Pseudo-PSSM (PsePSSM) is used to extract evolutionary information. These features are fed into the base model. These base models are different classification algorithms, such as random forest, XGBoost, k-nearest neighbor (KNN), and SVM. These models are loosely coupled. After combining the results of each classifier, logistic regression makes the final prediction. Finally, the experimental results prove that the model of stacking framework (MSF), which is built by SVM, XGBoost, and KNN, has the best effect. We compare MSF with the single classifier and majority voting on the same independent dataset and perform a t test, the MSF is significantly superior to other single classifiers. In addition, MSF also perform better than most existing methods for identifying electron transport proteins.
In our study, we utilize the benchmark dataset released by Le et al. [20], which contains 1324 electron transport proteins and 4569 general transport proteins. The data were initially taken from a previous study [17], and data from UniProt release-2018_05 (on 23-May-2018) [23] and Gene Ontology (GO) release-2018-05-01 [24] were also collected. Then, in order to avoid model overfitting, the data were removed the redundant sequences with similarities of more than 30%. To solve this binary classification problem, the dataset is randomly divided into cross-validation dataset and independent dataset in a ratio of 0.85:0.15. Table 1 shows the details of the datasets.
| Original | CV | IND | |
| Electron transport | 1324 | 1125 | 199 |
| General transport | 4569 | 3884 | 685 |
Using appropriate methods to extract protein characteristics is an important step to complete the task of classification. PSSM, which retains the evolutionary information of proteins in a matrix of L rows and 20 columns, is employed as the feature extraction approach in this work. PSSM was first introduced by Jones [25], which is a commonly used method in the field of bioinformatics. It has been used in a number of bioinformatics studies with positive results [26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37]. The PSSM profiles is derived from multiple sequence alignment and contains the evolutionary information of each residue in the protein sequence. Electron transport proteins belong to a class of proteins with a specific function. Compared with other protein families, the key evolutionary information of proteins can be captured by using PSSM matrix and related feature extraction method, and then the classifier can be used to effectively identify electron transport proteins.
FASTA sequences are searched against the Uniprot database to compile position specific scoring matrices (PSSMs) for two iterations using Position Specific Iterative BLAST (PSI-BLAST [38]). The options for using BLAST+ [39] are as follows:
In our study, Pseudo-PSSM (PsePSSM) is employed to retain information in PSSM, and its basic idea is to consider the pseudo-amino-acid composition in PSSM [40]. This operation aims to get vectors that satisfy the algorithms and involves two steps.
The first step is the process of standardizing the PSSM. The formula is shown below:
The second step is to use the standardized matrix to generate Pseudo-PSSM (PsePSSM).
where lag denotes the distance between one residue and its neighbors.
Finally, the standardized PSSM matrix is transformed into a 20 + 20
Support Vector Machine (SVM) [41] is a supervised learning
algorithm for classification. As a generalized linear classifier, the purpose of
the support vector machine is to find the maximum boundary hyperplane as the
decision boundary, so as to complete the classification task. SVM is widely used
in classification, regression, and other tasks [42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52]. Since the dataset is
linearly non-separable, the SVM with Gaussian kernel function (RBF) is employed
as the fitting algorithm. C is a crucial parameter in support vector machines.
The penalty factor, or error tolerance, is denoted by the C. The penalty SVM
receives in the case of classification error is positively connected with the C.
In addition, gamma (
Random forest (RF) [53] is an ensemble machine learning technique that trains and predicts samples using several trees, which has found many successful applications in the field of bioinformatics [54, 55, 56, 57, 58, 59, 60, 61, 62]. RF consists of many decision trees that are not related to each other. When a sample is input into RF for the classification task, the sample will be judged by each decision tree in the forest and the classification result will be obtained. The final output of the RF is a combination of the results of each decision tree in the forest. Plenty of parameters affect the RF, among which the most influential ones include the number of subtrees to be built, and the maximum growth depth of trees. In the same way, grid search is used to find suitable values for these parameters.
K-Nearest Neighbor (KNN) determines the categories of input samples based on the most similar samples in the feature space, which is one of the most commonly used machine learning algorithms. K value is the only parameter that the KNN algorithm needs to specify, so it has a significant effect on the result. In KNN algorithm, each sample can be represented by its nearest K neighboring values. The smaller the k value is, the less the approximate error of learning is, because a small k value can make the prediction result of the algorithm only be affected by the training instance that is close to the input instance. Again, we use grid search to find the appropriate K value.
Extreme Gradient Boosting (XGBoost) is another advance machine learning model [63]. XGBoost has been successful in a variety of machine learning competitions [64] as well as in other fields [65, 66, 67, 68]. It is a tool for large-scale parallel trees boosting. XGBoost has the advantages of higher accuracy and greater flexibility. The parameters that affect the XGBoost effect mainly include learning rate, minimum loss reduction required by leaf node splitting, maximum depth of the tree, and minimum weight of the leaf node. Again, use grid search to adjust these parameters.
Model stacking is a strategy of ensemble learning. The basic idea of ensemble learning is to combine multiple classifiers, and the errors encountered by one weak classifier are highly likely to be corrected by other weak classifiers. Combining multiple models can produce a model with better performance and stronger generalization ability. Typical integration strategies are bagging, boosting, stacking, and voting [69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80]. The use of bagging as an integration strategy is mainly to reduce the generalization error of models, which is achieved by combining multiple models. Bagging is implemented by using different bootstrap samples to train different models. When testing the sample input, the output of each model is voted to get the final result. Boosting’s idea is to combine a series of averagely performing models using particular cost functions. Majority voting includes both soft and hard voting. Hard voting is to make statistics on the predicted result label of the base model and take the result with more occurrences as the final result, while soft voting uses the predicted probability of the base model instead of the predicted result label to complete the voting mechanism. In our study, the integration strategy we choose is stacking. In order to prevent model overfitting, we use a simple model, logistic regression, to make the final prediction. The workflow of this method is shown in Fig. 1.
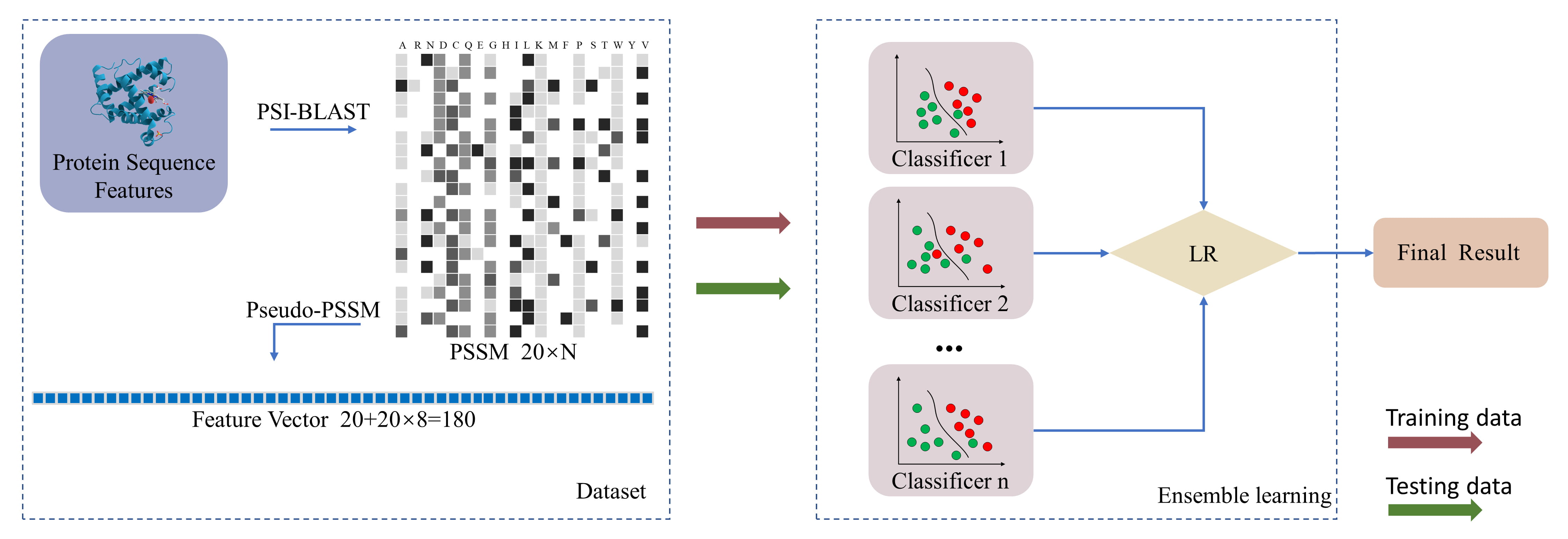 Fig. 1.
Fig. 1.The MSF workflow.
The evaluation of the result is shown in four standard measurements, Accuracy (ACC), Matthew’s correlation coefficient (MCC) [43, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92], Sensitivity (SN), and Specificity (SP). Their formulas are as follows:
where TP, TN, FN, and FP denote the number of true positive, true negative, false negative, and false positive, respectively.
We conduct two types of experiments to test model stacking frameworks. Firstly, predictors of four single classifiers are built by RF, XGBoost, KNN, and SVM, respectively. We compare their performance on the same independent dataset, and the results are shown in Table 2. Then, three classifiers using different combinations of base models are constructed: MSF (SVM, XGBoost), MSF (SVM, XGBoost, KNN), and MSF (SVM, XGBoost, KNN, RF). They employ SVM + XGBoost, SVM + XGBoost + KNN, SVM + XGBoost + KNN + RF as the base classifiers, respectively. Next, LR is employed as the last layer of the model and the results of these base classifiers are fed into the final prediction. The results are shown in Table 3. The ROC curve with AUC values and PR curve are shown in Fig. 2.
| SN (%) | SP (%) | ACC (%) | MCC | |
| RF | 93.20 | 91.58 | 91.86 | 0.7558 |
| XGBoost | 91.23 | 93.97 | 93.44 | 0.8057 |
| KNN | 90.50 | 94.75 | 93.89 | 0.8203 |
| SVM | 89.79 | 96.66 | 95.14 | 0.8599 |
| SN (%) | SP (%) | ACC (%) | MCC | |
| MSF (SVM, XGBoost) | 91.44 | 95.98 | 95.02 | 0.8549 |
| MSF (SVM, XGBoost, KNN) | 91.71 | 96.82 | 95.70 | 0.8756 |
| MSF (SVM, XGBoost, KNN, RF) | 91.24 | 96.81 | 95.59 | 0.8725 |
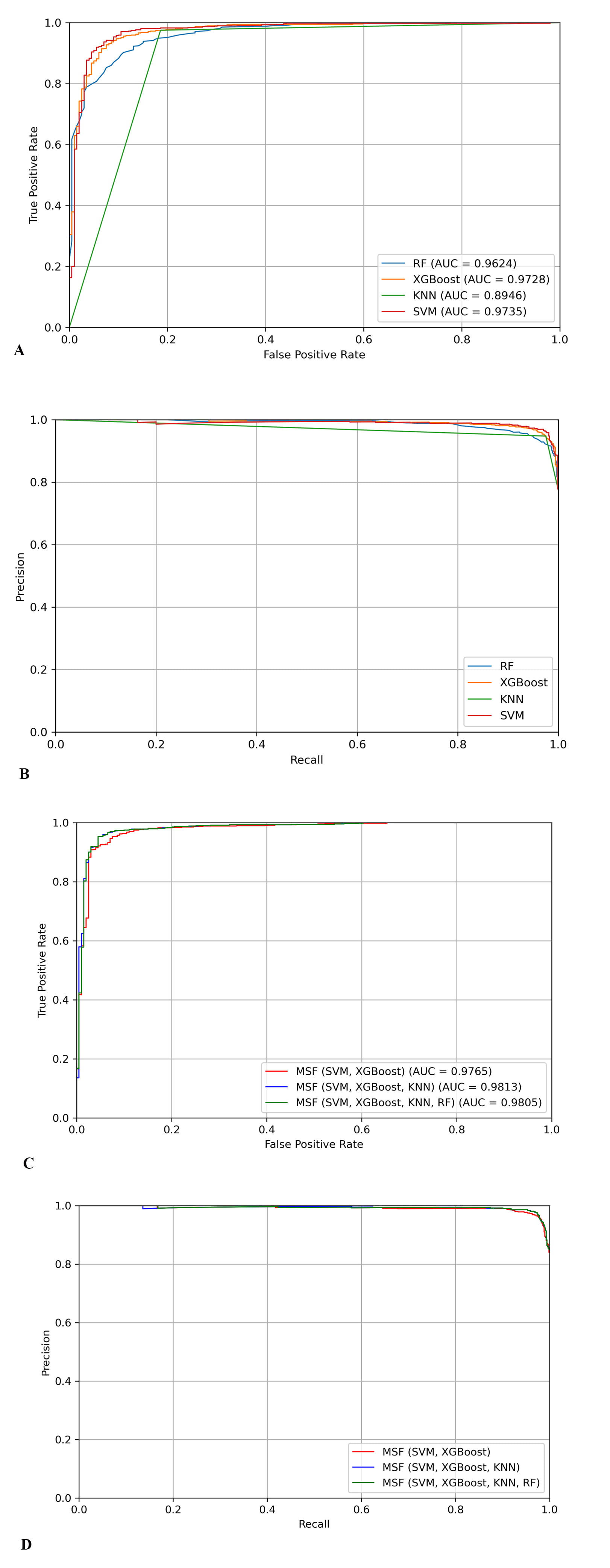 Fig. 2.
Fig. 2.The ROC and PR curves of different models. (A) The ROC curves of single classifiers. (B) The PR curves of single classifiers. (C) The ROC curves of using different base classifiers. (D) The PR curves of using different base classifiers.
Table 2 shows that the predictor that uses SVM, which achieves the best performance of MCC = 0.8599, ACC = 95.14, SP = 96.66, and SN = 89.79%. The RF performs the worst results of MCC = 0.7558, ACC = 0.9186%, SP = 91.58, and SN = 93.20. The gaps of MCC, ACC, SP, and SN between the worst and best ones are 3.28%, 0.1041, –3.41% and 5.08%, respectively. Fig. 2 also shows that SVM has the best performance and highest AUC values.
Table 3 shows that the fourth predictor is ahead of the others on four measures. In all evaluative measurements, ensemble model is better than a single predictor. In Fig. 2, comparing the AUC values of MSF and single classifiers, we also find that MSF has better performance. We believe that ensemble learning performs better than single classifiers because ensemble learning can combine different classifiers flexibly and make better use of the advantages of classifiers. In addition, different base classifiers may have different feature representations for the same data, resulting in the effect of mutual error correction, thus obtaining better performance. Compared to the first predictor, the second predictor obtain better performance by adding KNN which performs second-best among the four single classifiers. RF reduces the overall prediction performance of ensemble model. The results show that a weak base classifier may lead to the effect of model stacking.
To further demonstrate the statistical significance of MSF, we perform t-test analysis on MSF and single classifiers including RF, XGBoost, KNN, and SVM.
The p-value of the t-test are shown in Table 4. In addition, log (0.05/p-value) visually shows the difference between p-value and 0.05. The larger the value is, the more significant the difference is.
| p-value | log (0.05/p-value) | |
| RF | 3.8458 |
14.12 |
| XGBoost | 6.1513 |
4.91 |
| KNN | 9.6691 |
1.71 |
| SVM | 7.7435 |
0.81 |
The results show that the p-values of all algorithms are less than 0.05, indicating that the effects of stacked model framework and single classifier are significantly different and statistically significant. In addition, RF is the algorithm with the most significant difference from the stack model.
In order to further verify that model stacking is an appropriate integration strategy for protein classification problems, the majority voting-based method using SVM, XGBoost, and KNN is tested on the independent dataset. It contains two types of hard voting and soft voting. The results can be found in Table 5.
| SN (%) | SP (%) | ACC (%) | MCC | |
| Hard voting (SVM, XGBoost, KNN) | 91.89 | 95.85 | 95.02 | 0.8546 |
| Soft voting (SVM, XGBoost, KNN) | 92.86 | 95.73 | 95.14 | 0.8576 |
| MSF (SVM, XGBoost, KNN) | 91.71 | 96.82 | 95.70 | 0.8756 |
The gaps of MCC, ACC, SP, and SN between the Hard voting and Soft voting are 0.12%, –0.97%, –0.003%, and –0.12%, respectively. On the same independent dataset, MSF ranks first except for the SN measurements.
When the model is applied, the results of the system may fluctuate due to changes in the data. In order to judge the severity of the model changes, we conduct stability tests. We conduct five times of 5-fold cross-validation to test the stability of the algorithm, and calculate the mean and standard deviation of four metrics including SN, SP, ACC and MCC, as shown in Table 6.
| SN | SP | ACC | MCC | |
| RF | 92.96 |
91.48 |
91.70 |
0.7514 |
| XGBoost | 90.65 |
94.25 |
93.53 |
0.8090 |
| KNN | 89.06 |
95.22 |
93.91 |
0.8223 |
| SVM | 89.41 |
96.57 |
94.98 |
0.8554 |
| MSF (SVM, XGBoost, KNN) | 91.50 |
96.63 |
95.50 |
0.8697 |
The results show that MSF achieves the best mean performance except that the SN value is worse than that of RF. In stability, MSF’s four metrics rank 2,5,3, and 3 respectively among all classifiers.
To further test our method’s sophisticated and superior performance, we compare other existing works under the same data set. These models include ET-CNN [17] and ET-GRU [20].
Table 7 shows results of comparisons between our model and other approaches. It is easy to observe that MSF (SVM, XGBoost, KNN) ranks first with ACC = 95.7, MCC = 0.88, SN = 91.7%, and SP = 96.8%.
| SN (%) | SP (%) | ACC (%) | MCC | |
| ET-CNN | 80.3 | 94.4 | 92.3 | 0.71 |
| ET-GRU | 79.8 | 95.9 | 92.3 | 0.77 |
| MSF (SVM, XGBoost, KNN) | 91.7 | 96.8 | 95.7 | 0.88 |
We believe that the main reason why ET-MSF performs better than ET-CNN and ET-GRU may be that our algorithm is more suitable for the task. Due to the characteristics of the neural network, only a large number of samples can make the network better fitting. However, the dataset of electron transport protein is relatively small, which limits the growth of neural network model size and leads to poor model effect. Therefore, using the ensemble strategy to combine machine learning classifiers can have a better effect.
We provide a simple web server that can be freely accessible at http://82.156.89.65/ to allow readers to evaluate and use our approaches online. The online version of ET-MSF uses the Java language and the Spring Boot framework. ET-MSF can be used by biologists to identify electron transport proteins online. Biologists can obtain the model’s prediction probability by simply entering the protein’s amino acid sequence(s) in a standard FASTA file format. Biologists can also download our public datasets and models at https://github.com/Kinkou626/ET-MSF and run them on own computer.
In our study, a model of stacking framework is proposed to extract the features of protein sequences and identify electron transporter proteins by integrating multiple classifiers. Previously, the model of stacking framework has not been applied to electron transporter protein recognition tasks. We use an independent dataset to evaluate model. Model of stacking frameworks using different base model combinations are compared experimentally. Comprehensive experiments show that the model of stacking framework via SVM, XGBoost, and KNN is the best model, which achieve the ACC and MCC values of 95.70% and 0.8756 respectively. Compared with existing methods, MSF also achieves significant improvements in all measurements. Our method will be an effective bioinformatics tool. And it can also be used to recognize protein functions in other types of proteins.
YW did the experiments and wrote the manuscript. YW, QP, XL, and YD designed the method. YW, QP, XL, and YD revised the manuscript. All authors have read and approved the final manuscript.
Not applicable.
Not applicable.
The work was supported by the National Natural Science Foundation of China (No. 61922020), the Sichuan Provincial Science Fund for Distinguished Young Scholars (2021JDJQ0025), and the Special Science Foundation of Quzhou (2021D004).
The authors declare no conflict of interest.