
Background: Transposable elements (TEs) are the largest component of the genetic material of most eukaryotes and can play roles in shaping genome architecture and regulating phenotypic variation; thus, understanding genome evolution is only possible if we comprehend the contributions of TEs. However, the quantitative and qualitative contributions of TEs can vary, even between closely related lineages. For palm species, in particular, the dynamics of the process through which TEs have differently shaped their genomes remains poorly understood because of a lack of comparative studies. Materials and methods: We conducted a genome-wide comparative analysis of palm TEs, focusing on identifying and classifying TEs using the draft assemblies of four palm species: Phoenix dactylifera, Cocos nucifera, Calamus simplicifolius, and Elaeis oleifera. Our TE library was generated using both de novo structure-based and homology-based methodologies. Results: The generated libraries revealed the TE component of each assembly, which varied from 41–81%. Class I retrotransposons covered 36–75% of these species’ draft genome sequences and primarily consisted of LTR retroelements, while non-LTR elements covered about 0.56–2.31% of each assembly, mainly as LINEs. The least represented were Class DNA transposons, comprising 1.87–3.37%. Conclusion: The current study contributes to a detailed identification and characterization of transposable elements in Palmae draft genome assemblies.
Eukaryotic genomes are known to be densely populated with different types of repetitive elements, including tandem repeats [1] and transposable elements (TEs) [2]. TEs were first characterized in plant genomes over 65 years ago by B. McClintock, who discovered genes that move from one chromosome to another and, in so doing, affect the phenotype of the host organism [3, 4]. Thousands or even tens of thousands of TE families exist in plants [5]. They have conquered thousands of different families in the plant kingdom [6], making up anywhere from 14% of a plant’s genome (as in Arabidopsis thaliana[7]) to over 80% (as in maize [8, 9]. Plants are thus the front line for investigating the impact of TEs on genome structure and gene expression. Notably, TEs can generate genetic diversity upon which selection can act, and this can be leveraged for various purposes in plant breeding programs. Recent insertions of TE families have proven to be particularly helpful in better understanding the evolutionary mechanisms involved in species differentiation [10].
TEs are classified into two major categories based on the mechanism of transposition [11]. Both classes consist of assorted subdivisions, orders, and superfamilies, as described in [12]. Class I LTR retrotransposons (LTR-RTs) represent by far the majority of TEs harbored in plant genomes [13], primarily composed of two superfamilies, Ty1/Copia and Ty3/Gypsy [12], which are differentiated based on the order of their coding domains and evolutionary divergence [14]. Class I TEs replicate via a copy-and-paste mechanism involving an RNA intermediate, whereby TE mRNA is translated into its associated proteins, including a reverse transcriptase that converts the intermediate into DNA, which is then re-inserted into the genome to generate a new copy. Other retrotransposon lineages include long and short interspersed elements (LINEs, SINEs) and the less common Penelope elements [15]. Class II TEs, or “cut-and-paste” elements, mobilize themselves using an element-encoded transposase that mediates excision and transposition of the parent element from one position to another. Terminal inverted repeat (TIR) elements are the most common subclass of so-called cut-and-paste DNA transposons [16]. Other Class II elements common in plant genomes are Helitrons, which are generally less abundant than cut-and-paste TIR transposons and use a rolling circle form of replication [17].
TEs increase their copy number within a host genome through transposition, while the host often represses their activity through epigenetic mechanisms such as RNA and chromatin-mediated silencing [18]. Once integrated into a host genome, each element is subject to mutation and to a wide array of rearrangements including internal deletions, truncations, and nested insertions. Environmental stresses (cold, heat, UV light, pathogen attack, etc.), including tissue culture stress, can cause reactivation of a variable fraction of the TE population; such reactivation is thought to contribute to the host’s short-term response to changing environmental conditions [19, 20]. In tissue culture processes specifically, well-known triggers of LTR-retrotransposon remobilization [21] have been demonstrated in plants such as rice, tobacco, and barley [22, 23, 24]. Such investigations have confirmed that TEs can contribute to somaclonal variation and promote the emergence of altered phenotypes [25].
The major members of the palm family (Arecaceae or Palmae) are considered among
the tallest domesticated trees and the longest-lived monocotyledonous species
[26]. Palm trees are often used as landscape plants; they are also of
considerable economic importance, widely cultivated in arid and semi-arid regions
from North Africa through the Middle East and the Indus Valley. Among cultivated
palms, the greatest quantity of plantation area (17 million hectares) is given
over to oil palms in the genus Elaeis, producing 50 million tons of palm
oil annually. This genus comprises two species, Elaeis guineensis, and
Elaeisoleifera, which are responsible for about 33% of
vegetable oil and 35% of edible oil produced worldwide [27]. The earliest
recorded cultivation of the date palm Phoenix dactylifera occurred in
3700 BCE in the area between the Euphrates and the Nile River [28]. About 5000
date palm cultivars exist around the world [29], and they are an essential
species in drought and saline-affected regions, particularly Saudi Arabia, which
grows
In the present work, genome-wide annotation of TEs was conducted in the aforementioned species using their publicly available genome assemblies. This process involved combining several approaches for the identification and annotation of TEs based on structural features, inherent repetitiveness (de novo), and similarity to elements within existing reference libraries (homology-based) [33]. We additionally built a TE reference database to characterize the compositions of palm genome assemblies and compare their respective TE populations. The comprehensive detection and annotation of TEs is still an open topic in the area of bioinformatics [34], and this analysis provides insights into TE annotation, especially in complex genomes like those of plants.
Draft genome sequences for four palm tree species (P. dactylifera, Cocos nucifera, Calamus simplicifolius, and E. oleifera) were selected for the detection, annotation, and analysis of TEs. These sequences have been assembled at the scaffold level according to the genomic resources of the National Center for Biotechnology Information (NCBI). The assembly sizes were 555.61 Mb, 2102.42 Mb, 1960.81 Mb, and 1402.73 Mb, respectively. Genome sequences were downloaded in FASTA format from the Genomes FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/) and corresponded to the accession numbers GCA_000413155.1 [30], GCA_006176705.1 [32], GCA_900491605.1 [35] and GCA_000441515.1 [27], respectively. Draft completeness was assessed using the Benchmarking Universal Single-Copy Orthologs (BUSCO) v3.0.2 [36], which contained 4104 genes.
A combination of multiple approaches was employed to identify TEs in the four palm draft genome sequences: (i) signature-based identification of TEs, (ii) de novo identification of TEs, and (iii) similarity-based identification [33]. A flowchart describing our overall approach is given in Fig. 1.
 Fig. 1.
Fig. 1.Flowchart for de novo identification of canonical TE sequences using structural, similarity, and homology-based approaches.
In signature-based identification, candidate LTR-RTs were identified by the LTRharvest [37] from Genome-Tools v1.6.1 software [38], which searched the input sequences for direct repeats (LTRs) separated by at least 1000 bp and flanked by apparent target site duplications (TSDs). Default settings were employed with the following exceptions: -motif tgca -motifmis 1 -minlenltr 100 -maxlenltr 3500 -mintsd 2. The program LTRdigest [39] was applied to recognize coding regions and primer binding sites within the predicted LTR-RTs; this tool annotated protein-coding domains in the sequence bracketed by each putative element’s LTRs, specifically using HMMER3 [40] to identify homologs to a set of TE-related pHMMs from the Pfam [41] and GyDB databases [42]. Finally, the EMBOSS (v6.6.0) [43] utility getorf was used to annotate additional ORFs that did not overlap with LTRdigest predictions, considering only those longer than 100 amino acids.
For detecting non-LTR-RTs, we next masked the genome sequence to avoid hits with reverse transcriptase domains already identified. Next, the getorf tool from EMBOSS v6.4.0.0 [43] was employed to extract ORFs from the masked genome sequence. A minimum ORF size of 500 bp was used to accommodate the APE domain (97% of inspected non-LTR elements have sizes between 600 and 800 bp). Finally, we applied MGEScan-nonLTR (v4.0) [44] with default parameters. This program is a generalized hidden Markov model (GHMM) [44] that uses three states to represent two protein domains and the inter-domain linker regions encoded in non-LTRs, the scores for which are evaluated by Phmm (for protein domains) and Gaussian Bayes classifiers (for linker regions).
Putative Class II transposons can be divided into two subclasses: (1) terminal inverted repeat (TIR) elements, which are flanked by TIRs of various lengths and produce TSDs of various lengths upon successful integration into the genome sequence, and (2) non-TIR transposons such as Helitrons, which replicate via a rolling-circle mechanism and do not produce TSDs upon integration. Candidates in these subclasses were respectively identified using MiteFinderII [45] and HelitronScanner [46], both executed with default parameters.
The generated candidate LTR-RTs were interrogated for their inclusion in one of
the three recognized superfamilies: Ty1/Copia, Ty3/Gypsy, and Bel/Pao. The
evidence consisted of matches to hidden Markov models (HMMs) and BLAST results
against the Viridiplantae LTR-RT database (retrieved from Repbase and Dfam),
respectively, obtained via nhmmer (-incE 1
To exclude false-positive hits from our putative Class II elements, hits were
queried with BlastN (-evalue 1
Libraries generated as described above were filtered first for duplicates using SeqKit rmdup on the basis of sequence (-s) [48]. To further classify LTR retrotransposons into clades below the superfamily level, and Class II elements, the TEsorter hidden Markov model (HMM) profile-based classifier was used with default settings [49], taking as reference the protein domains found in the REXdb Viridiplantae version of the database [50]. Complete LTR elements were identified based on the presence and order of conserved domains, including capsid (GAG), aspartic protease (AP), integrase (INT), reverse transcriptase (RT), and RNase H (RH) as described in [12]. TEsorter was also used to filter the library of consensus sequences prior to genome sequence annotation, primarily by detecting chimeras or nested elements composed of drastically different types of protein-coding sequences (e.g., transposase and non-LTR reverse transcriptase).
LINE elements were further scrutinized by extracting RT coding regions as identified by TEsorter, over 200 aa were extracted, and fragments were aligned to a reference alignment from Kapitonov et al. [51]. Multiple members of LINE superfamilies could not be verified and thus were classified as unknown LINEs.
Reference TE sequences from palm species (Palmae) were extracted from Dfam (20170127) [52] and RepBase (20181026) [53] using the script ‘queryRepeatDatabase.pl’ supplied with RepeatMasker. After generation, the libraries were merged and used as an input to mask and annotate the assembled genomes. This masking employed (iii) similarity-based identification of TEs via RepeatMasker v.4.1.0 [54], with RMBlast as the search algorithm, Smith-Waterman for alignment, and -cutoff 225. We applied high sensitivity/low-speed search conditions to avoid spurious results: -s, -no_is, -lib, -norna, and exclusion of low complexity regions (-nolow); other parameters were default. Additionally, we counted the copy number of classified elements and determined genome sequence coverage from the RepeatMasker output files (.out), using the one code to find them all script [55] to estimate the fraction of the genome occupied by each TE family.
Finally, the unmasked portion of each genome sequence was scanned using (ii) de novo methodology for TE detection. Namely, RepeatModeler2 [56] was used with default parameters to identify any unclassified TEs missed by structure-based identification approaches. Results obtained will be merged to the reference library for filtration and Final Re-annotation.
The consensus sequences classified as belonging to Ty1/Copia superfamily
elements and containing all five protein-coding domains characteristic of LTR
elements (GAG, AP, INT, RT, RH) were selected for phylogenetic analysis. To
choose for elements more likely to have been recently active, amino acid
sequences translated from RT coding regions were screened for length (
To assess the completeness of each of the four genome assemblies, we adopted the Benchmarking Universal Single-Copy Orthologs (BUSCO) plant lineage dataset, which consists of 1440 single-copy orthologs for the Embryophyta lineage. Among surveyed genome drafts, C. nucifera had the highest BUSCO score (Fig. 2A), with 1335 complete BUSCOs (92.71%); another 2.40% of sequences were fragmented, and 4.93% were considered missing (71 BUSCOs). The BUSCO scores of the C. nucifera, P. dactylifera, and C. simplicifolius assemblies were comparable and higher than those of the E. oleifera assembly.
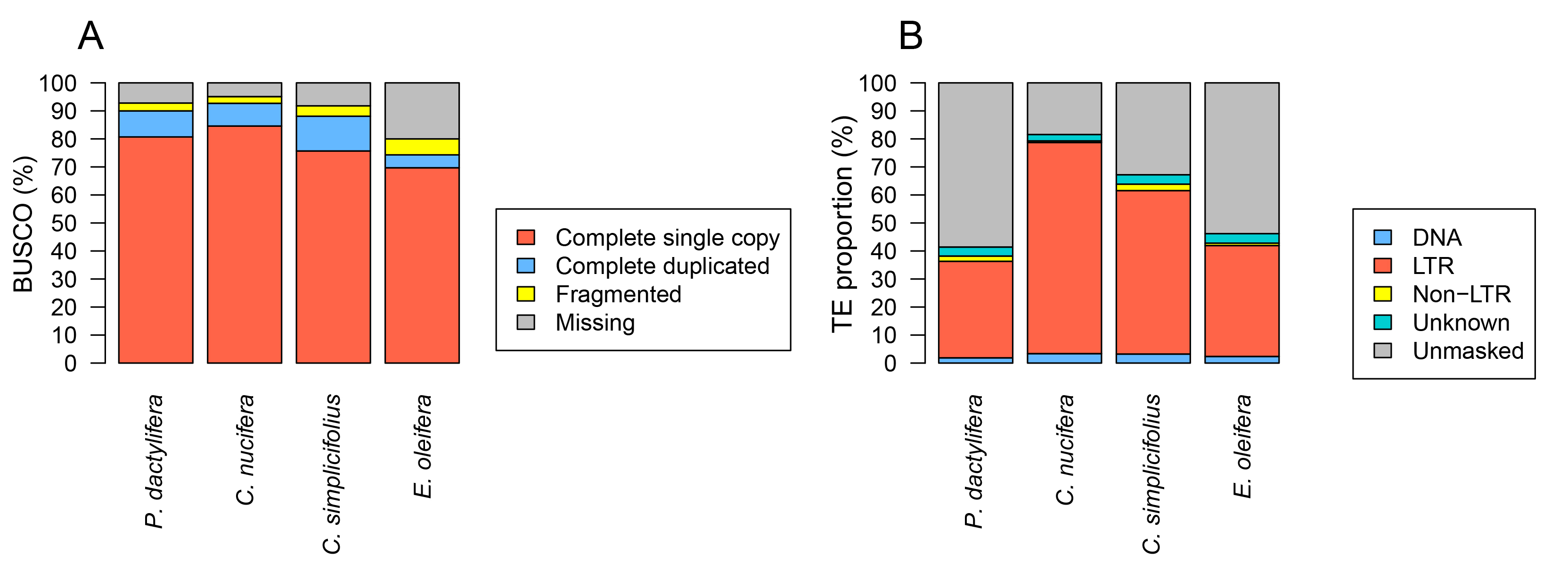 Fig. 2.
Fig. 2.Genome assembly evaluation and TE proportions. The four Palmae assemblies were evaluated using the BUSCO embryophyta_odb9 (A). Breakdown of TE types in the four studied genome drafts (B).
A reference TE library was created by applying a combination of structure-based and homology-based approaches to 335 P. dactylifera, 1473 C. nucifera, 1481 C. simplicifolius, and 777 E. oleifera scaffold sequences. After identifying, and filtering elements, we recorded 3526, 3563, 4542, and 2874 consensus sequences for each species classified as TEs according to the Repbase and Dfam reference libraries. Taken together, this library of TE candidates encompasses both Class I (LTR-RTs, non-LTR retrotransposons) and Class II elements (TIR elements, Helitrons, and MITEs), which are provided in Supplementary files1–4.
The contributions of TEs to the P. dactylifera, C. nucifera, C. simplicifolius, and E. oleifera assemblies were assessed by utilizing a similarity-based approach (RepeatMasker) to mask the assembled genomes with the generated TE libraries. The annotations produced with the merged Class I and Class II consensus libraries cover 229.91 Mb (41.42%), 1714.54 Mb (81.55%), 1314.23 Mb (67.20%), and 647.67 Mb (46.20%) of the respective assemblies (Table 1 and Fig. 2B). Something to be mindful of in this case, and for other genome sequences, is that assembly size can often differ considerably from the genome sequence size as measured using cytological methods [61]. These palm assemblies range from 70.5% (in P. dactylifera) to 95.65% (C. simplicifolius) of the estimated genome size, indicating that our values may be underestimates of the repetitive content in each genome assembly [62, 35].
| P. dactylifera | C. nucifera | C. simplicifolius | E. oleifera | ||||||
| Type | superfamily | Length (bp) | % | Length (bp) | % | Length (bp) | % | Length (bp) | % |
| Retroelements | 201856186 | 36.33 | 1595980008 | 75.91 | 1190037849 | 60.62 | 567206558 | 40.44 | |
| SINEs | 398931 | 0.07 | 385641 | 0.02 | 128925 | 0.01 | 435797 | 0.03 | |
| Penelope | 0 | 0 | 0 | 0 | 0 | 124114 | 0.01 | ||
| LINEs | 9993702 | 1.81 | 11284518 | 0.54 | 46540438 | 2.30 | 11420847 | 0.81 | |
| L1/Tx1 | 6173483 | 1.11 | 8681691 | 0.41 | 17421596 | 0.89 | 9917998 | 0.71 | |
| RTE/Bov-B | 1593639 | 0.29 | 1507386 | 0.08 | 3998649 | 0.20 | 1290079 | 0.09 | |
| Unknown LINE | 2226580 | 0.41 | 1095441 | 0.05 | 25120193 | 1.21 | 212770 | 0.01 | |
| LTR Elements | 191463553 | 34.46 | 1584309849 | 75.36 | 1143368486 | 58.31 | 555225800 | 39.58 | |
| BEL/Pao | 67751 | 0.01 | 283875 | 0.014 | 816730 | 0.04 | 218620 | 0.016 | |
| Copia | 121027091 | 21.78 | 1145720152 | 54.5 | 613029994 | 31.26 | 460711762 | 32.84 | |
| Gypsy | 68345431 | 12.30 | 378742685 | 18.01 | 512209691 | 26.12 | 53149036 | 3.79 | |
| Other LTR | 2023280 | 0.36 | 59563137 | 2.82 | 17312071 | 0.93 | 41146382 | 2.93 | |
| DNA transposons | 10375721 | 1.87 | 70824484 | 3.37 | 83012700 | 3.23 | 33270353 | 2.37 | |
| CMC | 1151924 | 0.21 | 5213372 | 0.25 | 4384176 | 0.22 | 5538032 | 0.39 | |
| hAT | 2903056 | 0.52 | 5501815 | 0.26 | 10644454 | 0.54 | 5976535 | 0.43 | |
| Mutator | 1228176 | 0.22 | 5940469 | 0.28 | 3756180 | 0.19 | 5488336 | 0.39 | |
| PIF-Harbinger | 514900 | 0.09 | 1211375 | 0.06 | 1935203 | 0.10 | 1134634 | 0.08 | |
| Tc1/Mariner | 54225 | 0.01 | 44857 | 0.002 | 484275 | 0.02 | 44351 | 0.003 | |
| Helitron | 4051817 | 0.73 | 52023577 | 2.47 | 60712540 | 2.12 | 14100492 | 1.01 | |
| Other TIR | 234964 | 0.042 | 827221 | 0.04 | 803905 | 0.04 | 627426 | 0.04 | |
| Unknown | 17922924 | 3.22 | 47804369 | 2.27 | 77188695 | 3.35 | 47554663 | 3.39 | |
| Total | 229918172 | 41.42 | 1714547063 | 81.55 | 1314236312 | 67.20 | 647671027 | 46.20 | |
Class I elements, or retroelements, formed the majority of the TE component in the aforementioned four assemblies, making up 36.33%, 75.91%, 60.62%, and 40.44%, respectively; this is in line with other plant genomes. The remainder consisted of DNA transposons (1.87%, 3.37%, 3.23%, and 2.37%), and unclassified elements (3.22%, 2.27%, 3.35%, and 3.39%).
The repetitive elements detected in P. dactylifera, C. nucifera, C. simplicifolius and E. oleifera were classified into five main categories: (1) LTR-RTs identified using Genome-tools (structure-based) supplemented with open reading frame (ORF) detection, which generated a total of 2078, 1101, 1586, and 665 elements in the respective genome sequence; (2) Non-LTR retrotransposons identified using MGEScan-nonLTR (structure-based) to align reference reverse transcriptase sequences with the ORFs predicted from masked genome sequence, which yielded 77, 15, 95, and 14 consensus sequences, respectively; (3) Non-autonomous DNA elements (MITEs and degraded DNA transposons) identified by MiteFinderII (structure-based) on the basis of TIRs and target site duplications (TSDs), yielding 116, 187, 382, and 139 consensus sequences, respectively; (4) Helitron-like sequences identified using HelitronScanner (structure-based), which resolved 51, 133, 131, and 69 consensus sequences, respectively, and (5) TE candidates left undetected by the above tools (structure-based) that were interrogated by RepeatModeler2 (de novo), which yielded 1179, 2026, 2232, and 1949 consensus sequences, including both classified TEs and unknown repeats.
The LTR-RT detection process, which identified elements consisting of two relatively intact LTRs and flanking TSDs, returned 11120, 94186, 116725, and 35452 raw hits for each of P. dactylifera, C. nucifera, C. simplicifolius, and E. oleifera. These candidates accounted for 7.45%, 28.46%, 28.35%, and 15.49% of the respective assemblies. Discarding false positive candidates decreased the counts of putative full-length LTR-RTs to 2078, 1101, 1586, and 665, respectively; these hits showed at least one specific protein domain and range in size from 202 to 18386 bp. Of predicted candidates, full-length LTR-RTs comprise 18.91% (7.843 Mb), 1.56% (9.36 Mb), 2.02% (11.28 Mb), and 1.83% (3.99 Mb) respectively.
The candidate LTR-RTs were classified into seven superfamilies according to the Wicker classification system [12] as represented in the RepBase and Dfam databases (Table 2). In all four studied assemblies, the most abundant LTR-RT superfamilies were Ty1/Copia and Ty3/Gypsy, respectively accounting for 574–1298 and 31–736 consensus sequences.
| Superfamily | P. dactylifera | C. nucifera | C. simplicifolius | E. oleifera |
| Ty3/Gypsy | 736 | 218 | 418 | 31 |
| Ty1/Copia | 1298 | 835 | 1148 | 574 |
| BEL/Pao | - | - | 1 | - |
| Unknown | - | 1 | - | 1 |
| Other LTR | 44 | 47 | 19 | 59 |
| Total | 2078 | 1101 | 1586 | 665 |
We then evaluated the distribution of predicted protein-coding sequences within LTR-RTs in order to gain insight into their possible associations. The total proteins identified in each assembly and their breakdowns by domain are summarized in Table 3. Most putative LTR-RTs featured the gag-integrase-reverse transcriptase protein domain order characteristic of Ty1/Copia elements.
| Protein | P. dactylifera | C. nucifera | C. simplicifolius | E. oleifera |
| gag-asp proteas | 32 | - | 2 | 1 |
| asp protease | 2 | - | - | - |
| asp protease 2 | 44 | - | 1 | 2 |
| Asp | - | - | - | - |
| AP2 | 3 | - | - | - |
| gag pre-integrs | 189 | 619 | 955 | 114 |
| Retrotrans gag | 152 | 129 | 57 | 51 |
| Retrotran gag 2 | 226 | 621 | 1206 | 102 |
| Retrotran gag 3 | 34 | 3 | 8 | 9 |
| zf CCHC | 96 | 94 | 524 | 27 |
| zf H2C2 | 12 | - | 2 | 5 |
| zf-CCHC 2 | 3 | - | - | 1 |
| zf-CCHC 3 | 1 | 1 | 64 | - |
| zf-CCHC 4 | 6 | - | 3 | - |
| zf-RVT | 20 | 4 | 17 | 1 |
| RNase H | 14 | 98 | 11 | 11 |
| Transposase 28 | 11 | - | 3 | 10 |
| RVP | 2 | - | - | 2 |
| RVP 2 | 44 | - | 5 | 7 |
| rve | 317 | 721 | 1143 | 137 |
| rve 3 | 36 | 35 | 510 | - |
| RVT 1 | 142 | 213 | 121 | 59 |
| RVT 2 | 346 | 1256 | 1971 | 381 |
| RVT 3 | 54 | 206 | 74 | 37 |
| Exo endo phos 2 | - | - | 1 | - |
| DUF4219 | 54 | 145 | 680 | 8 |
| DUF4413 | 3 | 1 | 1 | 1 |
| DBD Tnp Mut | 1 | 1 | - | - |
| SQAPI | 1 | - | - | - |
| Total | 1845 | 4147 | 7359 | 964 |
Ty1/Copia and Ty3/Gypsy consensus sequences were further classified into lineages using TEsorter. Within those groups, consensus sequences identified as complete (containing hits to each of the characteristic LTR proteins mentioned previously) were respectively classified into nine and six lineages. The most represented lineages in Ty1/Copia were Angela (2.24%–23%) and SIRE (0.74%–10.5%) (Table 4), while amongst the Ty3/Gypsy consensus sequences, Retand elements (0.85%–4.68%) were of the highest coverage in each assembly (Table 5).
| P. dactylifera | C. nucifera | C. simplicifolius | E. oleifera | |||||||||
| Lineage | Number | Length (bp) | % | Number | Length (bp) | % | Number | Length (bp) | % | Number | Length (bp) | % |
| Ale | 22 | 1715800 | 0.31 | 7 | 4371010 | 0.21 | 11 | 3718088 | 0.19 | 12 | 2875745 | 0.21 |
| Alesia | 0 | 0 | 0.00 | 0 | 0 | 0.00 | 0 | 0 | 0.00 | 1 | 152319 | 0.01 |
| Angela | 20 | 12427770 | 2.24 | 209 | 483611473 | 23.00 | 15 | 46492261 | 2.37 | 43 | 93423004 | 6.66 |
| Ikeros | 8 | 1494089 | 0.27 | 1 | 5827062 | 0.28 | 7 | 39942330 | 2.04 | 1 | 411161 | 0.03 |
| Ivana | 16 | 1155775 | 0.21 | 8 | 4216573 | 0.20 | 33 | 10837639 | 0.55 | 2 | 488189 | 0.03 |
| SIRE | 20 | 14518688 | 2.61 | 11 | 131534107 | 6.26 | 167 | 205800088 | 10.50 | 3 | 10437148 | 0.74 |
| TAR | 9 | 1616183 | 0.29 | 334 | 2736206 | 0.13 | 571 | 18592348 | 0.95 | 3 | 1535602 | 0.11 |
| Tork | 22 | 1835464 | 0.33 | 4 | 21440106 | 1.02 | 8 | 31829327 | 1.62 | 2 | 4275036 | 0.30 |
| Bianca | 0 | 0 | 0.00 | 9 | 653602 | 0.03 | 14 | 3292770 | 0.17 | 7 | 99955 | 0.01 |
| Total | 117 | 34763769 | 6.26 | 583 | 654390139 | 31.13 | 826 | 360504851 | 18.39 | 74 | 113698159 | 8.11 |
| P. dactylifera | C. nucifera | C. simplicifolius | E. oleifera | |||||||||
| lineage | Number | Length (bp) | % | Number | Length (bp) | % | Number | Length (bp) | % | Number | Length (bp) | % |
| Retand | 8 | 4697729 | 0.85 | 98 | 97895329 | 4.66 | 33 | 91750919 | 4.68 | 8 | 20841807 | 1.49 |
| Tekay | 6 | 1958062 | 0.35 | 1 | 4292793 | 0.20 | 8 | 32088493 | 1.64 | 2 | 2593648 | 0.18 |
| Galadriel | 3 | 267640 | 0.05 | 0 | 0 | 0.00 | 5 | 3364634 | 0.17 | 1 | 384216 | 0.03 |
| CRM | 16 | 2486306 | 0.45 | 5 | 7576046 | 0.36 | 2 | 12417699 | 0.63 | 3 | 1894920 | 0.14 |
| Athila | 8 | 4791147 | 0.86 | 0 | 0 | 0.00 | 1 | 5277174 | 0.27 | 3 | 5286853 | 0.38 |
| Reina | 10 | 394591 | 0.07 | 1 | 1631425 | 0.08 | 0 | 0 | 0.00 | 1 | 87725 | 0.01 |
| Total | 51 | 14595475 | 2.62 | 105 | 111395593 | 5.29 | 49 | 144898919 | 7.38 | 18 | 31089169 | 2.21 |
To build a phylogenetic tree of complete Ty1/Copia consensus sequences, we filtered their RT sequences for length, stop codons, and ambiguous regions. We selected the longest contiguous RT regions from consensus sequences categorized as complete by TEsorter. We selected representative lineages of LTR elements that are likely to be more recently active. This yielded 179 sequences for tree construction: 24 from P. dactylifera, 19 from C. nucifera, 136 from C. simplicifolius, and none from E. oleifera. Collectively, these represented 93 SIRE, 52 Ivana, 14 Angela, 8 Tork, 5 TAR, 5 Ikeros, and 2 Ale elements; those that classified into particular groups clustered in well-supported clades with their respective reference elements (SIR classified SIRE, Oryco1-1 for Ivana, Tork4 for Tork, Fourf for TAR, and Sto-4 for Ikeros). The one exception was the Angela and Ikeros complex, which is known to be paraphyletic [50]. Reference elements are denoted with dotted lines on the tree (Fig. 3). C. simplicifolius sequences in general, dominated the tree, but more specifically SIRE and Ivana; these groups featured several low-divergence clades, suggestive of recent activity (Fig. 3). In contrast, recently active SIRE clades are interspersed with more divergent lineages, some composed of elements from the other genomes, suggesting that SIRE, in general, has maintained more activity over evolutionary timescales. Of complete Ty1/Copia consensus sequences in C. nucifera, the bulk were Angela elements (see Table 4), but only a small number of these consensus sequences were represented on the tree, with several having low divergence.
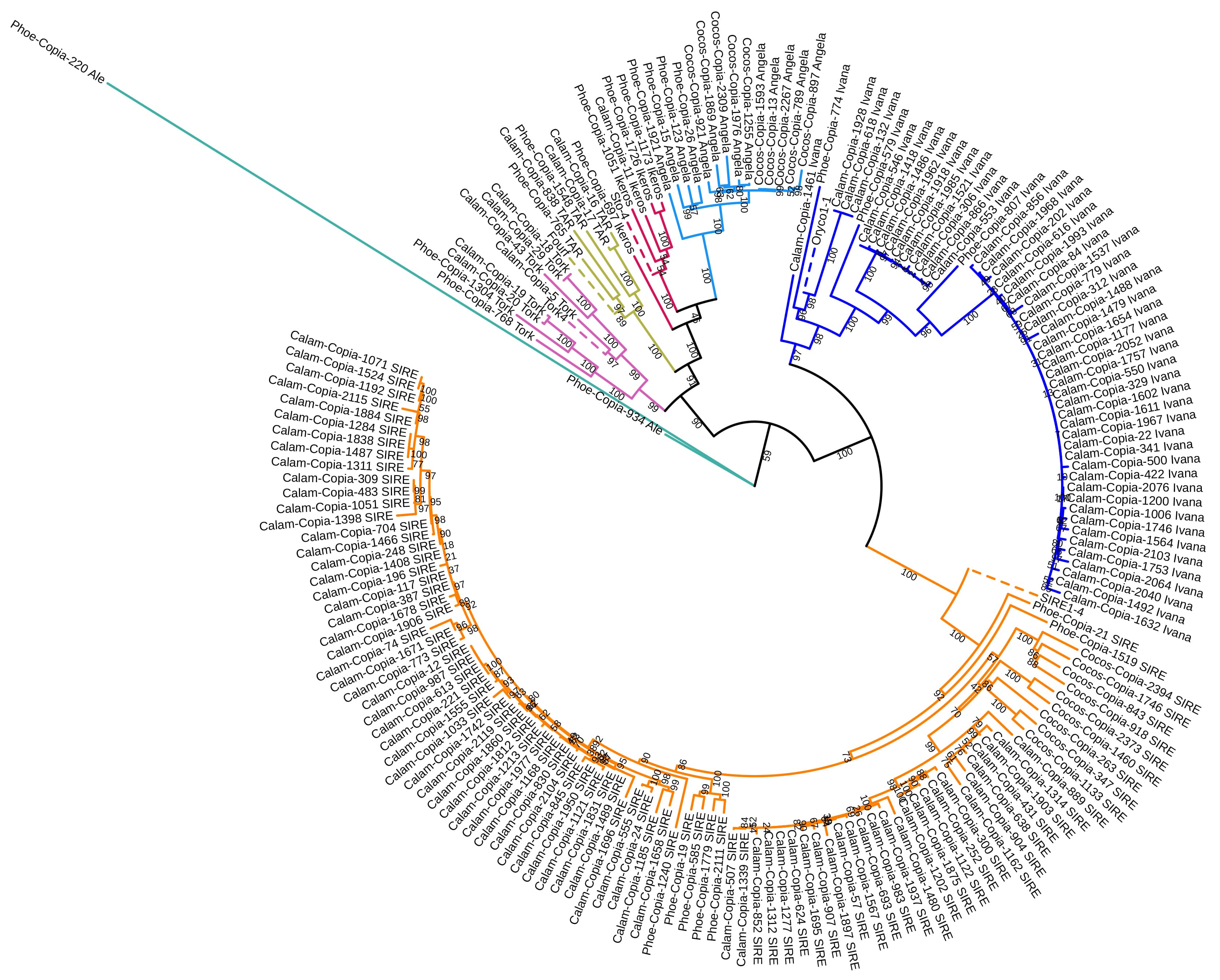 Fig. 3.
Fig. 3.Maximum-likelihood tree of 179 Ty1/Copia elements from the four palm genome assemblies. Major groups are denoted with different coloured branches (aquamarine = Ale, pink = Tork, mustard = TAR, red = Ikeros, light blue = Angela, dark blue = Ivana, Orange = SIRE); reference elements are denoted with dashed lines.
Non-LTRs were identified by applying MGEScan to the LTR-masked genome sequences. This tool discovers all known full-length elements and simultaneously classifies them into the following clades: CR1, I, Jockey, L1, R1, R2, and RTE. Previous studies have classified non-LTR retrotransposons into 11 clades based on the reverse transcriptase phylogeny [63]. The non-LTR retrotransposons we distinguished in palm species are summarized in Table 6. Seven superfamilies were represented in P. dactylifera, four in C. nucifera, seven in C. simplicifolius, and one in E. oleifera. R2 was the only superfamily present in all studied species, while the I superfamily was by far the most abundant when it was present, with 14 occurrences in E. oleifera. These full-length elements covered 205,245 bp, 13,932 bp, 241,692 bp, and 8925 bp of the associated genome sequences; the smallest counts of ORF-conserving elements were identified in E. oleifera.
| Superfamily | P. dactylifera | C. nucifera | C. simplicifolius | E. oleifera |
| CR1 | 9 | - | 8 | - |
| I | 3 | - | 25 | - |
| Jockey | 29 | 2 | 25 | - |
| L1 | 21 | - | 13 | - |
| R1 | 3 | 2 | 1 | - |
| R2 | 7 | 10 | 12 | 14 |
| Rex | - | - | - | - |
| RTE | 5 | 1 | 11 | - |
| Total | 77 | 15 | 95 | 14 |
In investigating Class II TEs, we first identified ubiquitous miniature
inverted-repeat elements (MITEs), characterized by essential structural features
such as TIRs and TSDs, AT-rich sequences, and a lack of transposase coding
capacity. Canonical MITE sequences with TIRs, TSDs, and perfect or near-perfect
structure (inverted repeats with some mismatches) feature a TIR pair (
| Superfamily | P. dactylifera | C. nucifera | C. simplicifolius | E. oleifera |
| hAT | 53 | 51 | 123 | 46 |
| CMC | 10 | 27 | 31 | 16 |
| PIF-Harbinger | 9 | 40 | 95 | 30 |
| Mutator | 34 | 47 | 95 | 33 |
| Tc1/Mariner | - | 1 | - | 1 |
| Other TIR | 10 | 21 | 38 | 13 |
| Total | 116 | 187 | 382 | 139 |
In the P. dactylifera draft assembly, we identified a total of 303 MITE elements, which accounted for 99,906 bp all told; only 116 elements showed significant homology to database entries (RepBase, PMITE), and these belonged to six different superfamilies. In C. nucifera, we identified 189 MITE elements, which accounted for 37,550 bp of the assembly; of these, 187 elements were collectively associated with seven different superfamily definitions. In C. simplicifolius, we identified 390 MITE elements, which accounted for 79,970 bp of the genome sequence; 382 of these elements returned database hits, encompassing six superfamily definitions. Finally, in E. oleifera, we identified a total of 140 MITE elements, which accounted for 27,750 bp; among those, 139 elements were associated with one of seven superfamily definitions. The most abundant superfamily of DNA transposons in all four evaluated assemblies was hAT, represented by 51–123 occurrences.
Finally, we identified Helitron-like sequences using the exhaustive structure-based approach of HelitronScanner, which predicts putative Helitrons based on definitive features by scanning for conserved structural traits: 5’ end with TC, 3’ end with CTAG, and a GC-rich hairpin loop 2–10 nt in front of the CTAG end. This method predicted 51, 133, 131, and 69 elements in P. dactylifera, C. nucifera, C. simplicifolius, and E. oleifera, respectively accounting for 0.09 Mb 0.26 Mb, 0.27 Mb, and 0.13 Mb in total.
After masking the four assemblies, we employed RepeatModeler2 to discover TEs not detected by previous methods, such as TIR elements, then merged those results with the otherwise-predicted libraries into a master library.
An overview of elements detected by RepeatModeler only, namely the number of families representing each superfamily in each assembly, is given in Table 8. The results reveal that retrotransposons, especially LTR-RT, dominate the masked genome sequences of these four palm species.
| P. dactylifera | C. nucifera | C. simplicifolius | E. oleifera | ||
| Type | Superfamily | ||||
| LTR Element | - | 3 | 1 | - | |
| Caulimovirus | 4 | 12 | 5 | 6 | |
| Ty1/Copia | 60 | 179 | 178 | 151 | |
| DIRS/Ngaro | - | 1 | 2 | 4 | |
| Ty3/Gypsy | 90 | 125 | 102 | 71 | |
| ERV1 | - | - | 1 | 3 | |
| ERVK | - | 1 | 2 | - | |
| ERVL | - | - | 1 | - | |
| BEL/Pao | 2 | 6 | 6 | 4 | |
| Cassandra | 2 | - | - | - | |
| Non-LTR/LINE | - | - | - | - | |
| I | - | - | 2 | 1 | |
| L1 | 39 | 111 | 48 | 96 | |
| L2 | 2 | - | 3 | - | |
| R1 | - | 1 | 2 | - | |
| R2 | - | - | 1 | - | |
| CR1 | 1 | 1 | 5 | - | |
| RTE-BovB | 6 | 5 | 9 | 6 | |
| Penelope | - | - | - | 2 | |
| Tad1 | - | 1 | 1 | 1 | |
| SINE | 1 | 1 | 2 | 2 | |
| DNA | 5 | 6 | 3 | 8 | |
| CMC | 16 | 50 | 37 | 56 | |
| PIF-Harbinger | 14 | 15 | 11 | 13 | |
| hAT | 72 | 68 | 45 | 76 | |
| Tc1/Mariner | 2 | 1 | 7 | 1 | |
| Mutator | 24 | 46 | 38 | 49 | |
| Maverick | 2 | - | 1 | 1 | |
| Dada | 1 | - | 1 | 1 | |
| Crypton | 1 | - | - | - | |
| Zisupton | 1 | - | - | - | |
| Kolobok | - | - | 1 | 1 | |
| Academ | - | - | 2 | - | |
| Sola | - | 1 | - | 3 | |
| Helitron | 14 | 17 | 34 | 17 | |
| Unknown | 820 | 1375 | 1681 | 1376 | |
| Total | 1179 | 2026 | 2232 | 1949 |
Overall, the studied palm draft genome assemblies contain different proportions and numbers of DNA-TIR and LTR elements relative to their respective genome sequence sizes. In absolute terms, for each of P. dactylifera, C. nucifera, C. simplicifolius, and E. oleifera, RepeatModeler2 respectively recovered 1179 (0.95 Mb), 2026 (2.11 Mb), 2232 (3.78 Mb), and 1949 (1.87 Mb) classified consensus sequences and 820, 1375, 1681 and 1376 unknown consensus sequences.
We generated repeat libraries for each of the four palm species with available genome sequences to investigate the abundance and characteristics of repeat-derived DNA within this family. This study also facilitates the repeat-masking of DNA and provides a first step towards constructing a comprehensive palm TE catalogue. Our analysis techniques were very conservative, which may have led to an underestimation of ancient and divergent elements; such elements may have been detected as unclassified. To ensure the reliability of our results, we employed a method incorporating both known TEs and signature-based repeat identification tools.
After merging all predicted repeats and performing validation and redundancy removal, we obtained libraries containing 3526, 3563, 4542, and 2874 consensus sequences, respectively, for P. dactylifera, C. nucifera, C. simplicifolius, and E. oleifera. We then merged them into a composite master reference library and re-annotated the four genome assemblies. Doing so revealed repetitive elements as comprising a total of 229.91 Mb (41.42%) in P. dactylifera, 1714.54 Mb (81.55%) in C. nucifera, 1314.23 Mb (67.20%) in C. simplicifolius, and 647.67 Mb (46.20%) in E. oleifera. Lastly, unclassified elements comprised a small proportion of each genome sequence, ranging from 2.27–3.39% in the four genome assemblies. A more detailed breakdown is given in Table 1. All told, the examined draft genomes were similar in terms of overall repetitive content (Fig. 2B), namely that retroelements dominated the assemblies.
A strong predominance of retroelements over DNA transposons is a common feature
of plant genomes [64]. Class I elements constituted 36–75% of the annotated
assemblies in the present study, with LTRs comprising 34–75%. This result was
expected; the larger the plant genome, the greater the chance it contains many
retroelements. For example, retroelements comprise 80–85% of barley and maize
genomes with size
In our phylogenetic investigation of Ty1/Copia elements likely to be recently active, C. simplicifolius dominated the tree with 136 sequences. Notably, although Ivana consensus sequences with full domains made up a small percentage of the assembly, they comprised a high percentage of elements on the tree, with at least two low-divergence clades suggesting recent transposition events. Similarly, C. simplicifolius also contained several low-divergence clades of SIRE elements, although these were interspersed with more divergent lineages. In C. nucifera, unlike the other assemblies, the bulk of complete consensus sequences were from the Angela group, comprising about 23% of the assembly. On the phylogenetic tree, Angela elements presented a low-divergence clade for C. nucifera specifically but otherwise do not seem to have many potentially active families based on consensus sequences, as defined by our filtering metric. Within C. nucifera, recent LTR activity has been reported as dominated by Ty1/Copia in the last 2 million years, with fewer and fewer elements showing evidence of activity when approaching the present [32]. In the P. dactylifera draft genome, we detected and classified TAR/Fourf, Orcyo/Ivana, and SIRE elements, supporting the work of Nouroz and Mukaramin [70]; we also identified four other Ty1/Copia groups. Despite these elements contributing less to the tree overall, the tree contains representatives of nearly all the Ty1/Copia groups detected in the P. dactylifera assembly, including the only two Ale consensus sequences. Both C. simplicifolius and C. nucifera represent the most complete and largest assemblies of the four palm species analyzed, thus probably contributing to their bias of complete consensus sequences analyzed on the tree.
Very few non-LTR retrotransposons have been reported in plants; such elements
appear more abundant in animal genomes [12, 71]. For example, SINEs may comprise
up to
We also found other classes of repetitive elements, such as Class II TEs, to be poorly represented in palm genome sequences, collectively making up 1.87–3.37% of the four annotated assemblies. The most prevalent DNA transposon super-families were the hAT, Mutator, and Helitron elements, likewise being the most abundant in previous studies [74]. In particular, members of the hAT superfamily are found in many monocots, such as the Ac-Ds family in maize [75]. Unlike other DNA transposons, Helitrons are challenging to identify because they require structural-based detection methods rather than homology. In the publication detailing HelitronScanner [46], Xiong et al. reanalyzed the genome sequences of 26 plant species and reported Helitron abundance to cover at most 2–6%, the highest percentage being in maize. In the present study, we observed Helitrons to comprise about 0.73–2.47% of each assembly, with the highest coverage being found in C. nucifera (2.47%) followed by the C. simplicifolius (2.12%) and the lowest percentage reported in P. dactylifera (0.73%).
The findings of this study will provide a valuable resource for further research into palm biology and genomics. While the investigated genome sequences were similar in terms of the content and distribution of the identified repetitive elements, differences were also observed that might be associated with factors such as different evolutionary origins or discrepancies in the assembly stages of these draft genomes. Additional research into repetitive elements in palm genome sequences, perhaps with more complete genome assemblies, would provide more information on and awareness of the genomic features of these economically important plants. Furthermore, the causes and consequences of the high degree of inter-genome variability in the distribution, amount, and relative proportion of TEs are still not wholly understood; it is essential to continue characterizing this critical fraction of eukaryotic genomes. Such characterizations can bring to light evolutionary phenomena, including genomic rearrangements and other dynamic events, that have occurred in the past and may also be underway in contemporary times.
MMM and FHA conceived and designed the experiments; MAI, TAE, BMA, SNA, MSA and MMM carried out the experiments; MAI, SNA, TAE, BMA, FHA and MMM analyzed the data; MAI, SNA, TAE, and MMM wrote the manuscript. All authors reviewed the manuscript.
Not applicable.
The authors would like to thank Guilherme Dias at the Department of Genetics and Institute of Bioinformatics, University of Georgia, for his valuable comments and suggestions to improve the quality of the manuscript. The authors would also thank Amer S. Alharthi at the General Directorate for Funds and Grants (GDFG), King Abdulaziz City for Science and Technology, for his technical support.
This study was supported by the National Centre for Biotechnology, Life Science and Environment Research Institute (Grant 37-1271), King Abdulaziz City for Science and Technology, Saudi Arabia.
The authors declare no conflict of interest.
All data generated or analysed during this study are included in this published article (and its supplementary information files).
TEs, Transposable elements; LINE, Long interspersed element; LTR, Long terminal repeat; MITE, Miniature inverted-repeat transposable element; SINE, Short interspersed nuclear element; TIR, Terminal inverted repeat.