



 , Caterina Politi 2, Arianna Delicati 1, Andrea Gabbin 2, Luciana Caenazzo 1
, Caterina Politi 2, Arianna Delicati 1, Andrea Gabbin 2, Luciana Caenazzo 11 Department of Molecular Medicine, Laboratory of Forensic Genetics, University of Padova, 35121 Padova, Italy
2 Department of Cardiac, Thoracic, Vascular Sciences and Public Health, University of Padova, 35121 Padova, Italy
Abstract
Numerous major advances have been made in forensic genetics over the past decade. One recent field of research has been focused on the analysis of External Visible Characteristics (EVC) such as eye colour, hair colour (including hair greying), hair morphology, skin colour, freckles, facial morphology, high myopia, obesity, and adult height, with important repercussions in the forensic field. Its use could be especially useful in investigative cases where there are no potential suspects and no match between the evidence DNA sample under investigation and any genetic profiles entered into criminal databases. The present review represents the current state of knowledge of SNPs (Single Nucleotide Polymorphisms) regarding visible characteristics, including the latest research progress in identifying new genetic markers, their most promising applications in the forensic field and the implications for police investigations. The applicability of these techniques to concrete cases has stoked a heated debate in the literature on the ethical implications of using these predictive tools for visible traits.
Keywords
- External visible characteristics
- Forensic DNA phenotyping
- SNPs analysis
- Phenotype prediction
- Review
In the last few decades, genetic markers have been widely used for forensic purposes and have revolutionized the field of forensic investigations, embodying what is probably one of the most meaningful breakthroughs of our times [1].
Numerous methods of DNA typing have been proposed over time, the first being represented by a variable number of tandem repeats (VNTRs) as used from the mid-1980s [2] for identity testing which were afterwards replaced by short tandem repeats (STRs) or microsatellite loci due to the more advantageous characteristics of the latter [3]. Nowadays, STRs are still the predominant forensic genetic markers for identity testing and kinship analysis and validated STR kits are readily available, and routinely employed, in most forensic laboratories around the world [4].
More recently, a third class of genetic markers has emerged, which is represented by single nucleotide polymorphisms (SNPs). SNPs are point mutations in the DNA sequence, occurring ubiquitously in coding and non-coding regions of the whole human genome (i.e., autosomal, sex-linked, and mitochondrial DNA) [5]. They encompass single-base substitutions, where one nucleobase is substituted by another, and single-base insertion and/or deletion (InDel), where one base is added or removed thus resulting in DNA length variation [6, 7]. Most SNPs are bi-allelic markers, which means that each locus usually has only two possible allelic variants (for example, A and B) and consequently, in the diploid human genome, there are only three possible genotypes (AA, BB, or AB). SNPs are classified as functional or neutral, depending on whether they influence gene expression and biological processes [8].
SNPs possess several characteristics that make them more valuable markers than STRs: smaller amplicon size (50–150 bp); higher occurrence in human genome (approximately 1 in every 1000 bp, millions per individual, thus representing the most common human genetic variation); lower mutation rate; and finally, an elevated amenability to high-throughput genotyping through multiplexed sequencing [1, 5, 7, 9, 10, 11, 12]. These features make SNPs particularly suitable for obtaining information in cases of aged, degraded/or low copy biological samples, (where DNA fragments may be smaller than the required length for STR analysis), in kinship and paternity testing (especially in cases where relationships are generations apart) and in population and evolutionary genetics research [4, 11, 12].
There remain some disadvantages and limitations, at least for the near future, to their routine use as primary markers in forensic investigations in place of STR, including their lower discrimination power (SNPs are predominantly bi-allelic, which implies that numerous loci must be tested to yield the same discriminative power as STRs) and the well-established utilization of STR kits and databases in global forensic communities [1, 13].
The forensic community has currently been utilizing SNPs for different purposes. According to their application, SNPs can be divided into four classes [1]: identity SNPs, employed for differentiating individuals from one another, lineage SNPs, which prove information for kinship/paternity testing and evolutionary studies, ancestry SNPs, used to predict the DNA owner’s biogeographical background, and phenotype SNPs, associated with the prediction of visible traits, such as skin, hair or eye colour, height, weight, facial morphology, etc., commonly knowns as External Visible Characteristics (EVCs). Since phenotypic traits are determined not only by environmental factors (including diet, exercise regimen, sunlight exposure, stress exposure, etc.) but also by the genotype, it could be possible to predict some physical appearance traits relying on a DNA specimen. This inferential process is referred to as Forensic DNA Phenotyping (FDP). The possibility of accessing and predicting phenotypic information from a DNA sample by using a precise selection of SNPs probably represents the most promising application of SNPs in the forensic field. SNPs can also be used to support forensic DNA analysis for the possibility of automation. In the “omic era”, different approaches to SNP genotyping have been developed. Among them, SNaPshot® mini-sequencing method (Applied Biosystems) has been commonly applied since it has the advantage of not requiring additional equipment to what is already used in forensic laboratories [10]. Other technologies like TaqMan® hybridisation probes, hybridisation microarrays and massive parallel sequencing (MPS) have been previously described in the literature [5, 10, 14, 15, 16, 17, 18]. Depending on the final purpose (e.g., sequencing of one single gene, sequencing of the whole exome, or sequencing of the entire genome), some Next Generation Sequencing (NGS) platforms are more suitable than others due to their different characteristics. Despite SNPs being predominantly bi-allelic markers, given that NGS can work in multiplex, a large number of SNPs can be studied simultaneously [19].
Current forensic DNA analysis is substantially based on comparison of profiles, i.e., biological traces left at the crime scene are analyzed and compared to that of a known person (a tested suspect) or with genetic profiles stored in forensic DNA databases. The new genetic technology consists of gaining information about phenotypic traits of the wanted person from the DNA sample itself [11, 12, 13, 19]. Its use could be especially useful in investigative cases where there are no potential suspects and no match between the evidence DNA sample under investigation and any genetic profiles entered in criminal databases. Through the phenotyping prediction starting from biological samples found at the crime scene, probabilistic information may be acquired as to the physical characteristics of the sample donor, such as the colour of the hair, eyes and skin, as well as on the biogeographical origin and age. The combination of these elements, therefore, narrows the circle of possible perpetrators and facilitates investigations. In particular, pigmentation and ancestry markers typically corroborate each other.
Moreover, other recent studies have developed tools in order to provide statistical support to the weight of information on the prediction of FDP. The VISAGE consortium has recently suggested, at least for now, the use of MLR (multinomial logistic regression) as the most appropriate method for predicting appearance traits from DNA, especially with regard to hair, eye and skin prediction [20].
The present review represents the current state of knowledge on SNPs regarding visible characteristics, including the latest progress in research in identifying new genetic markers, their most promising application in the forensic field, i.e., prediction of phenotypic traits from a DNA sample, and its implications in police investigations.
The review was made with regard to the prediction of physical characteristics that related to forensic applications. For some traits, like pigmentation, the restriction to the forensic field has worked well but for others, such as myopia or body mass, for which a lot of research has likewise been made, an incomplete picture has emerged since such traits have been poorly explored by forensic scientists. A further limitation of this review is that the details of the technical aspects have not been considered because our intent was to describe the state of the art of the inference possibilities of visible traits. Moreover, we have not considered the different regulatory approaches that, for example at a European level, are very heterogeneous in allowing or not to use this type of prediction in judicial cases. We have considered only the current possibilities, which are still arousing a scientific and sociological debate, without considering the future prospects of forecasting other visible traits.
This review was performed in adherence to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines [21].
In February 2021, a systematic literature review of English and non-English papers regarding EVC-SNPs was conducted by two authors (A.G. and C.P.) using a public electronic database (Scopus). The research strategy included the terms “SNP”, “indel”, “phenotype”, “forensic”, “forensic genetic”, and “visible characteristic” in the following combinations: “SNP [and] forensic”, “indel [and] forensic”, “SNP [and] forensic genetic”, “indel [and] forensic genetic”, “SNP [and] phenotype [and] forensic”, “indel [and] phenotype [and] forensic”, “SNP [and] visible characteristic [and] forensic”, “indel [and] visible characteristic [and] forensic”. The search terms were intentionally kept generic in order to include all potentially interesting papers about the topic.
A total of 1795 works were identified through database searching. Duplicates (314 works) were removed manually. Then, three authors (A.G., A.D. and C.P.), independently of each other, performed a first selection of the remaining articles according to the following inclusion criteria: (A) English language; (B) topic, i.e., only EVC-informative SNPs and/or EVC-informative indels; and (C) topic, i.e., only papers related to human beings. We first screened titles for inclusion criteria A–C, then abstracts, and only when necessary (i.e., the topic was not clear from the title and/or abstract reading) the authors undertook a full-text evaluation. In cases of disagreement or further doubts, the supervisors (L.C. and P.T.) were queried. Because of their indirect and limited informative value on phenotype, manuscripts concerning only ancestry-informative SNPs were not included in the review [1]. Similarly, although gender was reported as a kind of EVC, it was not included since it is usually assessed through standard STR analysis [19, 22].
After title and abstract evaluation, 1327 and 67 manuscripts (1394 in total), respectively, were excluded due to irrelevance. After a full text reading of the selected papers, only 66 were considered eligible using criteria A–C and included in the review. Additional pertinent manuscripts (17) were identified within the bibliography of selected papers. After external peer review, two articles were included in full text. A total of 90 articles were examined for the review and qualitative synthesis. For each article, the authors examined the full text and extracted the following data, managing them in Excel® (Microsoft® 365): title, authors, year of publication, type of visible trait considered, polymorphism(s) (SNPs or Indels) tested for association with EVCs, target population, and sample size.
The PRISMA flow chart in Fig. 1 summarizes the study screening and selection process as described above.
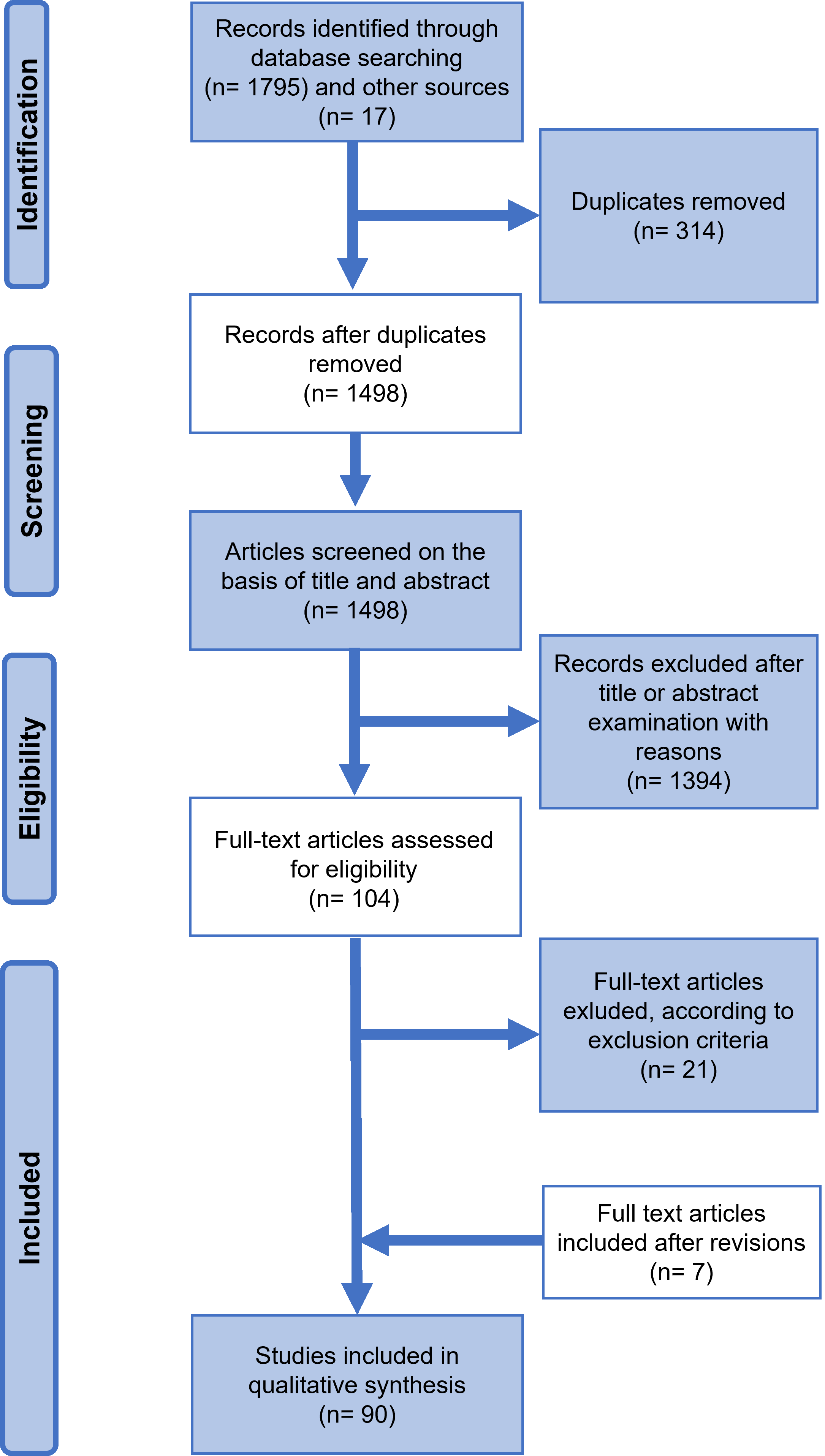 Fig. 1.
Fig. 1.Preferred Reporting Items for Systemic Reviews and Meta-Analyses (PRISMA) 2020 flow diagram.
So far, the following EVCs have been evaluated for forensic DNA phenotyping: hair colour, eye colour, skin colour (considered separately or in associations, e.g., eye and hair colour; eye and skin colour; eye, hair and skin colour), hair morphology, height, weight (obesity), facial morphology, presence of freckles, male-pattern baldness, and myopia.
Even ancestry-informative SNPs (AIMs) have been employed in DNA phenotyping, as exemplified in one selected study, where facial morphology prediction was found to be significantly associated with genetic ancestry information [22]. However, their contribution to the overall predictive power of human phenotype is limited to basic, ancestry-related information, such as light skin pigmentation in Northern Europeans or large noses and thick lips in African populations [1].
Amongst all EVCs, pigmentation traits (i.e., skin and/or eye and/or hair colour) have been reported as the least genetically complex traits, accurately predictable through analysis of only a few genes [13, 19]. In Fig. 2 we report a description of the melanin synthesis pathway, showing formation of various melanins (e.g., phaeomelanin, eumelanin) and the functions of important gene products and modifiers. Melanin is a pigment responsible for humans’ hair, eye and skin coloring. There are two main types of melanin involved in pigmentation pathway, a darker (brown/black) pigment called eumelanin and a lighter (reddish) pigment called pheomelanin. A person’s hair/eyes/skin colour depends both on the type and total amount of melanin. For instance, dark phenotypes (black hair, brown/black eyes, and dark skin) are determined by large amount of eumelanin; intermediate phenotypes (i.e., brown hair, hazel/green eyes, and intermediate skin colour) are determined by moderate concentration of eumelanin; lighter phenotypes (like blonde hair, blue eyes, and pale skin) come down from low amount of eumelanin, while red hair is the result of very little eumelanin and lot of pheomelanin. Melanogenesis is initiated by tyrosinase enzyme which converted melanin precursor, tyrosine, to DOPA (DihydrOxyPhenylAlanine) and then to DOPAquinone. Further transformations convert DOPAquinone to the final products, eumelanin or pheomelanin. OCA2, SLC24A5 and SLC45A2 encode for membrane transport proteins whose activity influenced the melanosome internal environment in terms of ion concentrations, which in turn influence the availability of tyrosine or the tyrosinase activity and thus, ultimately, the amount of melanin synthetized; HERC2 contains the promoter region for OCA2, affecting its expression.
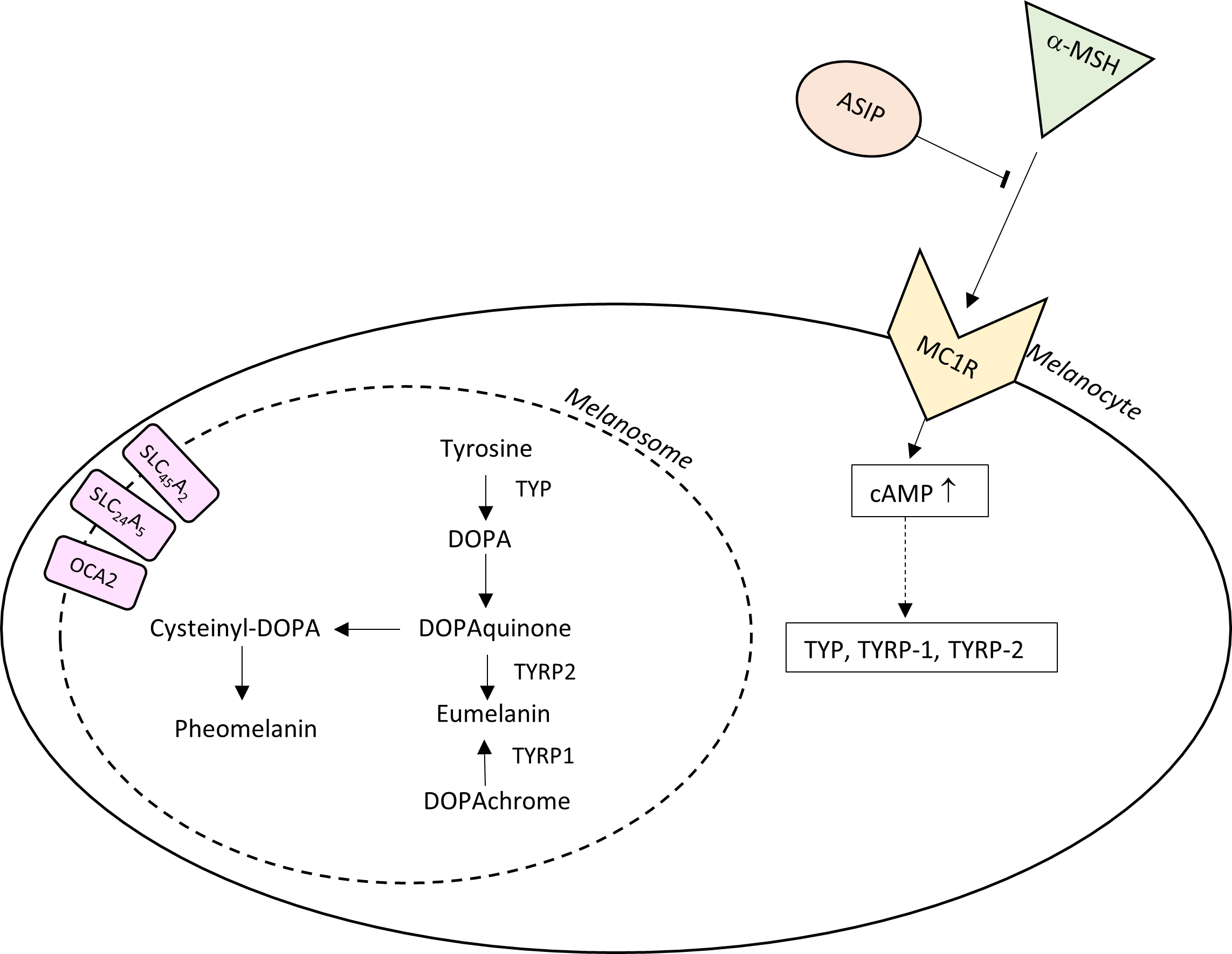 Fig. 2.
Fig. 2.Melanin biosynthesis pathway. A schematic representation of
eumelanin and pheomelanin synthesis is provided within melanocytes showing some
of the most important genes involved in melanogenesis regulation:
Most selected papers, 55 out of 85 (64.7%), concerned with pigmentation traits, are distributed as follows: 26 studies considered eye colour alone, seven studies considered skin colour only, three studies examined solely hair colour, while 21 studies examined more than one pigmentation trait (eye and/or hair and/or skin colour). Fig. 3 shows a diagram of the markers that make up the IrisPlex, HIris-Plex and HIris-Plex-S systems.
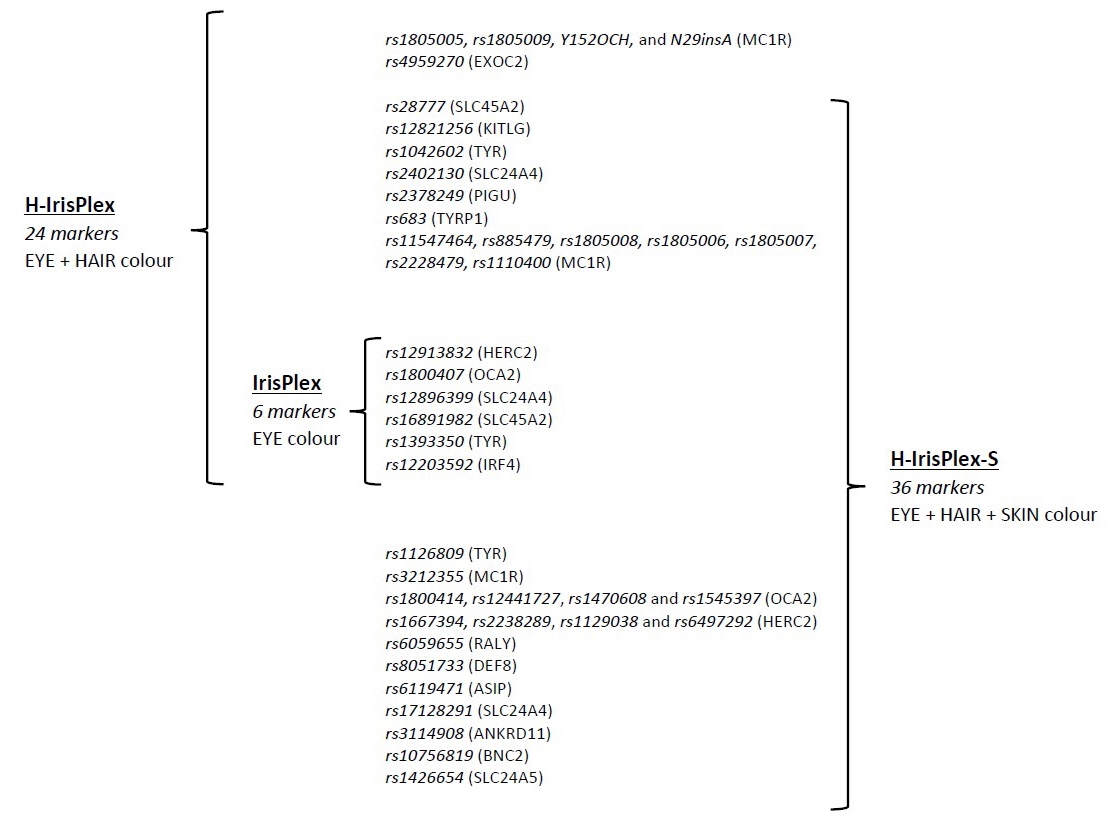 Fig. 3.
Fig. 3.The table displays the SNPs marker included in IrisPlex, H-IrisPlex and H-IrisPlex-S, respectively for eye, eye + hair, and eye + hair + skin colour prediction.
Other visible characteristics were found to be much less studied than pigmentation traits: head-hair morphology (eight studies), male-pattern baldness (three studies), presence of freckles (three studies), various facial features (eight studies, grouped together into one category named “Facial morphology”), high myopia (two studies), obesity (two studies), and adult height (three studies).
While less common than SNPs (meant as single-nucleotide substitutions) in the genome sequence, in our review we found that only one indel (N29insA, also denotes as rs86inA and rs312262906) has been so far identified in relation to visible characteristics and, more especially, to pigmentation traits [23, 24]. Insertion-deletion polymorphisms have been reported to be of increasing interest in a forensic context [7]; nevertheless, indels are frequently observed in the more severe cases of pigmentation variation–for example, albinism. Thus, they can be considered less relevant for EVC prediction, where the primary interest lies in the common pigmentary variation.
Table 1 (Ref. [25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75]) summarizes all the studies considered in this review with the SNPs that were studied and the type of predicted trait.
| SNP-ID | GENE | CHR. | PREDICTED PHENOTYPE |
| rs4845418 | Unknown | 1 | Hair morphology [25, 26, 27] |
| rs12130862 | Unknown | 1 | Hair morphology [25, 26, 27] |
| rs80293268 | ERRFI1/SLC45A1 | 1 | Hair morphology [25, 26, 27] |
| rs5781117 | LYPLAL1 | 1 | Facial morphology [28] |
| rs4648379 | PRDM16 | 1 | Facial morphology [28] |
| rs12565727 | TARDBP | 1 | Male Pattern Baldness [29] |
| rs11803731 | TCHH | 1 | Hair morphology [25, 26, 27] |
| rs17646946 | TCHHL1 | 1 | Hair morphology [25, 26, 27] |
| rs3827760 | EDAR | 2 | Hair morphology [26] |
| Facial morphology [28] | |||
| rs7559271 | PAX3 | 2 | Facial morphology [28, 30] |
| rs974448 | PAX3 | 2 | Facial morphology [31] |
| rs17479393 | TEX41 | 2 | Facial morphology [31] |
| rs7349332 | WNT10A | 2 | Hair morphology [25, 26, 27] |
| rs50663440 | CACNA2D3 | 3 | Facial morphology [28] |
| rs12635264 | MASP1 | 3 | Facial morphology [32] |
| rs1717652 | MASP1 | 3 | Facial morphology [32] |
| rs2977562 | RAB7A/ACAD9 | 3 | Facial morphology [31] |
| rs9995821 | DCHS2 | 4 | Facial morphology [31] |
| rs2045323 | DCHS2 | 4 | Facial morphology [31] |
| rs6555969 | C5orf50 | 5 | Facial morphology [31] |
| rs929626 | EBF1 | 5 | Male Pattern Baldness [29] |
| rs2074612 | HBEGF | 5 | Facial morphology [33] |
| rs10502861 | SLC12A2 | 5 | Male Pattern Baldness [29] |
| rs26722 | SLC45A2 | 5 | Hair colour [34] |
| Skin colour [24, 34, 35, 36] | |||
| rs28777 | SLC45A2 | 5 | Hair colour [37, 38] |
| rs13289 | SLC45A2 | 5 | Skin colour [39] |
| rs16891982 | SLC45A2/MATP | 5 | Eye colour [40, 41, 42, 43, 44, 45] |
| Hair colour [34, 46] | |||
| Skin colour [24, 34, 35, 36, 39] | |||
| rs4959270 | EXOC2 | 6 | Hair colour [37] |
| rs12203592 | IRF4 | 6 | Eye colour [40, 42, 42, 47] |
| Freckles [75] | |||
| rs227833 | SUPT3H | 6 | Facial morphology [28] |
| rs1852985 | SUPT3H/RUNX2 | 6 | Facial morphology [31] |
| rs756853 | HDAC9 | 7 | Male Pattern Baldness [29] |
| rs987525 | Unknown | 8 | Facial morphology [48] |
| rs10504499 | EYA1 | 8 | Facial morphology [31] |
| rs11782517 | MSRA | 8 | Facial morphology [31] |
| rs10756819 | BNC2 | 9 | Skin colour [49] |
| rs2153271 | BNC2 | 9 | Frekles [50] |
| rs1408799 | TYRP1 | 9 | Eye colour [24, 47] |
| Skin colour [39] | |||
| rs683 | TYRP1 | 9 | Hair colour [37] |
| rs1194708 | DKK1 | 10 | Facial morphology [28] |
| rs2219783 | LGR4 | 11 | Hair morphology [25, 26, 27] |
| rs644242 | PAX6 | 11 | High myopia [51] |
| rs35264875 | TPCN2 | 11 | Hair colour [38] |
| rs3829241 | TPCN2 | 11 | Skin colour [39] |
| rs35264875 | TPCN2 | 11 | Hair colour [38] |
| rs1393350 | TYR | 11 | Eye colour [40, 41, 42, 43, 45, 47, 52] |
| rs1042602 | TYR | 11 | Hair colour [37] |
| Skin colour [24, 35, 36, 53, 54] | |||
| rs2277404 | ABCC9 | 12 | Facial morphology [32] |
| rs7316271 | ABCC9 | 12 | Facial morphology [32] |
| rs12821256 | KITLG | 12 | Hair colour [37, 38] |
| rs10777129 | KITLG | 12 | Skin colour [39] |
| rs731223 | VDR | 12 | Hair colour [24] |
| rs7161418 | DICER1 | 14 | Facial morphology [31] |
| rs2224309 | GSC | 14 | Facial morphology [31] |
| rs8004825 | MIR495 | 14 | High myopia [55] |
| rs12896399 | SLC24A4 | 14 | Eye colour [40, 42, 42, 44, 52] |
| rs2402130 | SLC24A4 | 14 | Hair colour [37, 39] |
| rs17128291 | SLC24A4 | 14 | Skin colour [49] |
| rs1289399 | SLC24A4 | 14 | Eye colour [43, 44, 45, 56, 57] |
| rs8041414 | CEP152 | 15 | Skin colour [58] |
| rs12913316 | CTXN2 | 15 | Skin colour [58] |
| rs11637235 | DUT | 15 | Skin colour [58] |
| rs916977 | HERC2 | 15 | Eye colour [52, 59, 60, 61] |
| rs1667394 | HERC2 | 15 | Eye colour [43, 44, 45, 56, 57, 59] |
| Skin colour [49] | |||
| rs12913832 | HERC2 | 15 | Eye colour [40, 42, 42, 44, 46, 47, 52, 61, 62, 63, 64, 65, 66, 67] |
| Hair colour [38, 46] | |||
| Skin colour [49, 68] | |||
| rs1129038 | HERC2 | 15 | Eye colour [42, 43, 44, 45, 46, 47, 52, 62, 63, 64, 65, 66, 67] |
| Hair colour [38] Skin colour [49] | |||
| rs11636232 | HERC2 | 15 | Eye colour [43, 44, 45, 56, 57] |
| rs7183877 | HERC2 | 15 | Eye colour [43, 44, 45, 56, 57, 69] |
| rs7170852 | HERC2 | 15 | Eye colour [61] |
| rs12931267 | HERC2 | 15 | Hair colour [38] |
| rs1636232 | HERC2 | 15 | Skin colour [70] |
| rs1133496 | HERC2 | 15 | Skin colour [70] |
| rs2238289 | HERC2 | 15 | Skin colour [49, 70] |
| rs6497292 | HERC2 | 15 | Skin colour [49] |
| rs11070627 | MYEF2 | 15 | Skin colour [58] |
| rs1258763 | near GREM1 | 15 | Facial morphology [48] |
| rs7495174 | OCA2 | 15 | Eye colour [38, 61, 71] |
| rs6497268 | OCA2 | 15 | Eye colour [71] |
| rs11855019 | OCA2 | 15 | Eye colour [71] |
| rs17566952 | OCA2 | 15 | Eye colour [62, 72] |
| rs11638265 | OCA2 | 15 | Eye colour [62, 72] |
| rs1800411 | OCA2 | 15 | Eye colour [62, 72] |
| rs1900758 | OCA2 | 15 | Eye colour [62, 72] |
| rs1800407 | OCA2 | 15 | Eye colour [40, 41, 42, 43, 46, 47, 52, 62, 64, 65, 66, 67, 72] |
| Skin colour [36, 49, 66, 70] | |||
| rs749846 | OCA2 | 15 | Eye colour [62, 72] |
| rs74653330 | OCA2 | 15 | Eye colour [73] |
| rs121918166 | OCA2 | 15 | Eye colour [73] |
| rs1800416 | OCA2 | 15 | Eye colour [24, 60, 70] |
| Skin colour [36, 66, 70] | |||
| rs4778138 | OCA2 | 15 | Eye colour [52] Hair colour [38] |
| Skin colour [36, 66, 70] | |||
| rs1800404 | OCA2 | 15 | Eye colour [62, 72] |
| Skin colour [49, 70] | |||
| rs7170989 | OCA2 | 15 | Skin colour [36, 66, 70] |
| rs1375164 | OCA2 | 15 | Skin colour [36, 39, 58, 66, 70] |
| rs1448484 | OCA2 | 15 | Skin colour [39] |
| rs1800414 | OCA2 | 15 | Skin colour [49] |
| rs12441727 | OCA2 | 15 | Skin colour [49] |
| rs1470608 | OCA2 | 15 | Skin colour [49] |
| rs1545397 | OCA2 | 15 | Skin colour [49] |
| rs6059655 | RALY | 15 | Skin colour [49] |
| rs1426654 | SLC24A5 | 15 | Hair colour [46] |
| Skin colour [24, 35, 36, 39, 53, 54, 58] | |||
| rs2924566 | SLC24A5 | 15 | Skin colour [58] |
| rs4775730 | SLC24A5 | 15 | Skin colour [58] |
| rs3114908 | ANKRD11 | 16 | Skin colour [49] |
| rs8051733 | DEF8 | 16 | Skin colour [49] |
| rs1805007 | MC1R | 16 | Hair colour [37, 46] |
| rs11547464 | MC1R | 16 | Hair colour [37, 38] |
| Skin colour [53, 74] | |||
| rs885479 | MC1R | 16 | Hair colour [37] |
| Freckles [75] | |||
| rs1805005 | MC1R | 16 | Hair colour [37] |
| rs1805006 | MC1R | 16 | Hair colour [37, 38] |
| Skin colour [53, 74] | |||
| rs1805008 | MC1R | 16 | Hair colour [37, 38] |
| Skin colour [36, 53, 57, 70, 74] | |||
| rs1805009 | MC1R | 16 | Hair colour [38] |
| Skin colour [53, 74] | |||
| rs201326893 | MC1R | 16 | Skin colour [53, 74] |
| rs2228479 | MC1R | 16 | Hair colour [37] |
| Freckles [75] | |||
| rs1110400 | MC1R | 16 | Hair colour [37] |
| N29insA (or rs86inA or rs312262906) | MC1R | 16 | Hair colour [37] |
| rs3212345 | MC1R | 16 | Skin colour [53, 74] |
| rs228479 | MC1R | 16 | Skin colour [49] |
| rs1126809 | MC1R | 16 | Skin colour [49] |
| rs3212355 | MC1R | 16 | Skin colour [49] |
| rs33832559 | MC1R | 16 | Frekles [75] |
| rs228478 | MC1R | 16 | Frekles [75] |
| rs11150606 | PRSS53 | 16 | Hair morphology [25, 26, 27] |
| rs1268789 | FRAS1 | 17 | Hair morphology [25, 26, 27] |
| rs80067372 | TNFSF12 | 17 | Facial morphology [28] |
| rs12976445 | MIR495 | 19 | High myopia [55] |
| rs61374441 | Unknown | 20 | Male Pattern Baldness [29] |
| rs19980761 | Unknown | 20 | Male Pattern Baldness [29] |
| rs201571 | Unknown | 20 | Male Pattern Baldness [29] |
| rs6047844 | Unknown | 20 | Male Pattern Baldness [29] |
| rs913063 | Unknown | 20 | Male Pattern Baldness [29] |
| rs1160312 | Unknown | 20 | Male Pattern Baldness [29] |
| rs6113491 | Unknown | 20 | Male Pattern Baldness [29] |
| rs2180439 | Unknown | 20 | Male Pattern Baldness [29] |
| rs2378249 | ASIP/PIGU | 20 | Hair colour [37, 49] |
| rs6119471 | ASIP/PIGU | 20 | Skin colour [49] |
| rs2206437 | DHX35 | 20 | Facial morphology [31] |
| rs310642 | PTK6 | 20 | Hair morphology [25, 26, 27] |
| rs369378152 | GPR50 | X | Hair colour [34] |
| rs4827379 | AR | Xq12 | Male Pattern Baldness [29] |
| rs1385699 | AR | Xq12 | Male Pattern Baldness [29] |
| rs1352015 | AR | Xq12 | Male Pattern Baldness [29] |
| rs1041668 | AR | Xq12 | Male Pattern Baldness [29] |
| rs1397631 | AR | Xq12 | Male Pattern Baldness [29] |
| rs5919324 | AR | Xq12 | Male Pattern Baldness [29] |
| rs6625150 | AR | Xq12 | Male Pattern Baldness [29] |
| rs12558842 | AR | Xq12 | Male Pattern Baldness [29] |
| rs6625163 | AR | Xq12 | Male Pattern Baldness [29] |
| rs2497938 | AR | Xq12 | Male Pattern Baldness [29] |
| rs2497911 | AR | Xq12 | Male Pattern Baldness [29] |
| rs2497935 | AR | Xq12 | Male Pattern Baldness [29] |
| rs962458 | AR | Xq12 | Male Pattern Baldness [29] |
| rs6152 | AR | Xq12 | Male Pattern Baldness [29] |
| rs12396249 | AR | Xq12 | Male Pattern Baldness [29] |
| rs4827545 | AR | Xq12 | Male Pattern Baldness [29] |
| rs7885198 | AR | Xq12 | Male Pattern Baldness [29] |
The first evidence of a possible correlation between genetic variants and eye colour dates back to the early 2000s, when it was first suggested that the OCA2 gene was responsible for a great deal of normal eye-colour variation [76, 77, 78]. The following year, Duffy et al. [71] confirmed this assertion, finding a strong association between blue versus non-blue eye colour and three OCA2 SNPs (rs7495174, rs6497268, and rs11855019), with the TGT/TGT genotype explaining the 0.905 of total light eye colour (blue or green). A few years later, Branicki et al. [72] and Andersen et al. [62] explored in greater depth the contribution of the OCA2 gene to determining human eye colour. Significant association was found for eight new OCA2 SNPs (rs17566952, rs11638265, AY392134, rs1800411, rs1900758, rs1800404, rs1800407, and rs749846). Among these, the strongest association resulted between rs1800407 and intermediate (green/hazel) iris colour. Some years later, Andersen et al. [73] also identified two further important OCA2 variants (rs74653330 and rs121918166).
In order to evaluate the possible involvement of other genes in human eye-colour variation in addition to OCA2, Kayser et al. [59] performed GWAS (Genome Wide Association Studies) and linkage analysis with people of European descent. They showed that the HERC2 gene, located in the same region of OCA2 (15q13.1), is a new important determinant of human iris colour and identified up to 15 relevant loci in this region, among which rs916977 emerged as the most influential variant (the T allele, which represents the ancestral state of the marker, being predictive for brown iris colour, the C allele for blue iris colour), followed by rs1667394 [59]. In the same year, Eiberg et al. [63] identified the linkage disequilibrium (LD) of two tightly linked loci rs12913832 and rs1129038 with good predictive power for blue and brown eye colour. LD between alleles at two loci has been defined in many ways, but all definitions depend on the difference between the frequency of gametes carrying the pair of two alleles at two loci and the product of the frequencies of these alleles [79]. With regard to rs12913832, although located within the HERC2 region, it is part of a regulatory element upstream from OCA2 exerting an inhibitory effect on OCA2 itself. The decreased expression of OCA2, particularly within iris melanocytes, causes the blue eye-colour phenotype [47, 63]. Furthermore, Pośpiech et al. [24] found multiple epistatic interactions among genes affecting pigmentation phenotype, particularly eye colour.
A turning point came with the development by Walsh et al. and Liu et al. [40, 41, 42] of the first highly sensitive multiple genotyping assay for the prediction of blue/brown eye colour, named IrisPlex. It consists of the six SNPs currently identified as major eye-colour predictors in Europeans:rs12913832 (HERC2), rs1800407 (OCA2), rs12896399 (SLC24A4), rs16891982 (SLC45A2/MATP), rs1393350 (TYR), and rs12203592 (IRF4)—albeit two subsequent publications did question the usefulness of rs12203592 since they found very little or no predictive power linked to this locus [43, 56]. The model was based on initial genotype and phenotype data from a single European population (the Dutch) but its reliability for accurate eye-colour prediction was also demonstrated for individuals from several countries across Europe (including Norway, Estonia, the UK, France, Spain, Italy, and Greece), showing extremely high predictive power (0.96 AUC—Area Under Curve) for both blue and brown eye colour [41]. The prediction accuracy for blue/brown eye colour decreased when IrisPlex was applied to a more biogeographically diverse sample, including individuals of Asian, African, and South American descent [23].
Moreover, the intermediate eye colour could not be predicted by IrisPlex with as high accuracy as with blue and brown eyes, resulting in a higher rate of misclassified or unclassified predictions [40, 41, 42, 57]. Improved detection of intermediate phenotype (correct classification rate of 26.1%) was observed using a different eye-colour prediction model (Snipper5+1) elaborated by Freire-Aradas et al. [44] and based on five IrisPlex SNPs (rs1800407, rs12896399, rs16891982, rs1393350, and rs12203592) plus rs1129038 counted in combination with IrisPlex rs12913832 (HERC2 haplotype). Among these, particularly noteworthy is the effect on green eye-colour determination made by OCA2rs1800407 when considered in haplotype with rs12913832 [47]. Likewise, the interaction between rs12913832 and TYRP1rs1408799 also allowed for a slight increase in the accuracy of green-colour predictivity [47].
Ruiz et al. [43] confirmed the remarkable effect, as already described in other studies, of rs12913832, rs1129038, rs11636232, rs1289399, rs1800407, rs16891982, and rs1393350 and proposed to add three additional SNPs (rs1129038, rs1667394, and rs7183877 from HERC2) into the six-SNP panel of IrisPlex, aiming at increasing its discriminative power in predicting Europeans’ iris colour, especially for intermediate phenotypes (green and hazel) [43, 45].
Among all the SNPs discovered, HERC2rs12913832 was found to be the most strongly associated with iris colour, although HERC2rs1129038 and OCA2rs1800407 have been reported to be of predominant importance as well [42, 46, 47, 52, 62, 64, 65, 66, 67]. Indeed, many studies have observed that most eye-colour variation (ranging from 68.8% to 74.8% depending on the study) could be explained by rs12913832, while additional SNPs, although found to be associated with eye colour, allowed for only a slight improvement in predictive ability (75.6–76%) [46, 62]. Similarly, the five additional SNPs within the IrisPlex also turned out to produce a small predictive value to that determined by rs12913832 [42].
However, the selection of the best SNPs for EVC determination seems to be highly population-dependent, suggesting that panels for eye-colour prediction should be adjusted for different geographical regions: for instance, a relevant effect on eye colour was observed for rs1408799 in the Polish population, and for rs916977 in the Czech population, while rs1800416 was found to be significantly associated with eye colour only in the Brazilian population [24, 60, 70].
Although developed based on a European database, IrisPlex’s usability has also been tested in individuals outside of Europe. For example, Rahat et al. [80] showed the reliability of IrisPlex for brown and blue colour prediction in Pakistan’s population, but also pointed out the need for the inclusion of more SNPs in the model to increase prediction accuracy especially for intermediate colour. Al-Rashedi et al. [81] came to similar conclusions with the Iraqi population. Some other studies have demonstrated instead the existence of a significant bias linked to the geographical origins of the population to which it was applied, especially in cases of admixed populations or intermediate eye colour [56, 57, 82]. Yun et al. [56] observed a higher proportion of uncertain prediction, besides some inconsistences, when IrisPlex was applied in the Eurasian population, which presents an admixed genetic structure (European and Asian), except for East Asian populations (such as Koreans and Chinese), in which predictions were always consistent for brown eye colour. Bulbul et al. [57] also observed a worse performance of IrisPlex on the Turkish population compared with European results. Dembinski et al. [82] obtained only a moderate predictive power with the IrisPlex essay in a North American sample (US population), which is highly admixed compared to the European population.
To overcome this limit, further models were later developed for prediction of eye colour in other populations. Allwood et al. [52] developed a predictive model for New Zealanders. They first evaluated the effect of preselected SNPs upon eye-colour phenotype in a collection of dichotomous tree models (i.e., blue vs. non-blue, brown vs. non-brown, and intermediate vs. non-intermediate) and, finally, in a multiple-response tree considering all eye colours, i.e., blue vs. brown vs. intermediate. The SNPs chosen for use in the different models were: rs1129038 (HERC2), rs1393350 (TYR), and rs12896399 (SLC24A4) for blue vs. non-blue; rs1129038, rs1800407 (OCA2), rs12913832 (HERC2), and rs1393350 for brown vs. non-brown; rs1800407, rs916977, rs1393350, and rs4778138 (OCA2) for intermediate vs. non-intermediate; and rs1129038, rs1800407, and rs1393350 for the final model. It is worth noting that part of the SNPs employed in the binary response models (except for rs1129038, rs12896399, rs916977, rs4778138), and all SNPs composing the multiple response model, are also included in the IrisPlex system. Both models performed well though with differences for each specific eye-colour group, (i.e., considerably better for brown and blue than for intermediate eye colour). The “all-eye-colour” model predicted blue colour with an accuracy level of 89%, brown colour at 94%, and intermediate eye colour at only 46%, with an overall accuracy of 79% (obviously conditioned by the latter rate). As for IrisPlex, and in similar proportion, most prediction errors were generated by intermediate eye colour. Similarly, Alghamdi et al. [61] proposed an eye-colour prediction model for Saudi individuals containing five SNPs (rs12913832, rs7170852, rs7183877, rs7495174, and rs916977) that showed a high accuracy for both brown and intermediate eye colours (no participant was categorized as blue iris). Gettings et al. [69] developed a 50-SNP assay to predict eye phenotype among European-Americans, which was able to accurately predict eye colour in 61% of individuals tested.
Another discrepancy factor in eye-colour prediction of the Irisplex was seen to be gender. Martinez-Cadenas et al. [83] noticed that, given a specific IrisPlex genetic profile, males had lighter eye colours than predicted by genotype, while females tended to have darker eye colours, suggesting the possible existence of an unidentified gender-related component contributing to human eye-colour variation.
It is worth noting that, although developed on a European (Dutch) database, IrisPlex was also found to be a little less predictive in some European subpopulations (Italians, Spanish, and Portuguese) than in others (Germans and Dutch) [45, 84, 85]. It has been hypothesized that this result may reflect a greater degree of genomic admixing in the Southern European populations, just as reported by Dembinski et al. [82] in the US population. Another study highlighted that, in contrast to Northern European populations, not all six IrisPlex SNPs had significant association with eye colours in individuals from Mediterranean Europe, and this could be traced back to a lower frequency of blue iris colour in these populations, classically presenting a darker pigmentation phenotype [83].
Human hair colour depends on the combined amount of two types of melanin, eumelanin (dark pigment) and pheomelanin (light pigment). The first studies on hair-colour predictability were based on the search for genetic variations in people with a far-away biogeographical origin (individuals of Asian, African, Australian, and Caucasian descent), or among individuals from the same geographic region but presenting multi-coloured phenotypes (e.g., the Brazilian population). Among genes showing significant association with pigmentation variation, polymorphisms in ASIP, MATP (then renamed SLC45A2), SLC24A5, and MC1R—all implicated in the melanin biosynthesis—showed significant association with hair colour [34]. For instance, specific allelic polymorphisms in SLC45A2rs16891982, SLC452rs26722 and ASIPrs369378152 were found at a higher frequency in non-Caucasian dark-haired populations, such as African-Americans and Asians, as well as in dark-haired people of Caucasian descent [34]. Moreover, Pośpiech et al. [24] identified epistatic interactions between MC1R variants and both the HERC2 gene and rs731223 in VDR (Vitamin D Receptor), having an advantageous impact on red versus non-red hair colour prediction.
The publication of Valenzuela et al. [46] represented an important step forward, since they indicated four SNPs, sited in as many genes, as major genetic contributors to hair pigmentation: rs1426654 (SLC24A5), rs16891982 (SLC45A2), rs12913832 (HERC2), and rs1805007 (MC1R). Specifically, two of them (SLC24A5rs1426654 and SLC45A2rs16891982) showed the strongest association with both total hair-melanin amount and eumelanin/pheomelanin ratio, while HERC2rs12913832 turned out to be the third most significant contributor to total hair melanin and MC1R rs1805007 the third major contributor to eumelanin/pheomelanin ratio [46]. However, as for eye colour, some studies suggested that the strongest association with pigmentation traits, including hair, was with SNPs from the OCA2-HERC2region [70, 73, 86].
New acquaintances were used to integrate IrisPlex with the 22 most predictive SNPs currently identified for hair-colour determination, creating a system for simultaneous eye and hair-colour prediction named HIrisPlex. Among the 22 SNPs involved in hair-colour determination, four variants were already included in IrisPlex (being predictive for eye colour as well), while 18 were of new introduction, for a total of 24 SNPs (including one InDel) making up the model: SLC45A2rs28777, KITLGrs12821256, EXOC2rs4959270, TYRrs1042602, SLC24A4rs2402130, ASIP/PIGUrs2378249, and TYRP1rs683, 10 SNPs from MC1R (rs11547464, rs885479, rs1805008, rs1805005, rs1805006, rs1805007, and rs1805009, Y152OCH (later renamed as rs201326893), rs2228479 and rs1110400), and one InDel N29insA (also denotes as rs86inA and rs312262906). As its forerunner, HIrisPlex was initially developed using DNA samples collected from 1551 European subjects living in Poland (n = 1093), the Republic of Ireland (n = 339) and Greece (n = 119). The hair-colour prediction component of the HIrisPlex tool applied to individuals from different parts of Europe yielded prediction accuracies of 69.5% for blond hair colour, 78.5% for brown, 80% for red and 87.5% for black independently from bio-geographic ancestry. To verify its use outside of Europe, Walsh et al. [37] performed HIrisPlex analysis on worldwide DNA samples from the HGDP-CEPH panel relative to 952 individuals from 51 populations. Although with minor accuracy, it was demonstrated to provide satisfactory eye/hair colour prediction in non-European individuals as well [87]. Likewise, it was revealed to be a suitable and sufficiently robust predictive system for human skeletal remains [88].
Söchtig et al. [38] indicated a subset of 12 SNPs as the best hair-colour prediction markers (OCA2rs7495174 and rs4778138; TPCN2 rs35264875; HERC2 rs1129038, rs12931267, rs12913832, and rs28777; MC1R-R rs11547464, rs1805006, rs1805007, rs1805008, and rs1805009), only seven of which were also included in HIrisPlex. Moreover, they indicated HERC2 rs1129038 as the strongest predictor for blond hair, HERC2 rs12913832 the strongest predictor for black hair, while TPCN2 rs35264875 and HERC2 rs12931267 as key markers for brown hair colour. However, the predictive performance of the 12 SNPs set in the European population remained lower than with HIrisPlex [38].
In an effort to explain the higher rate of inaccuracy observed in HIrisPlex’s
blonde-hair prediction, Kukla-Bartoszek et al. [89] focused on
age-dependent hair-colour darkening, i.e., some individuals with blonde hair
colour in early childhood may experience a hair-colour darkening during advanced
childhood or adolescence. They observed that the number of incorrect blond
hair-colour predictions given by HIrisPlex was significantly higher in adult
individuals with brown hair who were blond in early childhood (2–3 years old),
compared to those who had always had brown hair (only one third of individuals
who experienced hair-colour darkening from childhood to adulthood were correctly
predicted by HIrisPlex) [89]. Still in regard to age-related hair-colour changes,
Pośpiech et al. [90] investigated the genetics underlying the
hair-greying process but concluded that most predictive power was given by age
alone, while genetic variants had only a small impact on hair-greying variation
(
Finally, with regard to red-hair-colour prediction, Keating et al. [23] observed that by removing four MC1R SNPs from the 22 HIrisPlex DNA variants, there was more red hair missed (nearly 60% compared to the 14% of HIrisPlex). This result confirmed the important role of the MC1R gene in red hair determination, as previously reported [23, 66].
As for other pigmentation traits previously argued, the first genes to be
associated with skin colour were those encoding for proteins which are involved
in the melanin production within melanocytes, e.g., MC1R (encoding for a
membrane receptor whose activation by
Significant associations with skin colour were found for MC1R gene variants,
including rs1805006, rs1805007, rs1805008,
rs1805009, rs11547464, rs201326893, N29insA
(InDel), and rs3212345. It has been hypothesized that rs3212345:
C
Maroñas et al. [39] described the 10 best predictive SNPs linked to skin colour in European and non-European individuals, among which are: SLC45A2rs16891982 and SLC24A5rs1426654 (the two most important markers, the former for intermediate, the latter for black and white skin colour); ASIP rs60580017 (the second major important marker for classifying black skin and fourth for classifying intermediate skin); TYRP1rs1408799 (for distinguishing intermediate skin); OCA2rs1448484 (important contributor to black versus white); SLC45A2rs13289 (olive skin colour in Europeans); KILTGrs10777129, TPCN2rs3829241, and SLC24A4rs2402130 (not previously described). Remarkably, the first two SNPs described above account for most of the classification success (respectively, 77.6% for intermediate, 87.6% for black, and 95.7% for white), the remaining eight SNPs enhancing classification success by only a few percentage points (2–3%) each.
However, as happened for eye colour, biogeographical divergences were observed here too: in the Indian population, in contrast to Maroñas’ results, the nine major contributors to skin pigmentation (overall explaining the 31% variance) were found to be OCA2rs1800404 and rs1375164, SLC24A5rs2924566, rs4775730, rs1426654, MYEF2rs11070627, CTXN2rs12913316, DUTrs11637235, CEP152rs8041414 [39, 58]. In the Polish population, HERC2 rs12913832 seemed to be the strongest variant for skin phenotype [68].
In the footsteps of HIrisPlex, a combined tool for simultaneous prediction of eye, hair, and skin colour named HIrisPlex-S was introduced, where skin colour prediction was based on a set of 36 SNPs (of which 19 had also been included in the previous model, plus 17 novel markers): SLC24A5rs1426654, IRF4rs12203592, MC1Rrs1805007, rs1805008, rs11547464, rs885479, rs228479, rs1805006, rs1110400 and rs3212355, OCA2rs1800414, rs1800407, rs12441727, rs1470608, and rs1545397, SLC45A2rs16891982 and rs28777, HERC2rs1667394, rs2238289, rs1129038, rs12913832, and rs6497292, TYRrs1042602, rs1126809 and rs1393350, RALYrs6059655, DEF8rs8051733, PIGUrs2378249, ASIPrs6119471, SLC24A4rs2402130, rs17128291, rs12896399, TYRP1rs683, KITLGrs12821256, ANKRD11rs3114908, and BNC2rs10756819. The model has proved capable of skin-colour prediction on a global scale with prediction accuracies of 0.74 for very pale, 0.72 for pale, 0.73 for intermediate, 0.87 for dark, and 0.97 for dark black [49]. More recently, another tool named VISAGE BT A&A (PSeq) for contemporary eye, hair, and skin-colour prediction was developed by Palencia-Madrid et al. [16], consisting of 41 phenotype SNPs plus 115 markers for biogeographical ancestry inference (three overlapping with the EVCs’ SNP set) for a total of 153 markers.
Freckles, including both lentigines and ephelides, consist of brown or reddish spots of the skin that can show up on different body areas (face, neck, arms, shoulders, back, and legs) predominantly in individuals with fair skin and light hair (therefore, people of European descent). Their presence, especially on exposed areas such as the face, represents a particular, and easily visible, phenotypic characteristic. Although different studies, as seen above, have focused on skin pigmentation, only four of them have investigated freckles as a separate trait.
Cao et al. [75] investigated the association between genetic variation on melanocortin-1-receptor (MC1R) gene and the presence of freckles in 225 Chinese subjects. Although MC1R was indicated as a major genetic determinant of freckle phenotype, no statistical difference was observed in individuals with freckles compared to controls, at least for the four SNPs tested (rs33832559, rs2228478, rs2228479, rs885479).
Conversely, Zaorska et al. [68] demonstrated a strong association between freckling and a different genetic locus, rs12203592 in IRF4, in 222 Polish individuals. No association was observed with MC1R gene (rs1805007) in this case either.
Based on genetic predictors previously correlated with human pigmentation, Kukla-Bartoszek et al. [94] developed a predictive model for freckle presence, divided into three categories, non-, medium-, and heavily-freckled, and obtained a moderate accuracy (respectively, AUC = 0.75, 0.66 and 0.79).
The model proposed by Hernando et al. [50] in 2018 for freckle prediction considered five genetic determinants: R variants of the MC1R gene (rs1805006, rs11547464, rs1805007, rs1110400, rs1805008 and rs1805009, defined as “R” alleles for their strong association with the red hair colour phenotype in population), IRF4rs12203592, r variants of the MC1R gene (rs1805005, rs2228479 and rs885479, defined as “r” alleles for their lower association with the red hair colour phenotype), ASIPrs4911442 and BNC2rs2153271). It leads to a cross-validated prediction accuracy of up to 74.13% [50].
On the sidelines of studies focused on the prediction of hair colour, different
authors investigated scalp hair morphology, which is known to be heritable, in
terms of shape and degree of the curl [25, 26, 27]. The first study evaluated the
predictive capacity of six SNPs (rs17646946, rs11803731, rs4845418,
rs12130862, rs1268789, and rs7349332) in 670 Europeans, identifying three of
them (rs11803731) as the most informative. Among these,
rs11803731 on TCHH (gene of the trichohyalin, a structural
protein of the hair follicle) showed the strongest effect on hair morphology,
particularly on straight hair (and, to a lesser degree, also on wavy and curly
hair). Weaker correlation with straight hair was also found for
rs7349332 (WNT10A) and rs1268789 (FRAS1).
Together, these three SNPs explained about 8% of total hair-shape variability,
with the highest predictive ability for the (rare) TTGGGG genotype (present in
only 4.5% of individuals, giving
Male pattern baldness (MPB), also named androgenetic alopecia (AGA), is a common form of hair loss in adult men, characterized by a receding hairline and/or a hair loss on the top or front of the head, thus determining a significant alteration in a person’s physical appearance. This condition is affected by both male sex hormones (androgens) and genetic predisposition, hence the name androgenetic.
Marcińska et al. [29] confirmed 29 SNPs’ role in MBP determination in European people of different ages: rs12565727 (chr1), rs929626 in EBF1, rs756853 in HDAC9, 8 SNPs on chromosome 20 (rs61374441, rs19980761, rs201571, rs6047844, rs913063, rs1160312, rs6113491, and rs2180439), rs10502861 in SLC12A2, and 17 SNPs on Xq12 (rs4827379, rs1385699, rs1352015rs1041668, rs1397631, rs5919324, rs6625150,rs12558842, rs6625163, rs2497938, rs2497911, rs2497935, rs962458, rs6152, rs12396249, rs4827545, and rs7885198). Among them, 2 SNPs, rs5919324 near AR/EDAR2 genes (chrX) and rs1998076 (chr20), showed the strongest association, followed by three other SNPs: rs929626 in EBF1, rs12565727 in TARDBP and rs756853 in HDAC9.
On this basis, they created a predictive model for MPB made up of 20 SNPs, which showed higher specificity (90%) but lower sensitivity (67.7%) in the population of men over 50 years old as compared to men under 50 years old (sensitivity of 87.1% and specificity od 42.4%), and a better predictive power for early-onset MBP (AUC = 0.761) rather than late-onset MPB (AUC = 0.657) [29]. Li and collaborators [101] conduct a large-scale meta-analysis of seven genome-wide association studies for early-onset AGA in 12,806 individuals of European ancestry demonstrating unexpected association between early-onset AGA Parkinson’s disease, and decreased fertility.
Liu et al. [102] built another predictive model including 14 SNPs and achieved a similar accuracy value for predicting early-onset MPB (AUC = 0.74). However, in 2017, Hagenaars et al. [103] studied genetic variants in a cohort of 52,000 English men, finding over 250 genetic loci to be associated with severe hair loss, and developed a prediction algorithm for identifying those at greatest risk of hair loss (AUC = 0.78).
Facial morphology is probably the most discernible physical trait, whose accurate prediction would have highly relevant implications in forensic applications. Nevertheless, given that it is affected by a large number of genes, most of which are still unknown, it remains a daunting challenge to predict an individual’s facial appearance. Moreover, although a strong genetic effect, many other factors, such as age, sex, and environment, may play a relevant role in its determination.
Given its extreme complexity and variability, all studies that have tried to predict facial morphology from genetic information have broken down the human face into basic bi-dimensional phenotypes, each consisting of a Euclidean distance (e.g., eye distance, nose width, facial height) between anatomical landmarks located on the facial surface (such as palpebral commissures, alae nasi, oral commissures, ear lobules, etc.). In this way, each facial feature could be considered separately from other facial traits and analysed in an easier way.
Boehringer et al. [48] investigated whether genetic loci involved in the pathogenesis of cleft lip and cleft palate (a birth defect determining a pathologic facial trait) were also correlated to variation in normal facial morphology, specifically in nose width and/or bizygomatic distance, studying the effect of 11 SNPs in 3026 European individuals (from Germany and the Netherlands). They found a statistically significant association between rs1258763 (near the GREM1 gene) and nose width (but only in the German cohort, and stronger in males than females), and between rs987525 and bizygomatic distance (but only in the Dutch cohort). These markers were able to predict, respectively, ca. 2% of nose width variation (rs1258763) and 0.57% of bizygomatic distance variation (rs987525) [48].
In 2014, Paternoster et al. [30] identified an association between rs7559271, which is an intron of PAX3, and nasion position in a population of adolescents. Furthermore, common variants in this gene are also associated with prominence and vertical position of the nasion [30].
Claes et al. [104] investigated the effect of 24 SNPs showing a significant effect on normal-range facial morphology, spread over 20 genes: POLR1D, CTNND2, SEMA3E, SLC35D1, FGFR1, WNT3, LRP6, SATB2, EVC2, RAI1, ADAMTS2, ASPH, DNMT3B, RELN, UFD1L, ROR2, FGFR2, FBN1, GDF5, and COL11A1. They first reconstructed a ‘base-face’ using sex and ancestry information, both estimated from the same DNA sample through analysis of, respectively, amelogenin and AIMs. Then, they overlapped the effects of the 24 SNPs onto the ‘base-face’ to obtain the final predictive model. Although facial morphology turned out to be mainly affected by sex and ancestry, the SNPs’ effect could significantly increase the distinctiveness of facial prediction [22, 104].
Jin et al. [32] investigated the genetic association between four SNPs selected from facial-shape-associated genes (rs2277404 and rs7316271 on ABCC9, rs12635264 and rs1717652 on MASP1) and eyelid morphology (single vs. double eyelid) in a cohort of 96 Chinese individuals. Only one SNP, rs2277404 in ABCC9, demonstrated significant association with difference in the Chinese-eyelid phenotype [32]. Shaffer et al. [105] demonstrated that MAFB, PAX9, MIPOL1, ALX3, HDAC8, and PAX1 play roles in craniofacial development or in syndromes affecting the face. Li et al. [28] tested the effect of 125 facial-shape-associated SNPs on facial features in a European-Asian admixed population of 612 individuals. Eight SNPs showed a significant association with one or more facial traits (EDARrs3827760, LYPLAL1rs5781117, PRDM16rs4648379, PAX3rs7559271, DKK1rs1194708, TNFSF12rs80067372, CACNA2D3rs56063440, and SUPT3Hrs227833) and explained 6.47% of the facial variation, adjusted for sex, age, and BMI. For example, rs3827760 on EDAR (a gene involved in the development of ectodermal-derived tissues, including skin) showed an association with incisor-teeth shovelling, earlobe size and attachment, ear protrusion, and ear-helix rolling. All of the eight SNPs had a different allele frequency between Europeans and Asians, and four of them (rs4648379,rs3827760, rs7559271 and rs1194708) showed an inverse allele frequency in the two groups [28]. Li L. et al. [33] focused on two specific facial traits: the epicanthal fold (a skin fold covering the inner angle of the eye, which is typical of people of Asian descent but can also be present with lower frequency in other populations) and palpebral fissure height and width. They observed, in a Chinese cohort, a significant association between rs2074612 and palpebral fissure appearance, while no correlation was found for epicanthal fold [33].
Fagertun et al. [106] evaluated the genetic association between facial traits in an Icelandic population of 1266 individuals and a large number of SNPs selected from a genome-wide association analysis, instead of using a few markers previously chosen from candidate genes. Even with this innovative method, they could explain only 4.4–9.6% of facial shape. In particular, they observed that six facial features were predictable with statistically significant accuracy from genetic information, with some gender differences: face width (for both sexes); mouth width (only in men); lip fullness and, to a lesser degree, eye distance, eye size, and eyebrow width (only in women) [106].
In 2019, Mbadiwe et al. [107] proposed, through a literature search, a panel composed of 6,816 SNPs associated with the human face and called it FaceSNPs. This panel is available upon request. Moreover, they also identified chromosomes that promise better performance in genotype-to-phenotype prediction of human face characteristics (so, for example, chromosomes 10, 17, 1 and 5) [107].
Liu M. et al. [31] have also worked with the genetic basis of facial morphology in a Chinese population, confirming a significant association with 12 reported SNPs which are together responsible for up to 3.89% of age- and BMI-adjusted variance (EX41rs17479393, PAX3rs974448, RAB7A/ACAD9rs2977562, DCHS2rs9995821 and rs2045323, C5orf50rs6555969, SUPT3H/RUNX2rs1852985, MSRArs11782517, EYA1rs10504499, GSCrs2224309, DICER1rs7161418 and DHX35rs2206437) [31].
Recently, White and collaborators identified 203 genome-wide significant signals associated with multivariate normal-range facial morphology. Among which 53 genome-wide significant peaks are located in region with no previously known role in facial development or disease. Moreover, they demonstrated interaction between variants at different loci affecting similar aspects of facial shape variation, identifying gene sets that work in concert to build human faces [108].
High myopia is a condition characterized by a highly negative refractive error
(
Amer et al. [110] investigated several (sixteen) SNPs in the cytochrome b gene of mitochondrial DNA to test their association with obesity in 66 Saudi Arabian individuals. Contrary to a previous study, they found only a weak relation with two non-synonymous mutations (corresponding to nucleotide substitutions, A15043G and C15677A respectively, in two obese males and two obese females), concluding that further research is needed to provide evidence for the possibility of applying cyt-b gene in obesity diagnosis [111].
Adult height has demonstrated itself to be a highly hereditable trait, whose variability is affected by hundreds to thousands of contributing genes [112]. In 2014, Liu et al. [113] introduced a model for DNA predictability of tall stature in Europeans consisting of a subset of 180 height-associated SNPs previously identified in other studies. In 2017, Marouli et al. [114] identified 32 rare and 51 low-frequency coding variants associated with adult height. In 2019, the model was updated and expanded by the same authors with the addition of newly discovered polymorphisms, for a total amount of 689 SNPs, in order to increase the model’s prediction accuracy (AUC = 0.79, compared to the previous 0.75) [112]. Subsequently, Jing et al. [115] investigated the predictive power of the same SNPs, originally identified for European individuals, in the Uyghurs, a population presenting an admixture of European and East-Asian genetic traits. On the one hand, the study confirmed a moderate genetic correlation between Uyghurs and Europeans, since some European height-associated SNPs also showed significant correlation in the Uyghurs. However, on the other hand, the study emphasises a substantial difference in terms of genetics of human stature (allele frequencies, allele effect sizes, and allele effect directions) between the two different populations [115].
It should be noted that different methods have been used to evaluate and classify pigmentation trait phenotype (iris, hair and skin colour). In some studies, colour assignments were made in a subjective manner, through direct observation and visual identification [26, 45, 84]. Some studies made use of specific tools and equipment, such as professional cameras with top-quality lens and flash systems, but also colourimeters, spectrometers and spectrophotometers, usually ensuring standardized lighting and distance-to-subject conditions for each subject, so as to obtain a more objective and quantitative classification [39, 42, 43, 44, 46, 53, 57, 58, 61, 62, 66, 67, 73, 75, 84, 85, 92]. Sometimes trait colour was determined in both manners, subjectively and objectively. In some studies, participants were scrutinized directly by one or more investigator(s) or volunteer(s), who were not specialists but who had been specially trained for the task [39, 44, 46, 62, 66, 70, 83, 85]. However, especially for eye and skin colour, one of the main approaches consisted of seeking advice from specialists in the sector, usually ophthalmologists and dermatologists, but also anthropologists with experience in phenotyping [24, 26, 38, 61, 72, 73, 75, 87, 90]. In a few cases, phenotype data were collected from participants themselves, through self-declaration or self-administered questionnaires [60, 64, 69, 92, 93] or were not known and just inferred from ethnic background [53, 65]. Occasionally, the method used was not indicated [41, 60, 65, 74, 76]. In one study, no pigmentation data were available for any of the participants [56].
One of the recent fields of research in forensic genetics has been focused on the analysis of EVCs such as eye colour, hair colour (including hair greying), hair morphology (including hair loss), skin colour, freckles, facial morphology, high myopia, obesity, and adult height, with important repercussions in the forensic field, in order to predict the appearance of EVCs of a trace left at a crime scene or of an unknown person represents an additional investigatory tool in the context of forensic investigative techniques.
So far, extensive knowledge has been attained for pigmentation traits, i.e., eye, hair, and skin colour, while phenotyping beyond pigmentation traits is still in its earlier stages and far from being deeply understood.
By now, one of the most widespread and applied predictive tools for simultaneous eye and hair colour prediction is HIrisPlex, which was initially developed based on a European database but shown to be suitable for people of non-European descent as well. Yet, despite this, there remains potential bias linked to geographical origins of the population to which it is applied, especially in the case of admixed populations.
In particular, as reported in Fig. 3, the IrisPlex consists of six SNPs currently identified as major eye-colour predictors. The selection of the best SNPs for EVC determination, indeed, seems to be highly population-dependent, suggesting that panels should be adapted for different geographical regions. One possible solution that has been suggested in the literature is to combine EVC prediction with DNA that allows for the inferring of the unknown person’s geographic origin with high accuracy. Although highly accurate for predicting the most extreme phenotypes, the model is not as informative for intermediate phenotypes. These findings highlight that DNA-based inference of a person’s physical appearance still needs to be cautiously applied in real-world casework since an erroneous prediction may lead an investigation down the wrong path. Further research is required that is aimed at broadening the pool of SNPs known for each phenotypic trait and for individuals from different geographic regions.
Considering all the SNPs that have been studied, those found to be the most strongly associated with iris colour are HERC2rs12913832, HERC2rs1129038 and OCA2rs1800407 [42, 46, 47, 52, 62, 64, 65, 66, 67].
When considering the prediction of hair colour, different polymorphisms in genes associated with melanin biosynthesis (Fig. 2) showed significant association with different phenotypes [34]. Following this new evidence, the IrisPlex system was integrated with new polymorphisms sets in order to develop and validate the HIrisPlex model as seen in Fig. 3 [42]. The questions that still remain open are those related to the predictivity of blond hair colour [89] due to the possibility that subjects born blond then turn brunette with growth and with the poor ability to predict, from a genetic point of view, the greying of hair with aging [90].
As for other pigmentation traits previously discussed, the first genes to be associated with skin colour were those encoding for proteins which are involved in the melanin production within melanocytes, and it has been observed that many genes found to be associated with hair and iris colour variations also seem to be associated with skin colour variation [19] (MC1R, SLC45A2/MATP, SLC24A5 and OCA2) [35, 36, 91]. Significant associations with skin colour were found for variants of MC1R, HERC2,OCA2, SLC45A2, SLC45A5, TYR, VDR, ASIP, TYRP1, KILTG, TPCN2 and SLC24A4 genes. In 2018, a combined tool for simultaneous prediction of eye, hair, and skin colour named HIrisPlex-S was introduced, where skin colour prediction was based on a set of 36 SNPs (of which 19 had also been included in the previous model, plus 17 novel markers) [18]. More recently, another tool named VISAGE BT A&A (PSeq) for contemporary eye, hair, and skin-colour prediction has been developed, consisting of 41 phenotype SNPs plus 115 markers for biogeographical ancestry inference (three overlapping with the EVCs’ SNP set) for a total of 153 makers [16].
In the examined literature, the most studied statistical models applied to IrisPlex, HIrisPlex and HIrisPlex-S system were based on MLR analysis in order to reach posterior probabilities for each trait (three for eye colour, four for hair colour and five for skin colour) [20, 24, 42, 45, 57, 76]. Some authors have proposed different statistical approaches, such as Principal Component Analysis (PCA) [92], Iterative naïve Bayesian approach [38, 39], other Bayesian classifiers such as Snipper [43], artificial neural networks and classification trees [40]. More recently, Katsara et al. [20] have proposed Machine Learning (ML) as a tool to be used for the prediction of visible traits. ML algorithms use mathematical-computational methods to learn information directly from data and ML algorithms improve their performance in an “adaptive” way as the “examples” learn from increase. Katsara et al. [20] applied some of the most popular machine learning systems to three datasets of samples for eye colour, hair colour and skin colour, from subjects belonging to populations around the world, comparing them with the predictivity provided by IrisPlex, HIrisPlex and HIrisPlex-S systems. This study concluded that there are no substantial differences in the ability to predict visible traits with ML or with MLR and, therefore, MLR, in the current state of knowledge, represents the best approach to evaluating the predictivity of these traits starting from DNA [20]. It should be noted that while MLR is a useful categorical classifier and has been successfully employed for the prediction of eye, hair and skin colour, there is a real risk for over-fitting data with small sample sizes when applying MLR. To avoid overfitting a regression model, the sample should be large enough to handle all of the terms that are expected to be included in the model, thus suggesting that a huge number of training data set should be settled up.
It should also be remembered that genetic background is not the only determinant at stake, since other factors such as age, gender and BMI, can affect a person’s physical appearance and should always be taken into account when dealing with predictive models in order to reduce prediction error and increase reliability. For instance, a gender divergence has been shown for eye colour, according to which, given the same genotypic background, males have lighter eye colours than females.
Alongside the main characteristics which are currently the most studied (pigmentation traits), there may be less-studied characteristics such as adult height, hair shape, facial morphology, male-pattern baldness, obesity, freckles, and high myopia that could act as auxiliary tools and help add information to reduce the size of the referring cluster. In particular, predictions on hair structure and male-pattern baldness are still under development, while the prediction of facial features remains among the most challenging goals of this field of research.
DNA analysis of genetic profiles is a comparative analysis, whose goal is to find a match between a trace found at a crime scene and a person (victim or suspect) through a direct comparison or through a DNA-database search. When the reference genetic profile is not available, this kind of approach may be useless because genetic profiles cannot be compared. Under these circumstances we consider that the forensic usefulness of EVC might consist of adding further information to criminal investigations in order to restrict the field of qualitative information useful for identifying the subjects potentially involved [19].
In fact, it is known that crime-scene stain analysis can play a crucial role in connecting a person to an object or a place with the possibility of investigating the visible characteristics that reduce the number of possible suspects of a crime, especially in cases where the police have little or no knowledge of the identity of the trace donor and how to find him/her, or in complex cases of missing persons or disaster-victim identification [116].
There is no doubt that EVCs can be altered in many different ways (e.g., cosmetics, coloured contact lenses, dyed hair colour, self-tanning skin procedures, plastic surgery, etc.) to alter one’s appearance in ID portrait images. There is always the possibility that the molecular inference of physical features might not correspond to body appearance [117]. Furthermore, even if today the detection of EVC has reached a good level of accuracy, future research activities have to be focused on reducing the limitations of available eye, hair and skin colour DNA testing in predicting intermediate categories [13].
Some critical issues that should be considered to correctly interpret the prediction studies of non-pigmented visible traits are the missing heritability in many GWAS studies used to find SNPs associated with phenotypes and the apparently inverse relationship between effect size and allele frequency (abundance) for complex traits like height [118, 119]. It has been demonstrated that for many complex traits there might be many SNPs (additional to those significantly associated with a certain phenotype) with small effects that together play a significant role in the phenotype variance. This hypothesis is also supported by data that tell us that GWAS conducted on ever growing sample sizes is able to find new hits, in particular for appearance traits. For instance, in the case of adult height, the number of associated variants has grown from about 40 in the first GWAS to about 700 when study sizes increased to 250,000 individuals and to 3290 in the latest study that included 693,529 participants [120, 121].
As seen, quite different methods of evaluation have been applied for colour assignments to different categories in studies dealing with pigmentation traits. Some authors have used quantitative and more objective methods, while in some others cases the evaluation has been based on the subjective judgment of one or more investigators. This means that different studies could be difficult to compare. In this regard, it is also worth noting that quantitative measurement is fundamental to obtaining an objective evaluation of the various colour categories. However, as observed by Andersen et al. [62], in real forensic caseworks, the categorization would be based on human interpretation and not on an objective method.
Despite the initial enthusiasm for pigmentation-related traits, and which continues to fuel research and training through international circuits, there is much information currently lacking in this field when facing with real applications in forensic caseworks. It should not be forgotten that, from a genetic point of view, physiognomic traits, precisely because they are multifactorial characteristics, are particularly difficult to identify unlike simple Mendelian traits. It should therefore not be forgotten that, at least at the present time and in the near future, only a few physiognomic characteristics, as mentioned, will be identifiable with a certain degree of accuracy. For all other conditions, estimates of greater or lesser probability of occurrence of this or that phenotype can only be provided, with the consequence of a reduced statistical weight of the information obtained. Relatively to the use of polygenic scores (largely applied for predicting therapeutic response in multi-genic diseases) for predicting complex phenotypes beyond pigmentation traits (for instance, adult height) numerous DNA variants previously implicated in normal height variation in Europeans have been demonstrated of being involved in determining tall stature. Nevertheless, it is necessary to improve the modeling of genetic interactions and allelic heterogeneities within height-associated loci [113]. This achievement is strictly related to future applications in real forensic caseworks where genomic height prediction can be applied.
Despite the potential usefulness of forensic DNA phenotyping, this innovative approach has not gained a socio-scientific consensus yet [122, 123]. Indeed, there still exists a lively debate concerning the legitimacy of its use in the criminal justice system, which is primarily focused on socio-ethical issues [117, 122, 123]. For instance, one of the main points under discussion is that information deriving from this technique does not identify a single or specific person (the potential suspect) but a cluster of individuals sharing similar visible traits, which leads to both legal and ethical concerns, such as the unfeasibility of massively screening the whole suspect population, or the risk of generating racial prejudice and stigmatization. In this framework, there is a need for further discussion regarding not only FDP applications and utility, but also related risks and, moreover, there is a need for new laws, especially considering that only a few countries have already enacted specific legislation on the matter, while in most other countries a legal vacuum exists or these techniques are specifically forbidden [124].
From a practical point of view, the application scenario for forensic DNA phenotyping in real caseworks should be that, at the moment, the STR profile has the first priority and phenotypic SNPs analysis should be performed on remaining DNA and the source tissue of the trace should be known. Given the nature of the prediction frameworks validated for forensic DNA phenotyping, single source or major/minor profiles are suitable for these analysis and multi-person mixtures should be avoided. Results should then be communicated as probabilities for a given phenotyping trait, or genetic (y-related or autosomal) ancestry, while the underlying genetic data should not be shared or stored in databases. The basis for any court proceeding is still the determination of STR profiles, and suspects will be excluded or identified based on traditional STR profile. In case of identification using STR profiles, phenotypic SNPs data are not relevant and can be deleted.
The ease of adding these types of analyses into the already overworked crime labs would be difficult to be set up in daily routine caseworks. While the instrumentation may be the same of “traditional” forensic genetics analysis, the training and integration into an already busy caseload would be difficult from a practical standpoint. It should be remembered that appropriate training is imperative to enable forensic practitioners to apply bio-informatic methods and given set protocols to analyse data generated and interpret such data in the context of case-related questions. Last but not least, it is essential that forensic geneticists are able to explain to their clients (police forces, judicial authorities) the meaning of the data obtained. After having spent years explaining the meaning of prepositions, at the source level and at the activity level, it will now be necessary to make us understand a different perspective, namely that these data do not give us likelihood ratios useful for identification purposes, but they give us a priori information on characteristics shared by the person of interest with many other people, and thus cluster the range of suspects. Results for EVCs prediction are communicated by threshold of probabilistic accuracy about each trait: this means that new knowledge and new cognitive and communicative skills are required both for scientists and investigators in order to request, understand and use such data. Therefore, in a potential future involving the forensic application of phenotype prediction, the involvement of agencies that set standards and accredit laboratories would be crucial to uphold high standards of admissibility of EVCs predictions to support criminal investigations.
The variations in the distribution of genetic differences on a geographical basis (based on the divergence and stabilization of migratory flows in the different geographical areas of the globe over the millennia), despite the awareness of their continuous distribution and without absolute characterizations, still allow us to provide information, albeit of a statistical nature, about the geographical origin of the sample. Indications that, added to those more directly connected with other physiognomic traits, can thus be usefully employed more in excluding a subject belonging to a certain ethnic-geographical group, rather than in positively associating him or her with it, in the awareness that the growing admixture between populations due to recent and current migratory flows may not allow for such distinctions in the future.
From an ethical point of view, the greatest concern linked to the use of such information is that it may cause a stigmatization of the group of subjects who share that visible characteristic. The risk is also associated with the likelihood that certain groups of individuals who share certain characteristics will be discriminated against as being statistically more frequently responsible for certain crimes. The passage from the stigmatization of single subjects to discrimination against whole groups could lead to a degeneration towards racial discrimination. The prediction of FDP on the basis of the genotype clusters the population groups on the basis of very frequent characteristics in that specific population, thus highlighting and enhancing the phenotypic differences that characterize the various ethnic groups by increasing the visibility of phenotyping differences. This could lead, as the cases in which investigations are initially guided by information of this type increase, to a collectivisation of suspicion for certain populations [125, 126]. Nevertheless, the discussion on the applicability of the prediction of visible traits cannot be detached from the discussion on the possible effects on minority groups that are already vulnerable to the action of police forces or judicial authorities. This risk was considered by some of the study participants by Granja et al. in 2020 [124] and has been highly debated in the literature [122, 127]. Worrying about the sociological and ethical implications of the application of these technologies does not mean denying their biological robustness and scientific soundness. Stimulating a greater bioethical, political, and social debate can also be of great help for forensic geneticists who do research in this area and who will apply these technologies in real caseworks, without necessarily entrenching oneself on contrary positions between detractors and promoters of the use of these technologies.
Although they are not yet widespread and systematically applied in different judicial systems among different countries, the diffusion and progress of research in the field of EVCs makes the techniques for determining, at least, pigmentation characteristics nonetheless promising. The application in real forensic cases of forensic phenotyping to achieving reliable investigative information is a promising tool that is particularly useful when other techniques cannot provide useful information. Moreover, certain limits should be taken into consideration to avoid overestimating the actual possibilities of this tool in forensics. In fact, although highly accurate for predicting the most extreme pigmentation phenotypes, the model is not as informative for intermediate phenotypes. Consequently, an improvement in the accuracy of determining intermediate colours is needed. Moreover, there still remains potential bias linked to the geographical origins of the population to which DNA phenotyping is applied, especially in the case of admixed populations as well as for eye and hair colour prediction.
Forensic DNA phenotyping of pigmentation traits (in particular eye, hair and skin colours) can now provide extraordinary information in the course of investigations. The real challenge for our scientific community will be to establish uniform rules of application at least at a European level, and continue to implement not only the analytical capacity but above all the interpretative one.
The issues raised by the use of these markers should be necessarily discussed, both to improve the awareness of forensic geneticists on the limits of these techniques, and to be able to think of technical and regulatory interventions that can make their applicability more concrete, widespread and uniform internationally.
LC and PT contributed to the study conception, design and supervision. CP, AD and AG dealt with data curation and formal analysis. The first draft of the manuscript was written by AG and CP; PT, AD and LC commented on previous versions of the manuscript the final draft of the manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
AGA, androgenetic alopecia;