














Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Discipline of Chemistry, Indian Institute of Technology Gandhinagar, Gujarat, India-382355
2 Discipline of Biological Engineering, Indian Institute of Technology Gandhinagar, Gujarat, India-382355
Abstract
Phosphatidylinositol-3 kinase-related kinases (PIKKs) is a class of six unique serine/threonine kinases that are characterized as high molecular mass colossal proteins present in multicellular organisms. They predominantly regulate the innumerable eukaryotic cellular processes, for instance, cell-signaling cascades related to DNA damage and repair, cell growth and proliferation, cell cycle arrest, genome surveillance, gene expression and many other important yet diverse functions. A characteristic PIKK member comprises of an N-terminal HEAT domain, followed by FAT domain, a highly conserved kinase catalytic domain, and a C-terminal FATC domain. In this comprehensive review, we reassess and discuss various established functions of all the six PIKK members with each function corroborated by their structural topology. In addition to the domain architecture of these atypical kinases, their specific inhibitors have been briefly deliberated. This review gives us the impression of the emergent importance of PIKKs, which, despite of their complexity, are the hub of research with respect to the inhibitor development.
Keywords
- PIKK
- Phosphatidylinositol-3 kinase-related kinases
- mTOR
- mammalian target of rapamycin
- ATR
- ATM- and Rad3-related kinase
- ATM
- Ataxia telangiectasia mutated kinase
- DNA-PKcs
- DNA dependent protein catalytic subunit
- TRRAP
- Transformation-transactivation domain-associated protein
- hSMG1
- Suppressor with morphological effect on genitalia family member
- DNA damage response
A human body comprises of ~1013 cells and these cells undergo thousands of DNA (deoxyribonucleic acid) damaging events leading to the formation of DNA lesions which can further hamper the genome replication and transcription (1). The cells have evolved themselves in an efficient way to generate a DNA damage response (DDR) which is activated on double-stranded break detection or any other exogenous/ endogenous damage followed by an intricate cascade of cell signaling pathways (2). In the 1990s, a series of high molecular mass atypical serine/threonine kinases were discovered, cloned and classified as phosphatidylinositol 3-kinase (PI3K)-related kinase (PIKK) (3). An interesting fact about this family of kinase being that their catalytic domain resembles more to PI3-kinase family of phospholipid kinases than to the classical protein kinase (4), (5). Despite their humongous size, the catalytic domain of PIKK members spans over a few 100 amino acid residues towards the C-terminal and contributes merely 5-10 % to the whole protein structure with hSMG1 (suppressor with morphological effect on genitalia family member) as an exception (6). This family of protein kinases is highly conserved in eukaryotes during evolution and few homologs are found in yeast as well, but there are no reports stating their expression in prokaryotes (7), (8). In Homo sapiens, six PIKK family members have been identified, which includes Ataxia telangiectasia mutated kinase (ATM), ATM- and Rad3-related kinase (ATR), DNA dependent protein catalytic subunit (DNA-PKcs), mammalian target of rapamycin (mTOR), suppressor with morphological effect on genitalia family member (SMG1), and transformation-transactivation domain-associated protein (TRRAP).
Over the past few years, there have been extensive studies on PIKK family of protein kinases resulting in the revelation of significant insights into their biological functions at the cellular level. Members of PIKK family participate in diverse set of cellular functions such as meiotic and V(D)J recombination, chromosome maintenance, DNA damage sensor and repair, cell cycle progression and cell cycle arrest, and their dysfunction results in a variety of diseases, including cancer and immunodeficiency neurological disorder (7), (9). On the structural level, all the six PIKK members share common features including α- helical rich HEAT (Huntingtin, elongation factor 3 (EF3), protein phosphatase 2A (PP2A), and the yeast kinase TOR1) domain towards N- terminal and a catalytic kinase domain towards C-terminal flanked by FAT (Frap, ATM, and TRRAP) and FATC (FAT C-terminal) domains on each side (Figure 1) (9). The FAT domain and the extended C-terminal kinase including the PRD (PIKK- regulatory domain) and FATC are also documented as a single ‘FATKIN’ unit (10). These atypical kinases have a themed regulation that depends on their subcellular localization and requires the interaction of their respective partner activator proteins or DNA to form a protein-protein or nucleic acid-protein complex (11). While ATR is rapidly activated by dsDNA break and stalled replication forks, ATM and DNA-PKcs signaling cascade are triggered during ssDNA break. These kinases are then recruited at the site of DNA lesion with the help of their activator proteins like ATRIP (ATR interacting protein), Mre11-Rad50-Nbs1 (MRN) and Ku70/80 for ATR, ATM, and DNA-PKcs respectively (12), (13), (14), (15). mTORC1 recruitment to the perinuclear compartment is assisted by RAG (recombination-activating genes) GTPases that interact with raptor and help in the localization of mTORC1 complex near to its activator protein i.e. Rheb (16). SMG1 also interacts with UPF1, UPF2, and UPF3 (UPF: up-frameshift) at premature termination codons and exon-junction complexes on mRNA, thus, regulating the nonsense-mediated mRNA (messenger ribonucleic acid) decay (17). TRRAP, an adaptor protein, is essential for the c-Myc- and E1A-mediated oncogenic transformation (18) (Figure 2-3). These features have attracted the tremendous interest of academic and pharmaceutical industries especially inhibiting the PIKK family proteins and their signaling pathways for effective cancer therapy. In this review, we comprehend the structure of reported PIKK family of protein kinases and discuss the strategic perspective for the discovery and development of PIKK kinase inhibitors for cancer therapy.
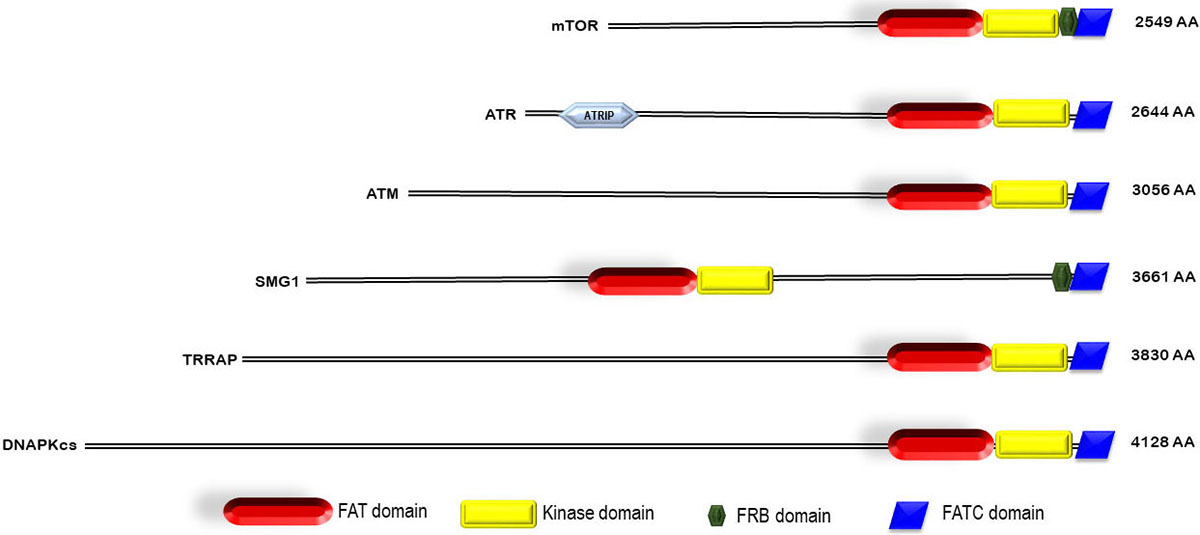 Figure 1
Figure 1Domain architecture of PIKK family.
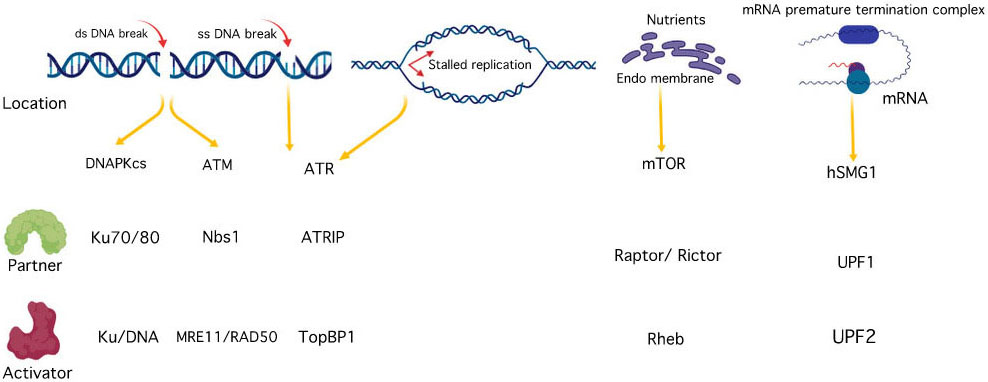 Figure 2
Figure 2PIKK family activation and interacting partners.
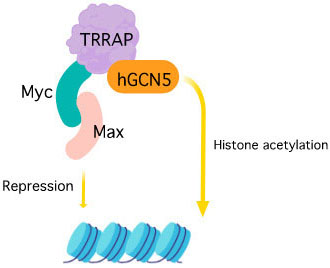 Figure 3
Figure 3TRRAP activation and interacting partners.
mTOR (mammalian target of rapamycin), also known as FRAB (FKBP-rapamycin associated protein) or RAFT (rapamycin and FKBP target ) was the first PIKK member to be cloned and studied in yeast (19) (Table 1). The discovery of mTOR is splendidly explained by Livi et al. in (20). mTOR is a mammalian serine/threonine kinase that is structurally and functionally conserved in all eukaryotes including fungi, worms, flies, plants, and mammals (21). It controls the cell growth in response to nutrients and growth factors and is frequently deregulated in cancer. mTOR exists as mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), first complex being rapamycin-sensitive whereas second complex being rapamycin insensitive (22). The N-terminal FAT domain of mTOR embraces a ‘C’-shaped solenoid-like structure made up of four sub-domains rich in α-helices. It comprises of 28 α-helices, α1 to α22 form three discontinuous domains (TRD1-3) and α23 to α28 form a single domain known as HRD that belongs to HEAT family. This is trailed by the kinase domain of the protein that adopts two-lobe architecture with a small N-terminal lobe (N-lobe) and a larger C-terminal lobe (C-lobe). A cleft formed in between the two lobes is the binding site for the ATP. The N-lobe begins with kα1 helix with FRB (FKBP-rapamycin-binding) domain followed by a β-strand and two short helices. The C-lobe of the kinase domain has mLST8 (mammalian lethal with SEC13 protein 8) binding site known as LBE (Lst8-binding element) that is specific to mTOR, a well-ordered activation loop with kαAL insertion, and a kα9b insertion (residues 2425–2436) followed by FATC domain. An important domain known as a negative regulatory domain (NRD) is also present between residues 2430–2450, which on deletion activates mTOR, both in vivo and in vitro. The active site (Figure 4) is highly restricted due to the presence of FRB, LBE, and mLST8 in addition to kα9b and the 55-residue unstructured segment that follows the helix (part of NRD) (23).
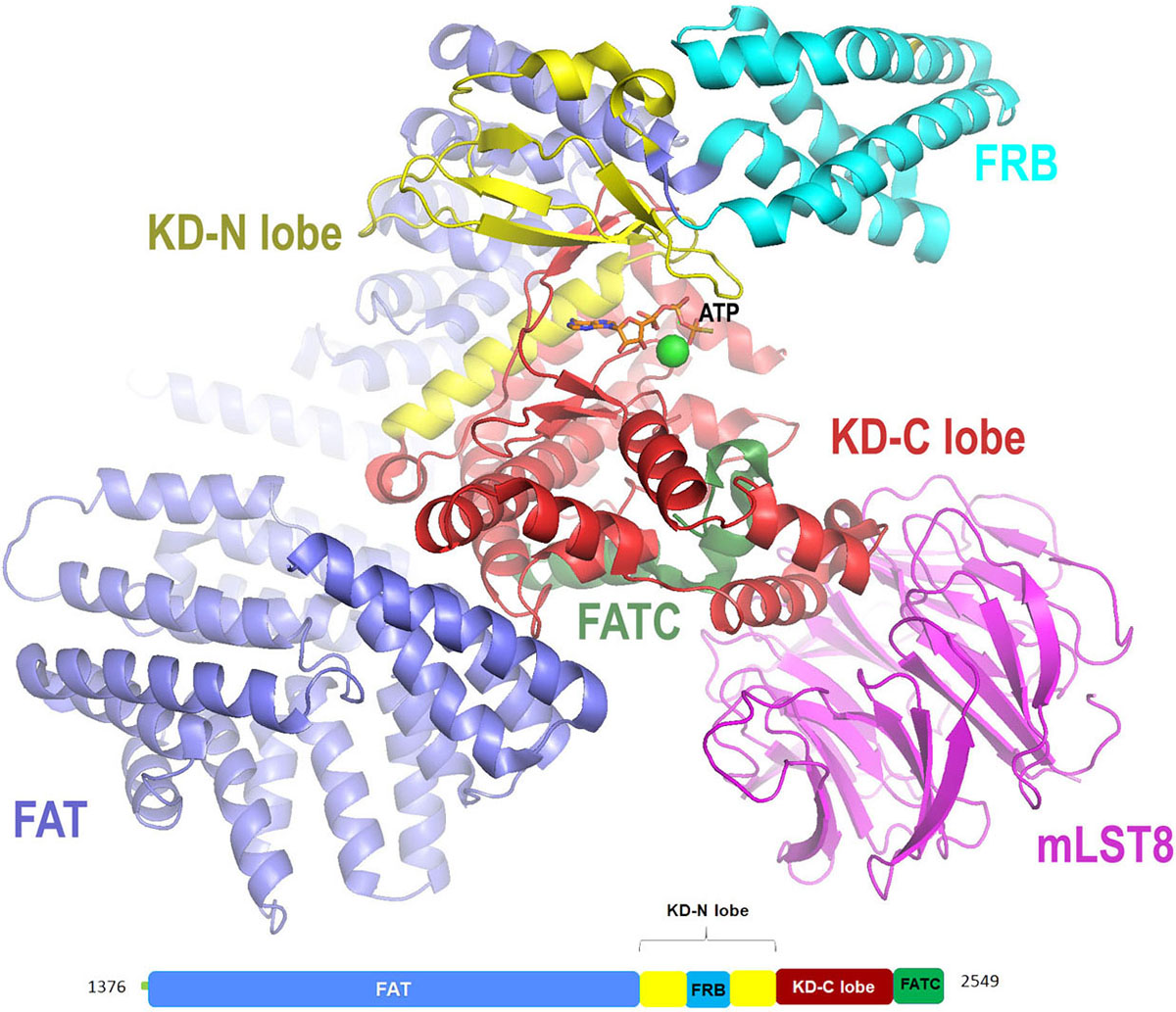 Figure 4
Figure 4Cartoon representation for X-ray crystal structure (at resolution of 3.5 Å) of mTORΔN-mLST8-ATPγS-Mg with color-coded domains (adapted with permission from PDB ID: 4JSX .
| PDB ID | PIKK protein name | Method | Resolution (Å) | Reported inhibitors | References |
|---|---|---|---|---|---|
| 4JSV | mTOR | X-Ray | 3.5 | Torin2(34), NVP-BEZ235(111), Temsirolimus(112), Everlimus(113), AZD8055(114), AZD2014(114), OSI-027(115),INK 128/MLN0128(37) | (23) |
| 4JSN | mTORdeltaN-mLST8 complex | X-Ray | 3.2 | (23) | |
| 4JSP | mTORdeltaN-mLST8 complex Mg2+ complex | X-Ray | 3.3 | (23) | |
| 4JSX | mTORdeltaN-mLST8 complex | X-Ray | 3.5 | (23) | |
| 5WBY | mTOR(deltaN)-mLST8-PRAS40(beta-strand) complex | X-Ray | 3.1 | (121) | |
| 5ZCS | mTOR complex 2 | Cryo EM | 4.9 | (122) | |
| 5FLC | mTOR complex 1 | Cryo-EM | 5.9 | (123) | |
| 2GAQ | FRB domain of mTOR | NMR | (124) | ||
| 2NPU | FRB domain of mTOR | NMR | (125) | ||
| 5NP0 | Closed dimer of ATM | Cryo-EM | 5.7 | Torin2(34), Compound 2(Cinnoline carboxamide)(58), AZ31, AZD1390 | (54) |
| 5NP1 | Open protomer of ATM | Cryo-EM | 5.7 | (54) | |
| 6HKA | FATC domain of ATM | NMR | (126) | ||
| 5YZ0 | ATR-ATRIP complex | Cryo-EM | 4.7 | Torin2(34),VE 821(70),VX 970(116), AZ20(117), AZD6738(118), NU6027(119), NVP-BEZ235(111) | (127) |
| 5LUQ | DNA-PKcs | X-Ray | 4.3 | Torin2(34), NU6027(119), NVP-BEZ235(111) | (79) |
| 3KGV | DNA-Pkcs | X-Ray | 6.6 | (78) | |
| 5Y3R | DNA-PK Holoenzyme | Cryo-EM | 6.6 | (128) | |
| 5W1R | DNA-Pkcs | Cryo-EM | 4.4 | (80) | |
| 5OJS | SAGA and NuA4 coactivator subunit Tra1 |
Cryo-EM | 3.7 | (98) | |
| 4ZRD | SMG1 mutant F278N | X-Ray | 2.3 | Compound 1 |
(129) |
Rapamycin is a macrocyclic antibody produced by Streptomyces hygroscopicus known to have antibiotic activity against various candida species (24), (25). It also exhibits an immunosuppressent effect to prevent kidney graft rejection by blocking T-cell activation. It is the first mTOR inhibitor (hence the name mammalian target of Rapamycin (mTOR)) discovered to have anticancer activity. It showed allosteric inhibition by binding to immunofilin domain of FK506-binding protein12 (FKBP12) and FKBP12-Rapamycin-Binding (FRB) domain (22).
Formation of this ternary complex inhibits phosphorylation S6K1 and 4E-BP1, a well-charactarised substrate of mTORC1. As a result, it specifically inhibits mTORC1, but not mTORC2. However, the clinical use of rapamycin as an anticancer agent is limited because of its poor physicochemical properties such as stability and solubility. To overcome these concerns analogs of rapamycin, such as Temsirolimus and Everolimus have been identified (Figure 5) (26). Temsirolimus is an ester analog of rapamycin approved by US food and drug administration in 2007 for the treatment of advanced renal carcinoma. Whereas Everolimus is the 40-O-(2-hydroxyethyl) derivative of rapamycin approved for the treatment of HER2-negative breast cancer in combination with exemestane (27), advanced hormone receptor-positive (28), and for tuberous sclerosis complex-associated partial-onset seizures (29), (30), (31), (32).
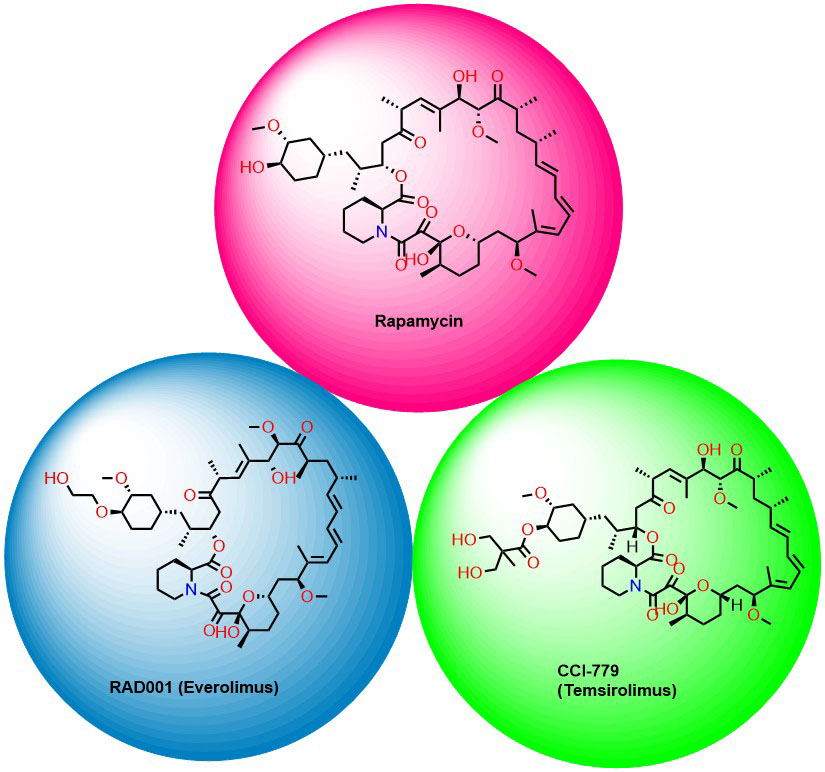 Figure 5
Figure 5Rapalogs as mTOR inhibitors.
Upon reviewing the ATP competitive binders for mTOR, inhibitors like Torin2, AZD8055, INK 128 and OSI027 have displayed promising anticancer activity (Figure 6)(33). Torin2 is a second-generation ATP (adenosine triphosphate) competitive mTOR inhibitor developed by Liu et al, that shows selective mTOR kinase inhibition with EC50 = 250 pmol/L (34), (35). Also, it has superior pharmacokinetic profile in comparison with other mTOR inhibitors. In addition, Torin2 also exhibits potent biochemical and cellular activity against ATM (EC50 = 28 nmol/L) and ATR (EC50 = 35 nmol/L) kinases with respect to radiotherapy in Hela and HCT116 (Human colorectal carcinoma cell line) cell lines (34). The preferential selectivity of Torin2 towards the mTOR kinase and its structural elucidation studies have been demonstrated by Haijuan Yan et al (23). According to the reports, the tricyclic benzonaphthyridinone ring binds to the adenine site of ATP and forms a hydrogen bond with the ‘hinge’ between the N- and C-lobes. In addition, benzonaphthyridinone ring stacks more effectively with the indole group of Trp2239 from the hinge than the ATP and absence of Trp2239 in canonical protein kinase makes Torin2 contribute towards ∼800-fold specificity for mTOR over PI3K. Further, structure interpretation also reveals that the trifluoromethyl group of Torin2 packs between Ile2163, Pro2169 and Leu2185 residues of N-lobe helping in the stabilization of Torin2 – mTOR complex. Strangely, the amino group of Torin2 extends to the ‘inner hydrophobic pocket’, an area at the back of the cleft, but fails to show any interaction with the amino acid residues.
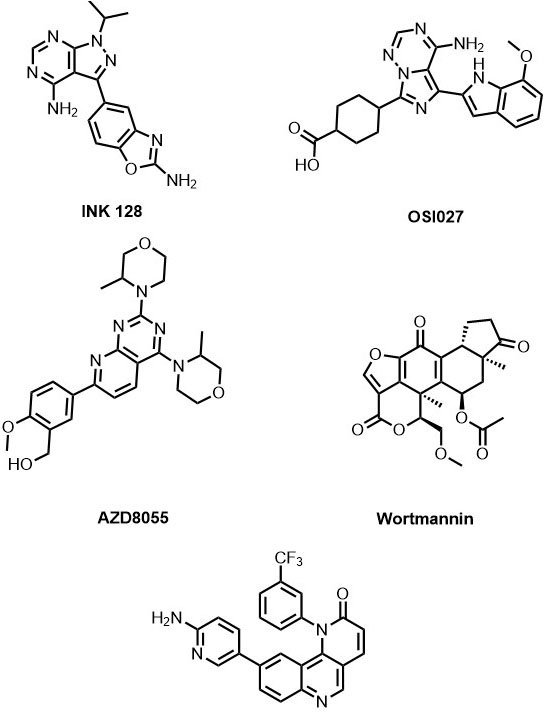 Figure 6
Figure 6List of mTOR inhibitors.
Due to its inability to form three predicted hydrogen bonds with Asp2195, Asp2357 and Tyr2225, provides an opportunity to develop next-generation Torin2 analogs to mimic these interactions. In our recent study, we have developed a series of Torin2 analogs for mTOR/ATR kinase inhibition According to the studies, compound-1, a piperazine analog, exhibited more selectivity towards mTOR kinase (EC50 = 50 nM in HCT 116) in presence of ATR kinase (EC50 = 250 nM in HCT 116). Further, the docking studies of one of the piperazine analogs (1) into the mTOR kinase structure revealed that the amino group of piperazine ring is able to extend further into the inner hydrophobic pocket and form hydrogen bonding in the inner region (Figure 7) and contributes to the selectivity of compound-1 towards mTOR.
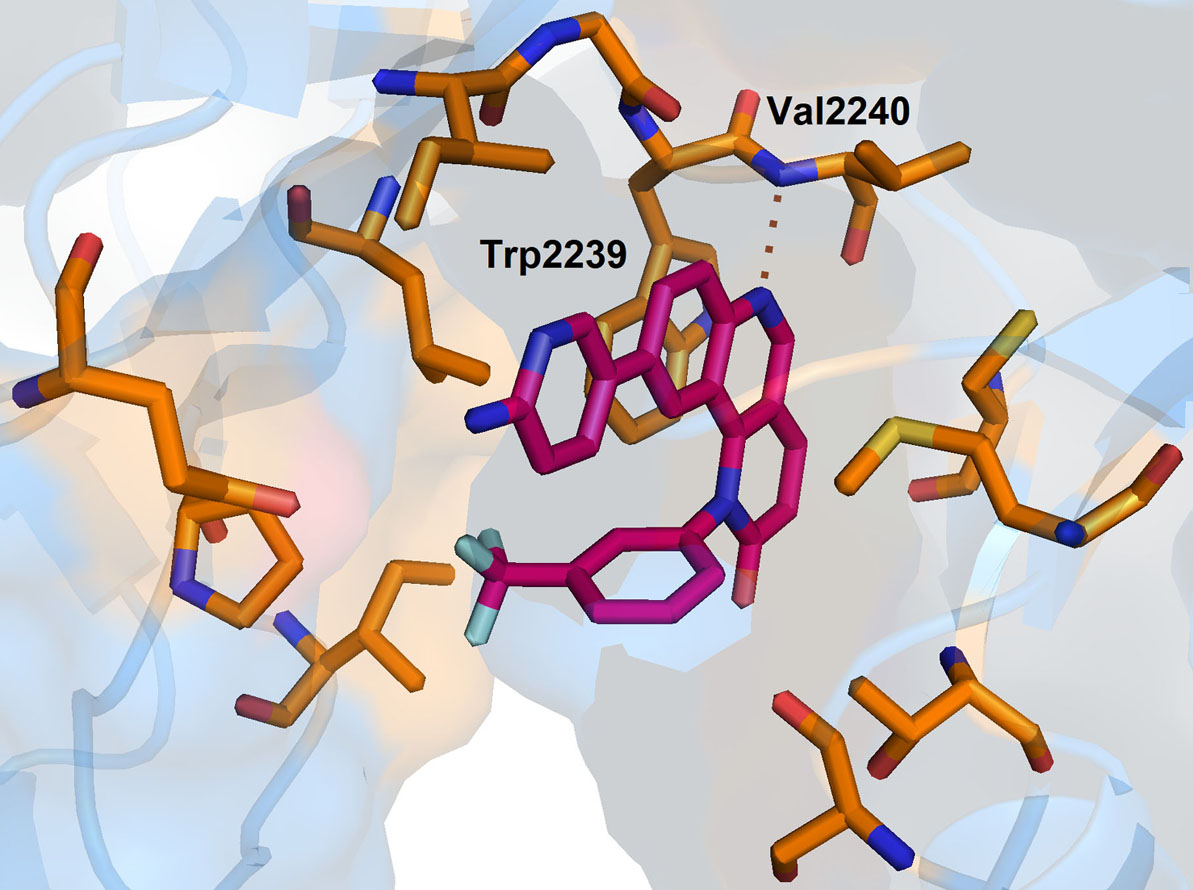 Figure 7
Figure 7Binding pose of Torin2 with mTOR. Protein structure is highlighted in blue colour and interacting residues are represented as tube model (orange colour). Inhibitor is displayed as tube model (magenta colour).
INK128 (Sapanisertib) is a 1H-pyrazolo[3,4-d]pyrimidine derivative developed by Interllikine. It is an ATP competitive inhibitor of mTOR and has a potent inhibitory activity against both mTORC1/mTORC2 (IC50 =1 nM). It has successfully inhibited colorectal cancer cell growth and survival, induced both apoptotic and non-apoptotic cancer cell death (37) and thus, has entered into clinical trials for the treatment of colorectal cancer. It is also in phase I/II clinical trials for the breast cancer, endometrial cancer, glioblastoma, neuroendocrine tumors, ovarian cancer, thyroid cancer, and urogenital cancer (38), (33).OSI027 (39) and AZD8055 (40) are the other examples of potent and selective ATP -competitive inhibitors of mTOR with IC50 values of 22 nmol/L and IC50 of 0.8 nmol/L respectively. OSI027 is in phase I clinical trials against advanced solid malignancy (41), (39). On the other hand AZD8055 is a morpholino substituted of pyrido[2,3-d]pyrimidin-7-yl derivative discovered by AstraZeneca. It has completed phase I trials for Adults With Recurrent Gliomas (NCT01316809), advanced tumors (NCT00731263 ), and entered into phase II clinical trials.
Wortmannin is a fungal metabolite produced by Penicillium wortmanni that acts against phosphatidylinositol 3-kinase (PI3K) (Figure 6). It is an irreversible inhibitor which binds covalently to the kinase (Lys residue) with the help of an electrophilic C-21 position on the furan ring (42). It is also known to inhibit mTOR kinase activity with IC50 = 0.2µM. However, the use of Wortmannin as a drug is limited because of poor solubility in a biological system. PX866 is a Wortmannin derivative developed by Ihle NT et al. (43)with improved stability and less toxicity in comparison to the parent compound. However, PX866 lost its potency against mTOR.
Replicating mammalian cell is constantly under exogenous and endogenous stress to duplicate and inherit the genetic material without any flaw, which subsequently affects the integrity of the genome. In order to overcome this adverse effect and maintain the genomic stability, eukaryotic cells developed a highly organized and coordinated network of cellular events called DNA damage and response (DDR) where they identify, and repair the DNA lesions by transiently halting the cell cycle events (44), (45). Ataxia-telangiectasia mutated (ATM); a 350-kDa apical protein kinase is one of the members of the PIKK family involved in the DDR pathway. Lesions in DNA structures induced by DNA damage agents or DNA replication stress activate DDR signaling pathway. DNA double-strand breaks: DSB (ionizing radiation) activates ATM, which can phosphorylate BRCA1 (Breast Cancer gene 1), and Chk2 kinase leading to activation of p53 followed by G1/S arrest or apoptosis (46). The Mre11-Rad50-Nbs1 (MRN) complex chiefly regulates the localization of ATM kinase to DSB (47). Studies have shown that MRN is one of the first factor sensed and recruited to DSBs (48). The unwinding of DNA ends by MRN complex stimulates the activity of dimeric/oligomeric ATM kinase ATM kinase was discovered in ataxia-telangiectasia (AT) patients, characterized by dilated blood vessels and progressive neurological decline. AT result in gait abnormality and lack of involuntary movement. AT patients are hypersensitive to IR radiation and are defective in DSB repair as well as the G1/S, intra-S, and G2/M checkpoints. The defective checkpoints in AT cells are mainly due to lacking ATM kinase (49). ATM is orthologous to Saccharomyces cerevisiae Tel1 (telomere maintenance 1) (50), a protein involved in controlling telomere length, DNA repair, and cell-cycle checkpoint control. ATM also phosphorylates the S139 in the C-terminal tail of histone variant H2AX in response to DSBs, resulting in discrete γ-H2AX foci at the DNA damage sites (51). H2AX phosphorylation is a general cellular response to processes involving DSB intermediates. In a most recent study, ATM inhibition is reported to suppress Epithelial-to-Mesenchymal transition (EMT) and metastasis in cisplatin resistant lung cancer cells by direct downregulation of JAK (Janus kinases)/STAT3 (signal transducer and activator of transcription proteins)/PD-L1(programmed death-ligand 1) signaling cascade (52).
In 2016, Xuejuan wang et al., carried out the structural analysis of homodimeric ATM/Tel1 using cryoelectron microscopy (Cryo EM) (52). The N-terminal of all PIKK family mainly composed of HEAT repeats forming a super-helix or solenoid. However, the N-terminal helical solenoid in cryo-EM of ATM/Tel1 has displayed a winding tertiary structure with two arms and segmented them as C-pincer and N-spiral. The C-pincer of the N-terminal helical solenoid is connected to the FAT domain of the ATM/Tel1 kinase and N-spiral of the N-terminal helical solenoid is positioned away from the dimer interface of ATM. Further, these two segments interact with FAT and kinase domain of ATM/Tel1. In which, one tip of the C-pincer binds to TRD2 region of the FAT domain, and another tip binds to the C-lobe of kinase domain as well as TRD1 region of the FAT domain. Whereas, the N-spiral connect to another region within the TRD2 domain. Hence, the entire catalytic core of ATM/Tel1 appears to be tightly combined to the N-terminal helical solenoids. Besides, in the dimer structure of ATM/Tel1 a rod-shaped protrusion leaned on the LST8-binding element (LBE)-like insertion in the C-lobe of the kinase domain has been observed and named as LBE interacting Domain (LID). In the dimeric state LID of monomer interact with LBE of another monomer and blocks the putative binding of ATM regulators to LBE. It is assumed that the LID could play a crucial role in the inhibition of ATM/Tel1 kinase activity in the presence of ATM regulators.
In another study, Domagoj et al., reported the cryo-EM structure of purified human ATM dimer at a resolution of 5.7 Å (54). Their structural work highlights the open and closed ATM dimer. Open dimer has a limited intermolecular interface and compatible with substrate binding, suggesting that the open dimer might be a more active form of ATM. Whereas, closed dimer has a large intermolecular interface, in which PRD binds as pseudo-substrate and block the closed dimer. The transition between open to closed dimers are mainly regulated by the compact arrangement of FAT C-terminal (FATC), LST8-binding element (LBE), activation loop, and PIKK regulatory domain (PRD; helices kα9b, kα9c, and kα9d) . Further in the ATM dimer the FAT domain wrapped around the kinase domain with polar interaction involving Glu1959-Arg2849, Arg2486-Glu2950, and Gln2522-Gln2730. These residues are highly conserved in ATM and mutation of R2849P was observed in the patient associated with A-T disease, suggesting that these interactions are important for stabilization of ATM.
NVP-BEZ235 is an imidazoquinoline derivative that has potent inhibitory activity against ATM and DNA-PKcs, both of these kinases being activated in response to IR radiation induced DNA damage (55). NVP-BEZ235 inhibits the phosphorylation of ATM and DNA-PKcs targets, thereby, blocking both nonhomologous end-joining and homologous recombination DNA repair pathways resulting in significant attenuation of DSB repair. In 2017, NVP-BEZ235 completed phase I study in patients with Advanced Solid Malignancies Enriched by patients with Advanced Breast Cancer and entered into phase II study (56).
3-Quinoline carboxamide derivatives are the potent, selective and orally active ATM kinase inhibitors developed by Astra Zeneca (57). They have reported the crystal structure of 3QH (IC50 = 0.049 µM) with PI3Kγ, a known homology of ATM (PDB Id :5G55). In the crystal structure, the quinoline nitrogen forms the hydrogen bonding with Val882 residue present in the backbone of ATP binding site (Figure 8). Additional hydrogen bond interaction between the cyano group and Tyr867 helps in further stabilization of the complex. This investigation gave the opportunity to perform SAR (structure-activity relationship) studies of compound-3 on ATM kinase activity and led to the discovery of compound-4 with excellent ATM kinase selectivity (IC50= 0.033 µM) and better rodent oral ADME (absorption, distribution, metabolism, excretion) properties.
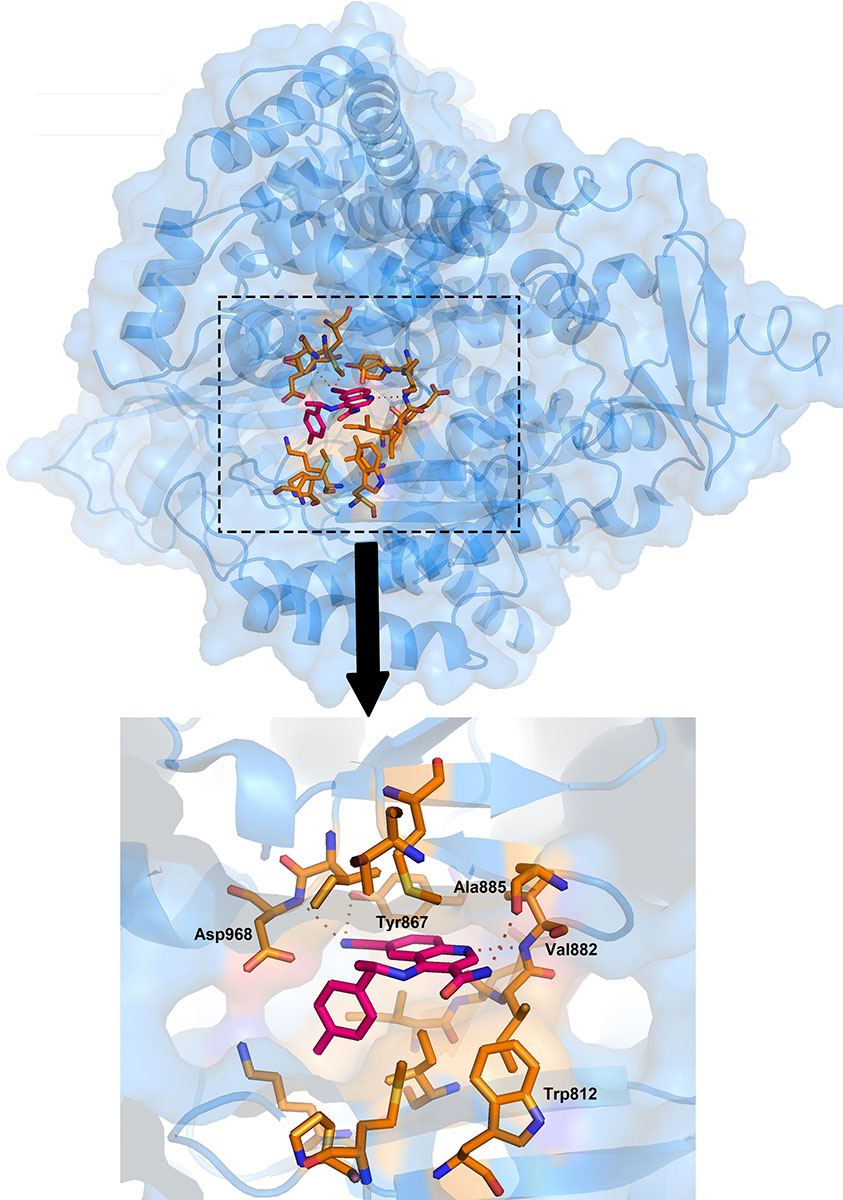 Figure 8
Figure 8Zoomed image showing the binding pose of 3QH with ATM. Protein structure is highlighted in blue colour and interacting residues are represented as tube model (orange colour). Inhibitor is displayed as tube model (magenta colour).
Recently, AstraZeneca has identified cinnoline 3-carboxamide derivatives as selective ATM inhibitor with IC50 values 0.0028 µM (compound 21) (58). When compared to quinoline 3-carboxamide derivatives, compound 21 showed improved physicochemical properties and pharmacokinetic properties. Further, when administered with irinotecan, compound 21 showed excellent tumor regression in the SW620 colorectal tumor xenograft model and superior inhibition to only irinotecan treated tumor xenograft model. Currently, compound 21 has enetered into preclinical trials for the treatment of colorectal cancer (Figure 9).
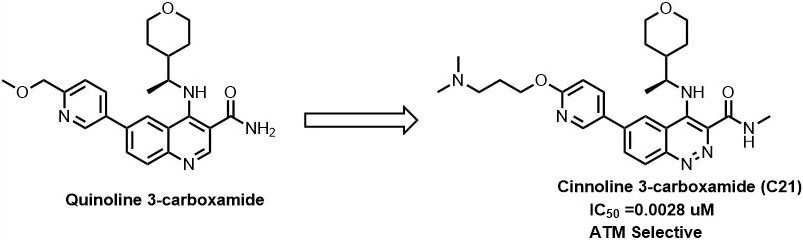 Figure 9
Figure 9Optimisation of quinoline derivatives for ATM inhibition.
Ataxia-telangiectasia mutated- and Rad3-related (ATR) is the important controller for checkpoint response to damaged DNA and incomplete replication through stalled replication forks (59). ATR is a 2644 amino acid residue long protein with a molecular weight of 300 kDa that is structurally and functionally quite similar to ATM (60). ATR activation is associated with UV (ultraviolet) light irradiation and replication fork stalling. When DNA damaging agents cause the DNA polymerase to stall during the replication and helicases continues to unwind the DNA, leading to the generation of a stretch of single-strand DNA (ssDNA). The ssDNA is recognized by replication protein A (RPA) and recruits the ATRIP/ATR complex (61). ATR-interacting protein (ATRIP) having a molecular weight of 85 kDa binds to the N-terminus of ATR. Further, ssDNA-RPA complex stimulates the loading of RAD9–HUS1–RAD1 (9–1–1) heterotrimer onto the DNA ends (62). Consecutively, the 9-1-1 complex recruits TopBp1 to activate ATR (63). Activated ATR phosphorylates the Ser317 and Ser345 on Chk1 kinase leading to S and G2 arrest (60), (64). Further ATR also phosphorylates replication factor C complex, RPA, and RPA2, the MCM (mini chromosomal maintenance) 2-7 complex, and BRCA1 (59), (65). The phosphorylation of these substrates is important for inhibition of replication, recovery of replication and activation of NHEJ (non-homologous end joining) and HR (homologous recombination) DNA repair pathway (66).
Recently, Qinhui Rao et al. solved the cryo-EM structure of the human ATR-ATRIP complex at 4.7Å resolution and constructed an atom model of the C-terminal catalytic core of ATR (residues 1 521-2 644) at 3.9 Å using homology model of mTOR crystal structure (Figure 10) (67). Similar to other PIKK family, ATR consist of N-terminal α-solenoid consist HEAT repeats, a FAT (FRAP, ATM, TRRAP) domain, a kinase domain (KD), and a C-terminal short segment referred to as FATC. The N-terminal segment (1-1636 amino acids) of ATR has two HEAT repeats, the N-HEAT and M-HEAT. N-HEAT consists of HEAT 1 – HEAT 26 repeats, in which HEAT1- 16 form a right-handed super-helical α-solenoid, whereas HEAT 17 to HEAT 26 repeats attain extended conformation, and the last HEAT-26 repeat of the N-HEAT binds directly to the FAT domain. On the other hand, the M-HEAT has 12 α-helices arranged together to adopt C-shaped conformation and binds to the FAT and the KD domains. Followed by HEAT repeats there are two tetratricopeptide repeats TPR1 and TPR2, present in the FAT domain. Similar to other PIKK kinases ATR kinase domain has two lobes, N-lobe, and the C-lobe. N-lobe consist of six-stranded anti-parallel β-sheet (kβ1-kβ6) stabilized by the helix Kα1 and the C-lobe consist of eleven α-helices (kα2-kα12). The N-lobe of ATR has a unique insert (residues 2270-2290) connecting strands kβ1 and kβ2. A similar insert was also noticed in ATM kinase with weak sequence homology, suggesting distinct inter-domain interactions. The key catalytic residues in the ATR kinase domain are highly conserved with respect to mTOR. Residues K2327 and D2330 in ATR helps in ATP binding, whereas residues N2480 and D2494 stabilize Mg2+ for catalysis. Residue D2475 and H2477 needed for activating the hydroxyl group of ATR substrates, and electrostatic stabilization of the transition state. Mutations of above-mentioned residues led to the reduced kinase activity of ATR in the ATR-ATRIP complex, suggesting their significant role in catalysis.
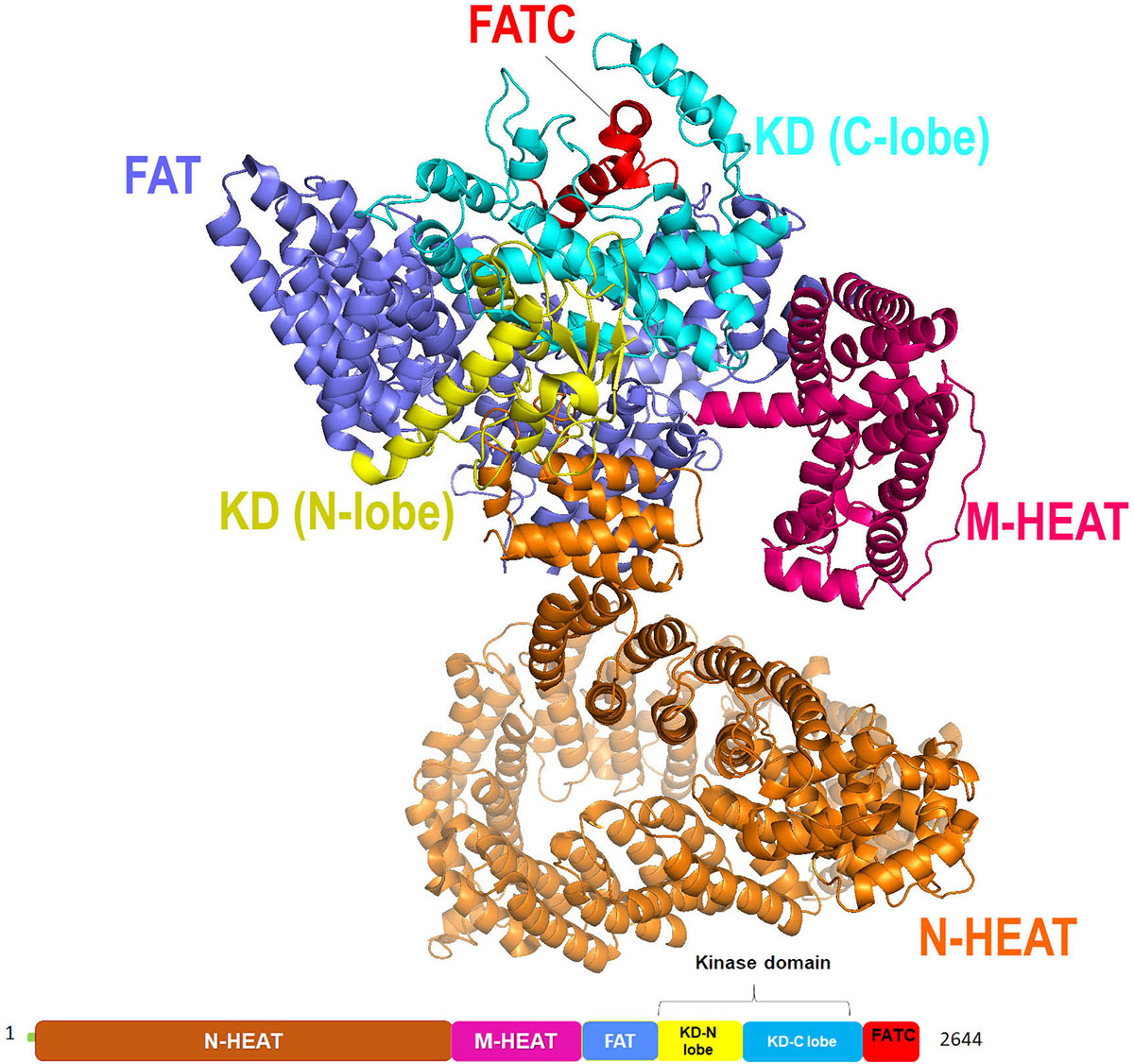 Figure 10
Figure 10Cryo-EM structure (at resolution 4.7 Å) of ATR kinase represented as cartoon with color-coded domain (adapted with permission from PDB ID: 5YZ0).
It has been proposed that targeting the kinase domain of ATR with small molecules, ATP competitive inhibitors could potentiate the effect of radio and chemotherapy for various human cancers. VX-970 was one such ATR kinase inhibitor currently in early-phase clinical trials for the treatment of solid tumors (68), (69). VX-970 is a potent and selective inhibitor of ATR developed by vertex pharmaceuticals. VX-970 inhibited ATR kinase activity with IC50 of 19 nM in HT29 cells and sensitizes various types of tumor cell lines and cancer Docking of VX-970 into the cryo-EM structure of ATR kinase displayed that pyrazine ring occupies the adenine of ATP, Oxazole ring extended close to the ribose pocket of ATP, and the sulfonyl group occupies the γ-phosphate group of ATP, thereby preventing the competitive binding of ATP to the ATR kinase catalytic site (67).
DNA-PKcs is a 470-kDa enormous protein kinase in PIKK family. The holoenzyme comprises of a DNA dependent protein kinase catalytic subunit (DNA-PKcs) and an autoimmune antigen heterodimer Ku70/Ku80 playing an important role in double-stranded break repair through non-homologous end joining (NHEJ) (71). In the NHEJ pathway, DNA-PKcs is known to phosphorylate its physiological substrates, preferably at SQ/TQ consensus sequences, including Ku70, Ku80, XRCC4 (X-ray repair cross-complementing protein 4), XLF (XRCC4-like factor), Artemis and PNKP (Polynucleotide Kinase 3'-Phosphatase) with few exceptions like SAF-A (scaffold attachment factor A) /hnRNP-U (heterogeneous nuclear ribonucleoprotein U), a non-NHEJ protein (72), (73), (74). It also exhibits autophosphorylation at multiple SQ/TQ (T2609, S2612, T2638 and T2647) as well as non-SQ/TQ sites (S2624, S3205) in vitro. DNA-PKcs interaction with protein phosphatase 6 (PP6) specifically through PP6R1 (protein phosphatase 6, regulatory subunit 1) subunit is required for its activation after DNA damage, also, siRNA(small interfering RNA) mediated depletion studies and inhibition studies of DNA-PKcs with inhibitor NU7441 highlights on the role of the protein in mitosis (75). In addition to the well-established functions, DNA-PKcs are also known to play important roles in transcription, in viral infections especially in HIV (Human Immunodeficiency Virus)-induced cell death in CD4+ T cells and in proteasome-mediated destruction in Herpes Simplex Virus-infected cells. DNA-PKcs is required for telomere capping and it also phosphorylates Golgi protein GOLPH3 (Golgi Phosphoprotein 3) resulting in Golgi fragmentation and dispersal in cytoplasm (76), (77).
DNA-PKcs structure is reported both through single-crystal X-ray diffraction as well as through cryo-EM (Figure 11). According to the crystal structure solved at a resolution of 4.3 Å, it is fractionated in three major parts a core, body and a tail. The ‘core’ region is a compact structure comprising of FAT domain, a kinase domain and FATC domain broadly labeled as Core DNA-PKcs. The ‘body’ is formulated of N-terminal α solenoids and ‘tail’ has KU70/80-DNA complex. The catalytic domain in DNA-PKcs is homologous to the kinase domain of the PI3Kγ of the classical PI3-kinases. While by the cryo-EM description of the protein structure, the comparative study was performed on Michigan Cancer Foundation-7 (MCF-7) cell line (siRNA knockdown of DNA-PKcs), MO59-J cells (lacks DNA-PKcs expression) and MO59-K cells (normal DNA-PK activity) which inferred that out of all the selective inhibitors, N-terminal region of the protein, subdivided into ‘arm’ and ‘bridge’, is L-shaped, followed by HEAT repeats forming a cradle-like structure. The cradle is connected to the C-terminal head region (FAT, FRB, KD, FATC) via ‘bridge’ (as mentioned earlier). As per the cryoEM model (4.4 Å) of DNA-PK and previous reports pertaining to the NMR model of Ku80, extra electron density towards the N-terminal ‘arm’ region hint towards the probable binding site of Ku80. In addition, there are two perpendicular apertures in the structure, first aperture formed due to the circular cradle and the second aperture formed by the arm and the bottom part of the circular cradle. Ku bound DNA is assumed to pass through these apertures during the DNA repair process (78), (79), (80).
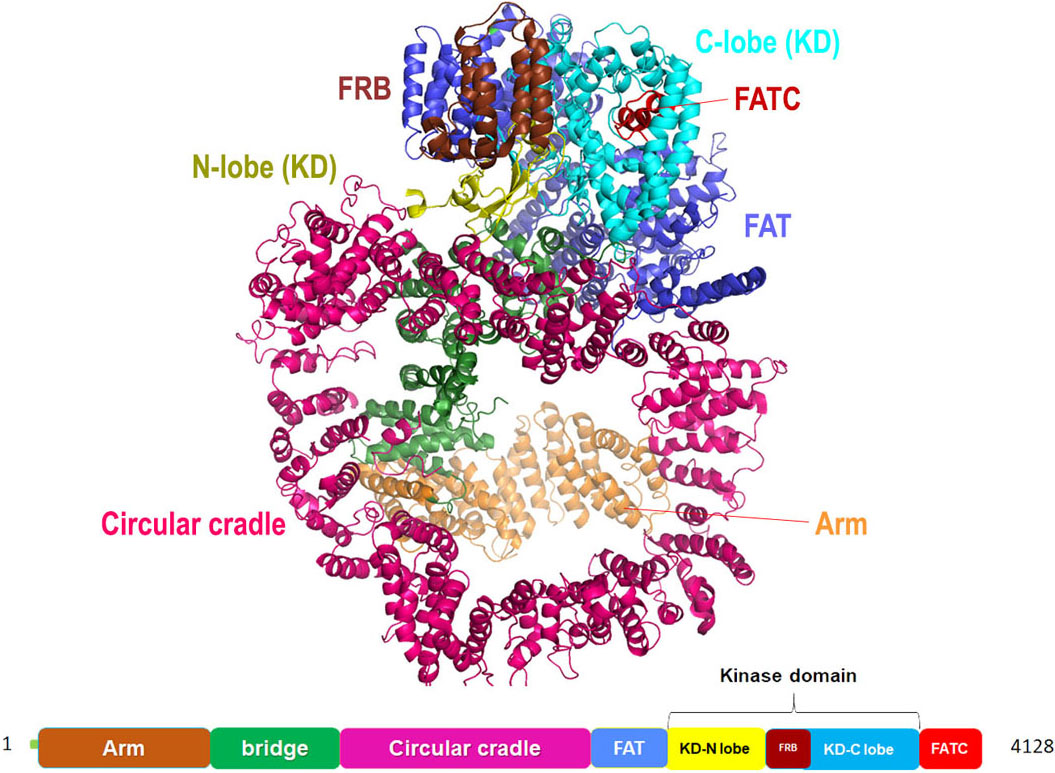 Figure 11
Figure 11Cartoon representation for Cryo-EM structure (at resolution 4.4 Å) of DNA-PKcs kinase with color-coded domains (adapted with permission from PDB ID: 5W1R).
There are several small molecule inhibitors identified with respect to their DNA-PKcs inhibition ability like wortmannin (IC50: 0.10 µM), SU11752 (IC50: 0.13±0.028 µM), LY294002 (Ki: 6 µM). While wortmannin is a non-competitive irreversible inhibitor, SU11752 and LY294002 act as competitive inhibitors for DNA-PKcs (81), (82). A NU7026 is 50 folds more selective towards DNA-PKcs than other PIKK members with an IC50 of 0.23 µM (83). The effect of drug exposure on the relatively radiosensitive CH1 human ovarian carcinoma cell line was also examined which exhibited that at least 4h exposure at 10 μM NU7026 is necessary with 24h exposure producing an even greater effect (84). However, the clearance rate of NU7026 was observed to be quiet fast due to monohydroxylation that occurs on position-2 of the morpholino group resulting in the opening of the ring (85). Hollick et al. also predicted that a 6-arylpyran-4-one or 6-arylthiopyran-4-one template, bearing the essential 2-morpholin-4-yl group, would offer more opportunities for introducing structural diversity in the aromatic region, while retaining the core pharmacophore elements common to NU7026 (86) (Figure 12). Similarly, the derivatives based on NU7026 scaffold show considerable interaction with Lys 3749, Arg3733, Leu 3802, Tyr3824 and Asp3937 forming hydrogen bond linkage (86).
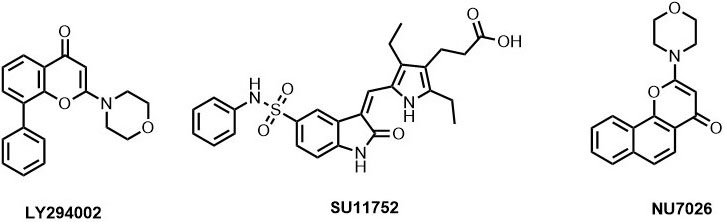 Figure 12
Figure 12ATP-competitive DNA-PKcs inhibitors.
Transformation/transcription domain-associated protein (TRRAP) is a 430 kDa adaptor protein encoded by TRRAP gene that acts as an essential cofactor for c-Myc and E1A/E2F oncogenic transcription factor pathways (18), (88). TRRAP is a highly conserved protein and the C-terminal of this protein shares homology with phosphatidylinositol 3-kinase (PI-3 kinase) family. However, unlike other members of the PIKK family, the key residues responsible for the kinase activity and required for the binding of ATP are missing in the sequence (89). It forms a stable component of MRN (MRE11, RAD50, and NBS1 ) complex where TRRAP-MRN complex are assumed to have a role at the DSB repair sites that promotes repair fidelity (90). TRRAP containing HAT complexes are known to participate in DNA repair by histone acetylation by chromatin remodeling (91). It is also reported to be an essential requirement along with TIP60 (Tat interactive protein) for the transcriptional activation of histone gene where they are recruited to histone gene promoters at the G1/S-phase boundary by NPAT (nuclear protein of the ATM locus). NPAT has a novel DLFD motif that is the interaction locus for TRRAP (92). The TRRAP forms a complex with hGCN5 (Histone acetyltransferase GCN5) that is reported to interact with the functionally important MbII (Myc homology box II) N- terminal domain of c-Myc which further modifies nucleosomal packaging at specific chromosomal targets (93). Also, it is a component of the TATA-binding protein (TBP)-free TAFII-containing complex (TFTC) and a common subunit of TIP60 and PCAF (P300/CBP-associated factor) complex that forms a connection between Myc/Max and/or E2F/DP (dimerization partner) and the basic transcriptional machinery and is assumed to modulate acetylation of nucleosomal histones by stimulating catalytic activity (94), (95). TRRAP, like other PIKK family of proteins, shows interaction with p53 tumor suppressor protein. The p53 protein employs TRRAP/HAT (histone acetyltransferases) complex to mdm2 (Mouse double minute 2 homolog) promoter resulting in increased histone acetylation (96). This protein also plays an important and distinctive role in regulating cell cycle progression where it affects embryonic development and cell proliferation. The cells with disrupted Trrap (the murine homolog of TRRAP), cease to undergo G2/M arrest even after unattached chromosome or spindle disruption (97).
There are several homologues of TRRAP in various other species like Trrap in Mus musculus, Tra1 (Transcription-associated protein 1) in Saccharomyces cerevisiae, trr-1 in Caenorhabditis elegans, and Nipped-A in Drosophila melanogaster however the structural report for only Tra1 is elucidated using cryo-electron microscopy at a resolution of 3.7 Å. As described in the reports, the Tra1 has a similar arrangement as in mTOR and ATM but it shows a striking similarity to human DNA-PKcs, especially with respect to their ‘diamond ring’ like topology. The Tra1 has three well-defined structural units namely N-terminal unit (1-824 amino acids), a circular cradle (825- 2630 amino acids), and C-terminal unit (2631- 3744 amino acids). Starting from the N-terminal, there are 49 HEAT repeats and 15 TPR (tetratricopeptide) repeats. This region forms two α-solenoids that forms 80% of the total mass of Tra1. Also, this is the domain that displays maximum variation when compared to DNA-PKcs. This is followed by the FAT domain which is similar to PIKK family members. The FRB domain that lies on the N-Terminal of kinase domain also shows conformation variation. Unlike DNA-PKcs and mTOR, FRB moves away from the N-lobe and directly packs against the enclosing the Tra1 active site. This is followed by kinase domain (KD) that has two lobes spanning ~500 amino acid residues and FATC domain. It also has a pseudokinase domain comprising of 17 residues that are reported to stabilize the active site.
While going by the diamond ring analogy, as reported, the entire protein can be sectioned into three parts; ring (HEAT domain), setting (FAT and FRB) and center stone (KD and FATC). Further, the ring part/ HEAT domain can be subdivided into three parts finger, clasp, and ring. The first α-solenoid starting from the ‘finger’ spans from H1 to H16 and is equivalent to the spiral region of mTOR/ATM and arm/bridge region of DNA-PKcs. After H16, the loop region traverses over along with the two-stranded β sheet. The second and the largest α-solenoid follow this from H17-H49 that closes the ring with a diameter of 125 Å. In addition, the FAT, FRB, KD and FATC regions form the head of the ring where the FAT domain has 15 TPR repeats (TPR1 – TPR 15). The kinase domain fold is very similar to the fold in DNA-PKcs and mTOR but interestingly the residues required for the binding of ATP/Mg and responsible for the kinase activity are not conserved in Tra1, rather it has an extra 18 residue insertion (98), (99).
The other significant member of the PIKK protein family is hSMG1. Similar to other PIKKs, it is a huge protein with 3661 amino acid residues and has a molecular weight of 410 kDa. It participates in nonsense-mediated mRNA decay surveillance mechanism by interacting with the highly conserved mRNA surveillance complex that includes hUPF1/ SMG2, hUPF2, and hUPF3. SMG1 is reported to phosphorylate the C-terminal of hUPF1 at SQ motifs forms an SMG1C complex in combination with SMG8 and SMG9 during the phosphorylation process. The SMG1C facilitates the NMD (nonsense-mediated mRNA decay) initiation by recruiting UPF1 and UPF2 in close proximity, thus improving their accessibility to the kinase domain (101). At the same time, the mutated SMG-1 with loss of function mutation in catalytic domain ceases to display commendable phosphotransferase activity, hence, confirming its role as a kinase (102). There is evidence that supports the fact that UPF1 phosphorylation by SMG1 is not a direct phenomenon, rather it is triggered independently by UPF2 or UPF3b in addition to the other members of SURF i.e. SMG-1-Upf1-eRF1-eRF3 complex and its association with exon junction complex (EJC) (103), (17). In addition to this, the hSMG1 activity is reported to be inhibited by wortmannin, a known inhibitor for mTOR. However, despite the presence of the FRB domain, the hSMG1 is not sensitive to rapamycin (104). It is reported to be activated during genotoxic stress and is functionally relatable to ATM kinase. Also, it is stimulated by exposure to UV or IR radiation (105). Other than its central role in NMD, hSMG1 also participates in protection against TNFα (Tumor Necrosis Factor-alpha) induced cell death by providing stability to FLIPL (FLICE-like inhibitory protein, long form) protein and diminishing the impact of caspase-8/10 dependent apoptotic cascade (106).
The molecular architecture of hSMG1 is similar to other six members of the PIKK family of kinases with a conserved kinase domain that participates in autophosphorylation as well as substrate phosphorylation. As described through cryo-EM studies, the hSMG1 structure comprises of a dense ‘globular head’ and a ‘thinner arm’. The globular head corresponds to the C-terminal FAT, FRB, KD and FATC domains that form a cleft for ATP binding, when compared to mTOR structure. Whereas, the thinner bent arm corresponds to the N-terminal region populated with HEAT repeats similar to DNA- PKcs (101). The super-helical HEAT repeats in hSMG1 are comparatively shorter when compared to other PIKK members (107), (108). In addition to the signature domains of PIKK family, hSMG1 has an additional insertion of ~1200 residues between the catalytic domain and FATC domain that is absent in other family members like mTOR, ATM, ATR, DNA-PKcs or TRRAP. This C-terminal insertion has predominant α helices and it functions as the scaffold to engage the substrate in close vicinity to the kinase domain On comparing the structure of SMG1 with SMG1C, it is evident that the HEAT repeats in the arm region rotate on interaction with the SMG8-SMG9 proteins (101).
Owing to its important role in genome surveillance non-sense mediated mRNA decay, hSMG1 is predicated as an important target for cancer therapy.
In 2012, Ariamala Gopalsamy et al. (110), investigated the pyrimidine derivatives as hSMG1 kinase inhibitors. In their study, they have identified compound-5 as potent hSMG1 inhibitor with IC50 = 0.45µM. Docking of compound-5 into the PI3K, a homology of hSMG1 has revealed that compound-5 attained a ‘U’ shaped conformation. The nitrogen atom of the pyrimidine ring is required for hydrogen bonding with Val 882 located in the hinge region of ATP binding pocket (Figure 13).
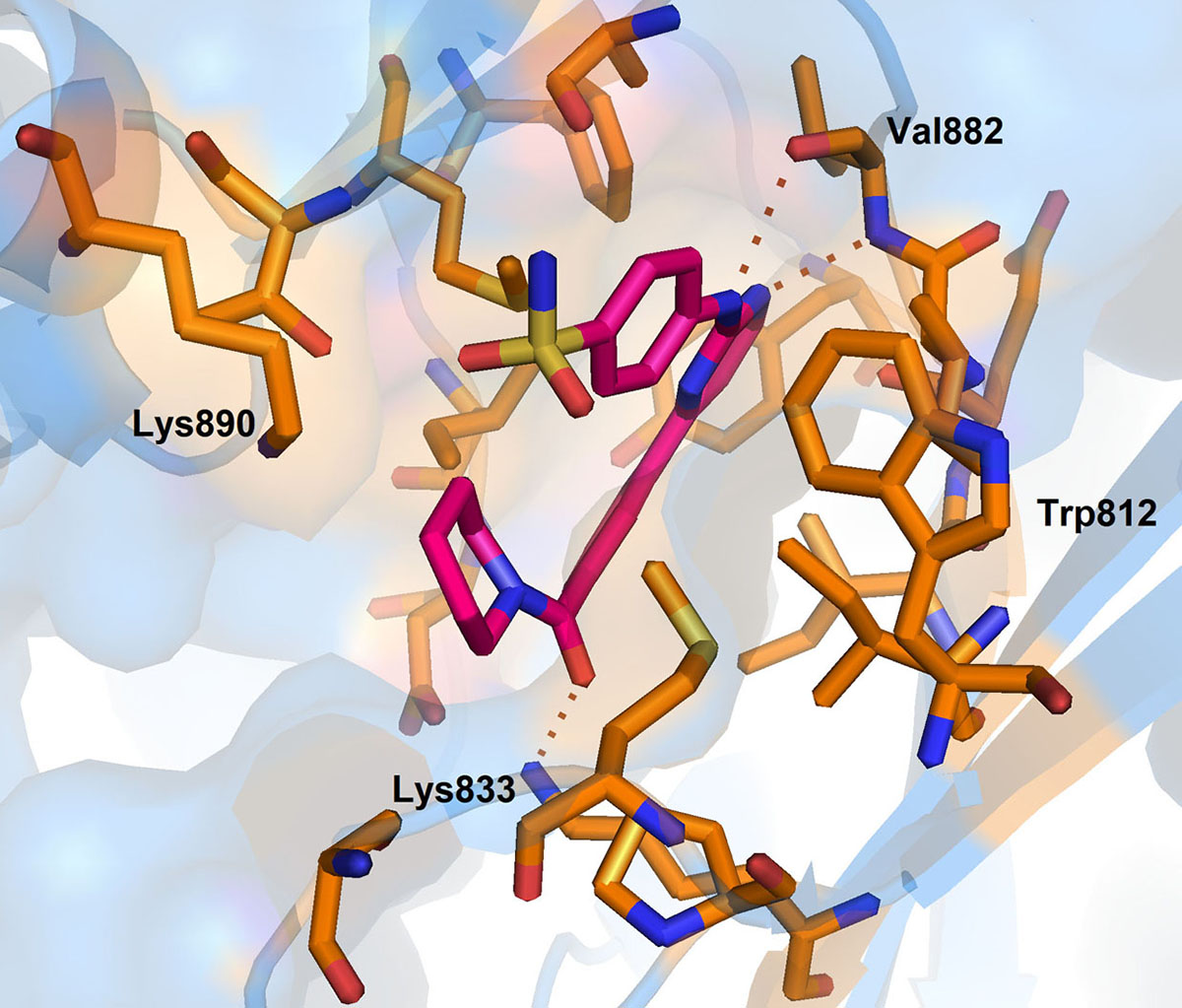 Figure 13
Figure 13Binding pose of 0VU with hSMG. Protein structure is highlighted in blue colour and interacting residues are represented as tube model (orange colour). Inhibitor is displayed as tube model (magenta colour).
Further, the complex is stabilized by multiple hydrogen bonding interaction between the amino group of pyrimidines amine scaffold and carbonyl oxygen amide group with Val 882 and Lys 833. Surprisingly, the p-sulphonamide ring did not make any interaction with active residues in hSMG1. In addition, compound-5 showed certain off-target reactivity towards CDK1 (cyclin dependent kinase 1) and mTOR kinase. Further optimization of compound-5 led to identification of compound-6 with excellent hSMG1 selectivity (IC50 =0.003µM) (Figure 14). Changing the sulfonamide substitution from para to meta position has led to the formation of hydrogen bonding interaction with the backbones of Gly884 and Ala885(110). This additional hydrogen bonding could be reasoned for excellent selectivity of compound-6 towards hSMG1 in presence of other kinases. These key residues in the hSMG-1 binding site can be explored to improve kinome selectivity of other chemical scaffolds targeting hSMG1.
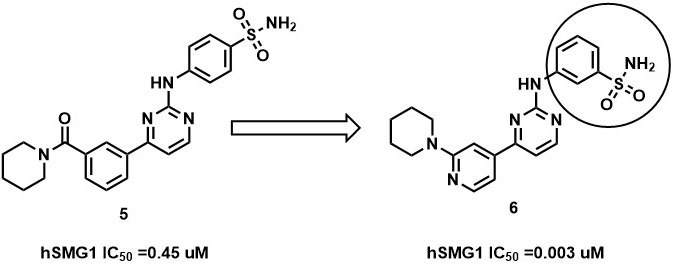 Figure 14
Figure 14SAR of hSMG1 inhibitors.
For the last two decades, there has been extensive research on the PIKK family of the kinase in understanding their diverse role in various signaling pathways. Several biological aspects of PIKK family members have been revealed and gave valuable insights with respect to biochemical, cellular and their mechanistic function. This review contributes as an overview of six PIKK family members with particular emphasis on the domain architecture, activators, themed localization, phosphorylation of downstream substrates, and their roles in signaling pathways that trigger cancer cell proliferation and survival. Six human PIKK family of kinases have been identified till date, which includes Ataxia telangiectasia mutated kinase (ATM), ATM- and Rad3-related kinase (ATR), DNA dependent protein catalytic subunit (DNA-PKcs), mammalian target of rapamycin (mTOR), and suppressor with morphological effect on genitalia family member (SMG1) and transformation/transactivation associated protein (TRRAP). All these kinases exhibit structural similarities with PI3K with respect to the mechanisms of regulating kinase activity.
The insights in the architectural and functional aspects of these kinases have unveiled numerous portentous questions for future. Despite exhausting research on the varied cellular aspects of the PIKK family of kinases, the studies elucidating the structure of these atypical kinases is meager. This can be attributed to the fact that due to their enormous sizes, the expression and purification of these complexes to a concentration sufficient for X-ray crystallographic studies is apparently a challenging assignment. Their organizational inflexibility hampers the structure elucidation studies. In addition to this, their tendency to interact with protein and DNA macromolecules further adds to the intricacy in their characterization. Although drug discovery is at its technologically and genomically advanced era referred to as ‘precision medicine’(120), yet structure-based drug design can help researchers to synthesize inhibitors with minimized cross-reactivity, hence, it needs more thoughtfulness and specific approach for the divulgence.
Deekshi Angira, Althaf Shaik contributed equally to this paper. We gratefully acknowledge the Indian Institute of Technology Gandhinagar and DST-FIST Department of Science and Technology, India (Project No. SR/FST/CSI-277/2016) for financial support and Schrodinger suite. The authors declare no conflict of interest related to the work.
PIKKs
Phosphatidylinositol-3 kinase-related kinases
deoxyribonucleic acid
DNA damage response
Suppressor with morphological effect on genitalia family member
Ataxia telangiectasia mutated kinase
ATM- and Rad3-related kinase
DNA dependent protein catalytic subunit
mammalian target of rapamycin
Transformation-transactivation domain-associated protein
Huntingtin elongation factor 3 (EF3)
protein phosphatase 2A
and the yeast kinase TOR1
Frap ATM and TRRAP
FAT C-terminal
PIKK- regulatory domain
ATR interacting protein
Mre11-Rad50-Nbs1
up-frameshift
messenger ribonucleic acid
FKBP-rapamycin associated protein
Rapamycin and FKBP target
Mammalian lethal with SEC13 protein 8
Negative Regulatory Domain
FK506-binding protein12
FKBP12-Rapamycin-Binding (FRB) domain
Human colorectal carcinoma cell line
DNA double-strand breaks
Breast Cancer gene 1
Telomere maintenance 1
Epithelial-to-Mesenchymal transition
Janus kinases/ Signal transducer and activator of transcription proteins
Programmed death-ligand 1
LST8-binding element
LBE interacting domain
absorption distribution metabolism excretion
Mini chromosomal maintenance
Non-homologous end joining
Homologous recombination
X-ray repair cross-complementing protein 4
XRCC4-like factor
Polynucleotide Kinase 3'-Phosphatase
Scaffold attachment factor A
Heterogeneous nuclear ribonucleoprotein U
Protein phosphatase 6
Protein phosphatase 6 regulatory subunit 1
Small interfering RNA
Human Immunodeficiency Virus
Golgi Phosphoprotein 3
phosphatidylinositol 3-kinase
Tat interactive protein
Nuclear protein of the ATM locus
Histone acetyltransferase GCN5
Myc homology box II
TATA-binding protein
TBP-free TAFII-containing complex
P300/CBP-associated factor
Histone acetyltransferases
Mouse double minute 2 homolog
Transcription-associated protein 1
Tetratricopeptide
Nonsense-mediated mRNA decay
Exon junction complex
Tumor Necrosis Factor-alpha
FLICE-like inhibitory protein long form
Cyclin dependent kinase 1